
Abstract
Cerebral ischemic preconditioning protects against stroke, but is clinically feasible only when the occurrence of stroke is predictable. Reperfusion plays a critical role in cerebral injury after stroke; we tested the hypothesis that interrupting reperfusion lessens ischemic injury. We found for the first time that such postconditioning with a series of mechanical interruptions of reperfusion significantly reduces ischemic damage. Focal ischemia was generated by permanent distal middle cerebral artery (MCA) occlusion plus transient bilateral common carotid artery (CCA) occlusion. After 30 secs of CCA reperfusion, ischemic postconditioning was performed by occluding CCAs for 10 secs, and then allowing for another two cycles of 30 secs of reperfusion and 10 secs of CCA occlusion. Infarct size was measured 2 days later. Cerebral blood flow (CBF) was measured in animals subjected to permanent MCA occlusion plus 15mins of bilateral CCA occlusion, which demonstrates that postconditioning disturbed the early hyperemia immediately after reperfusion. Postconditioning dose dependently reduced infarct size in animals subjected to permanent MCA occlusion combined with 15, 30, and 60 mins of bilateral CCA occlusion, by reducing infarct size approximately 80%, 51%, and 17%, respectively. In addition, postconditioning blocked
Introduction
Stroke is the third leading cause of mortality in America. Despite extensive research for stroke treatment in the past several decades, few neuroprotectants have been successfully translated into clinical application from basic research. Ischemic preconditioning, a brief nonlethal ischemia performed 1 or several days before onset of a subsequent severe ischemia, reduces ischemic damage, an effect also referred to as ischemic tolerance (Schaller and Graf 2002; Dirnagl et al, 2003; Huang 2004; Sharp et al, 2004). Preconditioning is considered to be the most robust endogenous neuroprotectant identified to date for decreasing ischemic damage in the brain. A major purpose of studying preconditioning is to understand the mechanisms underlying its protection against stroke-induced neuronal death, therefore providing clues for acute stroke treatment. However, its clinical application is only possible for cases in which the occurrence of stroke is predictable and controllable.
Revascularizing the occluded blood vessels allowing timely reperfusion is one of the strategies currently being pursued for acute ischemic stroke treatment, for example, by using thrombolytic reagents like t-PA (Fisher and Brott 2003), or mechanical devices (Smith et al, 2005). However, reperfusion itself generates overproduction of reactive oxygen species (ROS) or free radicals, leading to reperfusion injury (Chan, 1996). Despite this well-recognized fact, surprisingly, to our knowledge, no attempt has been made to directly alter the patterns of reperfusion to block reperfusion injury by, for example, controlling the extent of reperfusion or by interrupting the early hyperemic response after reperfusion.
In the research field of myocardial ischemia, it was reported that postconditioning with a series of mechanical interruptions of reperfusion reduces infarct after cardiac ischemia (Zhao et al, 2003b). In addition, a recent exciting clinical report demonstrated that postconditioning after coronary angioplasty and stenting protects the human heart during acute myocardial infarction (Staat et al, 2005). Despite such significant progress made in the treatment of cardiac ischemia, whether ischemic postconditioning protects against cerebral ischemia has not been reported previously. Thus in the current study, we tested our hypothesis that ischemic postconditioning reduces cerebral ischemic damage after stroke using a focal ischemia model in rats.
Materials and methods
Focal Cerebral Ischemia
Experimental protocols were approved by the Stanford University Administrative Panel on Laboratory Animal Care. Focal cerebral ischemia was generated as described previously (Zhao et al, 2003a, 2005). Male Sprague-Dawley rats (350 to 450 g) were used. Anesthesia was induced by 5% isoflurane and maintained with 2% to 3% isoflurane during surgery and early reperfusion. Core body temperatures were monitored with a rectal probe and maintained at 37°C during whole experiments. The bilateral common carotid artery (CCA) were separated and a suture was circled around each artery. The distal middle cerebral artery (MCA) was exposed. For ischemia induction, the bilateral CCAs were occluded first, and then the distal MCA was cauterized above the rhinal fissure 2 mins later (Figure 1, Figure 2A). In a subset of animals with the same experimental condition, the left femoral artery was cannulated with PE-50 tubing to monitor arterial blood gases, which indicated that pCO2 and pO2 of the arterial blood were within a normal range during experiments.
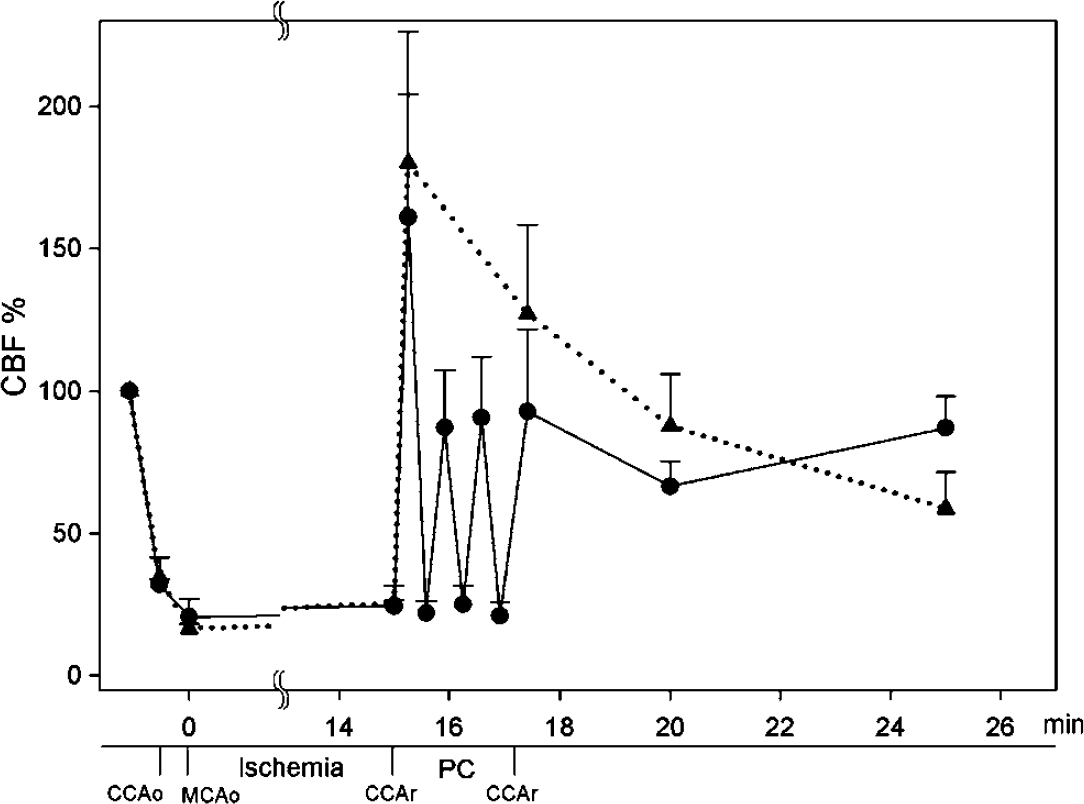
Changes in CBF in animals subjected to 15 mins of transient bilateral CCA occlusion after permanent MCA occlusion with and without postconditioning. Bilateral CCA occlusion reduced CBF to approximately 30% of the baseline, and additional MCA occlusion further decreased CBF to approximately 20%. After CCA release, a transient hyperemic response was observed, which was interrupted by postconditioning of three cycles of 30 secs reperfusion and 10 secs occlusion. Dashed line with triangle, no postconditioning (n = 3); solid line with circle, postconditioning (n = 3). CCAo, CCA occlusion; MCAo, MCA occlusion; CCAr, CCA release; PC, postconditioning.
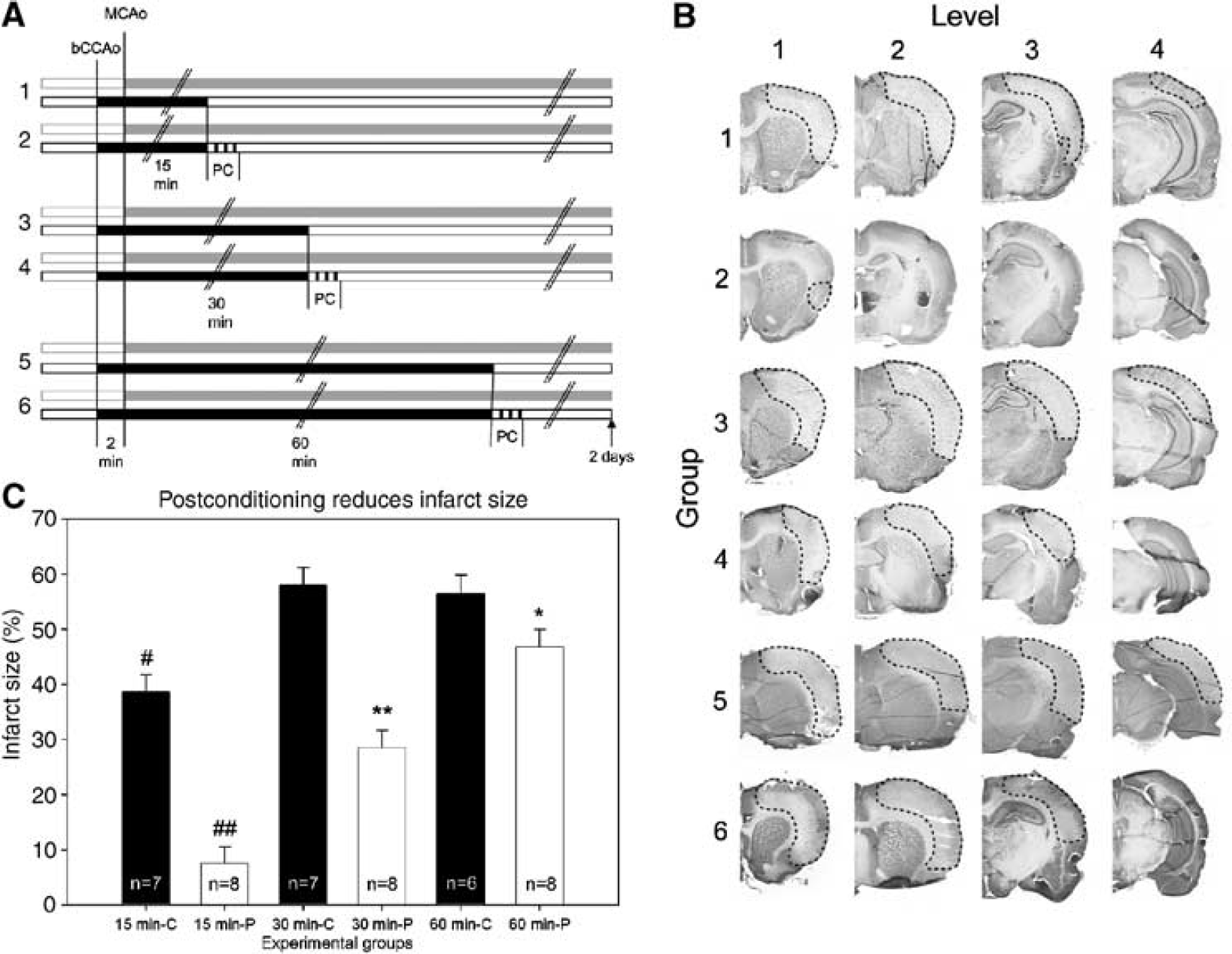
Postconditioning reduced infarct size. (
Postconditioning
Animals were randomly assigned into six groups (Figure 2A). The operator who performed the surgery for ischemia was blinded without knowing which animal would be subjected to postconditioning. A second person randomly assigned and performed postconditioning after reperfusion. For the postconditioning study, CCAs were occluded by aneurysms clips for 15, 30, or 60 mins after MCA occlusion and then released, while the distal MCA remained occluded permanently. After 30 secs of CCA reperfusion, the CCAs were occluded again by tightening sutures around the CCA for 10 secs, followed with another 2 cycles of 30 secs reperfusion and 10 secs occlusion (total of 3 cycles), and then allowed reperfusion for 2 days. Another 3 groups subjected to bilateral CCA occlusion for 15, 30, or 60 mins and combined with permanent MCA occlusion, but without postconditioning, served as controls. All animals were maintained under anesthesia for approximately 5 mins after the initial CCA release for postconditioning and/or wound closure.
Cerebral Blood Flow Measurement
Another six rats were used to detect cerebral blood flow (CBF) by using a laser Doppler flowmeter (laser FLO, TSI Inc., Shoreview, MN, USA) in animals subjected to transient 15 mins bilateral CCAs occlusion after permanent MCA occlusion. After the CCAs and MCA were exposed, a 2mm diameter hole was drilled (2 mm behind the Bregma, and 2 mm lateral to the midline). The laser probe was attached to the surface of the brain through the hole to detect CBF values. Cerebral blood flow was measured at 2 mins before CCA occlusion (base line), immediately after CCA occlusion, immediately after MCA occlusion, 7 and 15 mins after ischemia onset. After CCA reperfusion, for postconditioning, CBF was detected during three cycles of 30 secs of reperfusion and 10 secs of occlusion, and then at 2 mins 30 secs, 5 and 10 mins after CCA reperfusion. For control animals, CBF was detected at 30 secs, 2 mins 30 secs, 5 and 10 mins after CCA reperfusion. Cerebral blood flow values were expressed as percentages relative to the baseline (100%).
General Histology and Infarct Size Measurement
Two days after ischemia, rats were perfused transcardially with normal saline followed with 4% paraformaldehyde (PFA). Brains were post-fixed with 4% PFA, 20% sucrose for 24 h, and sectioned into 4 coronal blocks rostral (level 1) to caudal (level 4). Thirty-micrometer sections were cut onto glass slides in the coronal plane using a cryostat. A section from each block was selected and stained with cresyl violet, Infarct size was measured at all four levels. The percentage of infarct cortex at each was measured using the imaging system of MCID-M4 (Imaging Research Inc., Ontario, Canada), and then normalizing to the entire ipsilateral cortex as described previously (Zhao et al, 2003a, 2005).
Terminal Deoxynucleotidyl Transferase-Mediated Uridine 5′-Triphosphate-Biotin Nick End Labeling Staining
To detect spatial distribution of stroke caused DNA fragmentation, a terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) staining protocol was employed with minor modifications (Saito et al, 2003). Sections prepared above were used. The sections were incubated with Neuropore (Trevigen, MD, USA Cat no. 48206061) for 30 mins and incubated in 1 × terminal deoxynucleotidyl transferase (TdT) buffer (Invitrogen, Carlsbad, CA, USA Cat no. 16314-015)) for 30 mins, and then reacted with a mixture of TdT enzyme (1:40, Invitrogen, Cat no. 10533-073) and a biotinylated 16-dUTP (1:16, Roche Diagnostics, Indianapolis, IN, USA Cat no. 1093070) diluted in the TdT buffer at 37°C for 90 mins. After washed in phosphate-buffered saline (PBS), Streptavidin solution (1:100, Molecular Probe, Carlsbad, CA, USA, Cat no. S-869) was applied onto the sections for 60 mins at room temperature. The slides were then mounted with 4,6,diamidino-2-pheny-lindole (DAPI)-containing medium and assessed under the confocal microscopy.
In Situ Detection of Superoxide Radicals
Superoxide radicals are detected using a method that detects hydroethidine (Het) (Kim et al, 2000; Maier et al, 2002). Before stroke onset, animals were injected intravenously with Het solution (200 μL; 1 mg/mL in PBS) under anesthesia. Rats were subjected to 30 mins of CCA occlusion plus permanent MCA occlusion, with or without postconditioning. Animals were killed 30 mins after CCA release by overdose of isoflurane and transcardially perfused with icy PBS and 4% PFA. Animals with sham surgery without ischemia were also injected with Het solution, which served as a control for baseline of superoxide products. In addition, a subset of ischemic rats were not injected with Het solution, which were used to confirm that ischemic brains did not generate Het-like autofluorescence. Brains were fixed with 4% PFA overnight. One hundred micrometer coronal sections were cut on a vibratome. A slice near the Bregma was mounted with medium containing DAPI and observed under a LSM510 confocal laser scanning microscope (Zeiss LSM 510, Thornwood, NY, USA). For collecting Het-positive signals, excitation wavelength was 543 nm (HeNe laser), the emission spectra was between 560 and 590 nm. Pictures corresponding to the three regions as shown in Figure 3 were taken with the × 40 objective under fixed conditions. Optical densities of Het signals of up to nine positive individual cells in each field were measured by Photoshop, and subtracted by the local background. The Het intensity was expressed as mean value per cell from the three fields in each animal.
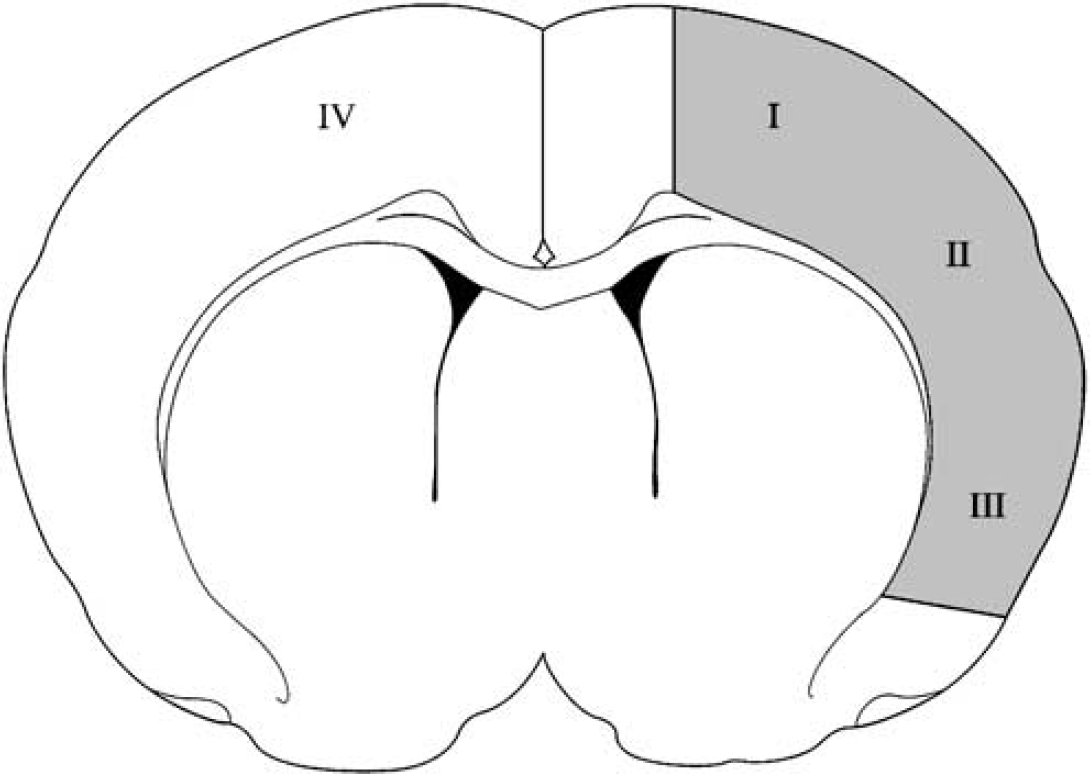
Diagram indicates regions for analysis of TUNEL and superoxide staining by confocal microscopy. A coronal brain section near the Bregma was selected for photomicroscopy. Four regions, including the infarct margin or ischemic penumbra (I), the region II and III in the ischemic core and the nonischemic cortex (IV) are indicated.
Double Immunofluorescence Staining with Het
Slices of 100 μm prepared above for detecting Het signal were used. Free-floating immunostaining was performed as described (Zhao et al, 2005). After washing three times in PBS, sections were blocked in PBS containing 5% donkey serum (Sigma, St Louis, MO, USA) and 0.3% Triton X-100 for 2h at room temperature and then incubated in the primary mouse monoclonal antibody of MAP (1:500, Sigma, Cat no. M4403) diluted in blocking solution at 4°C overnight. Sections were washed with PBS and incubated for 2 h at room temperature (light shielded) in the fluorescein isothiocyanate-conjugated anti-mouse secondary antibodies diluted in blocking solution (1:200, West Grove, PA, USA). The results were examined under the confocal microscope.
Statistical Analyses
Two-way analysis of variance (ANOVA) was used to compare the protective effect of postconditioning on infarct size. Pairwise comparison was performed followed by Student—Newman–Keuls post hoc test. Het intensities were compared by one-way ANOVA followed by LSD post hoc test. Tests were considered statistically significant at P-values < 0.05. Data are presented as means ± s.e.m.
Results
Postconditioning Interrupted Reperfusion
In this study, bilateral CCA release only allows partial reperfusion. Since postconditioning was performed by CCA occlusion and release, CBF was first measured and it was validated that postconditioning significantly changes CBF after reperfusion. Cerebral blood flow was monitored in the ischemic penumbra, which was defined as the region saved by postconditioning, in animals subjected to transient 15 mins of bilateral CCA occlusion after MCA occlusion with and without postconditioning (Figure 1). Bilateral CCA occlusion reduced CBF to equivalent extents (32.1% ± 1.7% and 34.5% ± 7.0%; NS, respectively) in animals with and without postconditioning. Additional MCA occlusion further reduced CBF to equivalent extents (20.7% ± 6.2% and 16.6% ± 1.7%; NS, respectively) in animals with and without postconditioning. Such reduction in CBF is consistent with the previous study which first used this model (Chen et al, 1986). After CCA release, acute hyperemia response was observed in all animals. Cerebral blood flow in animals without postconditioning reached 180.1% ± 46.0% immediately after CCA release and gradually decreased to 58.7% ± 12.7% at 10 mins after CCA reperfusion. A similar hyperemia response was also observed in animals with postconditioning, which was interrupted by three cycles of reperfusion and occlusion. Each occlusion reduced CBF to approximately 20% to 24%, and reperfusion allowed CBF to increase to approximately 60% to 100%. These results suggest that controlling bilateral CCA release and occlusion has robust effects on CBF supply to ischemic brain.
Postconditioning Reduced Infarct Size
We then measured infarct size 2 days after cerebral ischemia with various paradigms followed by post-conditioning. Animals were divided into six groups (Figure 2A). This model generated ischemic damage in the cortex (Figure 2B). In control animals without postconditioning, infarct size caused by 15 mins of bilateral CCA occlusion after permanent MCA occlusion (group 1) is significantly smaller compared to infarct caused by 30 (group 3) and 60 mins (group 5) of bilateral CCA occlusion (Figure 2C). However, similar infarct sizes were detected in the latter two groups. Two-way ANOVA analysis demonstrated that postconditioning reduced infarct size in all four levels of brain section in rats subjected to 15 mins of CCA occlusion after permanent MCA occlusion (P ≤ 0.001 in all four levels, data not shown). In addition, two-way ANOVA analysis also allowed a comparison of infarct size between different levels, which showed that post-conditioning reduced infarct in the level 3 and 4 in animals with 30 mins of bilateral CCA occlusion (P < 0.001, data not shown) and reduced infarct in the level 4 in animals subjected to 60 mins of CCA occlusion (P = 0.008, data not shown). Overall, postconditioning reduced the mean infarct size approximately 80%, 51%, and 17%, respectively, in animals with 15 (P 0.001), 30 (P 0.001), and 60 mins (P =0.044) of bilateral CCA occlusion. Thus, the extent of protection depends on the severity of ischemia.
Postconditioning Blocked Apoptosis in the Penumbra and Reduced Superoxide Products After Stroke
At 2 days after stroke, TUNEL-positive cells were detected in both the ischemic penumbra (the region I, Figure 3) and core (the region III) in animals subjected to 30 mins of CCA occlusion plus permanent MCA occlusion without postconditioning (Figure 4). However, in the ischemic brains treated with postconditioning, no TUNEL-positive cells were observed in the ischemic penumbra (Figure 4), suggesting postconditioning blocked stroke-induced apoptosis in the penumbra but not in the core.
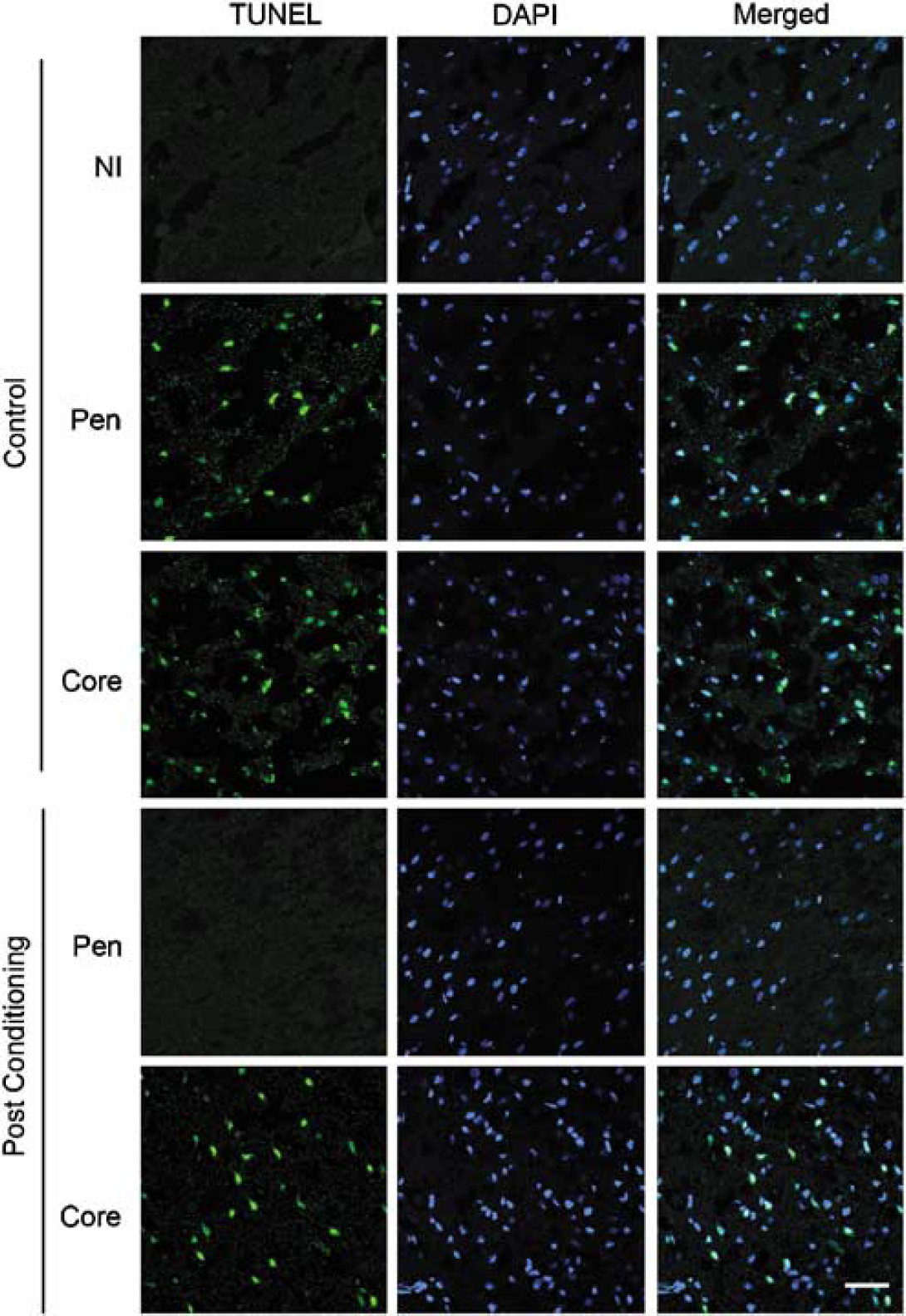
Postconditoning blocks TUNEL-positive staining in the penumbra after 30 mins of CCA occlusion plus permanent MCA occlusion. As indicated in Figure 3, the regions analyzed by confocal microscope are the region I and IV. TUNEL staining (green) was counterstained with DAPI (blue). No TUNEL-positive cells were observed in the non-ischemic cortex (IV). However, numerous TUNEL-positive cells were detected in regions I and III (n = 3) after stroke. No positive staining was observed in region I in rat brains treated with postconditioning (n = 3). Postconditioning did not inhibit TUNEL staining in region III. Scale bar = 20 μmol/L. NI, non-ishemic cortex (the region I); Pen, penumbra.
At 30 mins after CCA reperfusion, no Het-like signals were detected in the ischemic brains without Het treatment (data not shown), suggesting specific Het staining in Het-treated brains. Minimal Het signals were observed in brains from sham surgery animals without ischemia (Figure 5). Nevertheless, Het signals significantly increased in ischemic brains after reperfusion, which were attenuated by postconditioning.
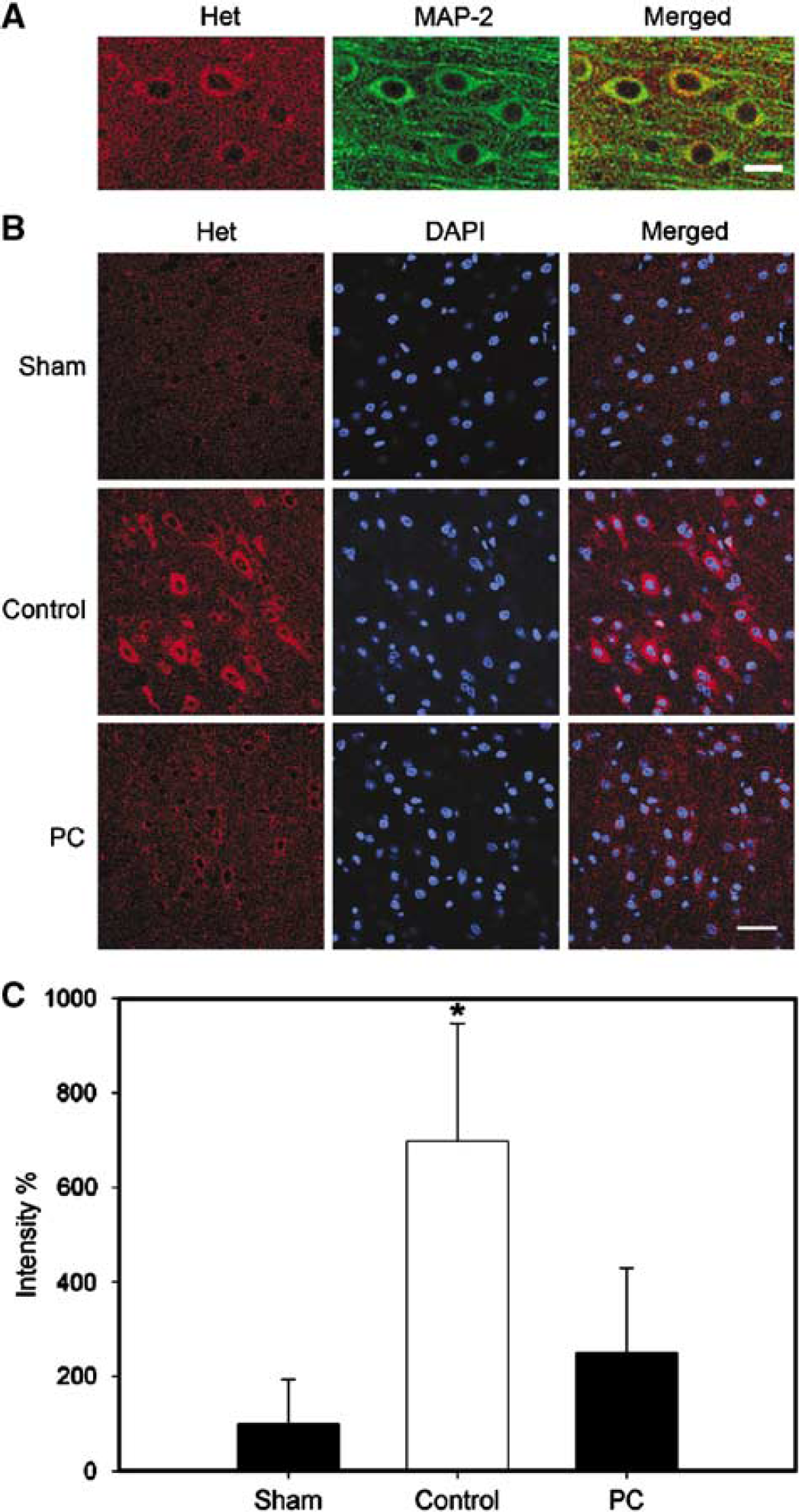
Postconditioning attenuated superoxide generation after 30 mins of CCA occlusion plus permanent MCA occlusion. As shown in Figure 3, the regions analyzed by confocal microscope for superoxide were the regions I, II, and III. (
Discussion
This is the first demonstration that postconditioning with a series of interruption of reperfusion reduces ischemic damage, and that this protection is dependent on the severity of stroke. Postconditioning robustly reduces infarct size when bilateral CCAs are occluded for 15 and 30 mins after permanent MCA occlusion, but mildly attenuates ischemic damage when CCA occlusion is extended to 60 mins. In addition, we found that postconditioning blocks TUNEL-positive cells in the penumbra but not in the core 2 days after stroke. Furthermore, postconditioning inhibited products of superoxide at 30 mins after CCA reperfusion. In this study, a focal ischemia model with permanent MCA occlusion plus transient CCA occlusion was employed. Bilateral CCA release allowed partial reperfusion, which models clinical cases in which recanalization is incomplete.
The brain is intrinsically more vulnerable to ischemia than any other organ (Lee et al, 2000). For example, while 20 to 40 mins of ischemia is needed to cause ischemic injury to heart and kidney, a few minutes of cerebral ischemia is sufficient to generate significant damage to brain (Lee et al, 2000). This may be attributable to the fact that the brain consumes more metabolic substrates, such as glucose and oxygen, than other organs. In addition, glucose is the only source for adenosine triphosphate (ATP) generation in the brain, and stores of metabolic substrates in the brain are limited and are not available during ischemia. Despite such differences in preconditioning between brain and heart, postconditioning still protects brain against stroke after reperfusion, similar to that observed in the setting of myocardial ischemia/reperfusion.
Little is known about the protective mechanisms mediating postconditioning against cerebral ischemia. Nevertheless, free radicals contribute to brain injury after ischemia/reperfusion. We found that postconditioning robustly attenuated the amount of superoxide at 30 mins after reperfusion in the model of 30 mins of CCA occlusion plus permanent MCA occlusion, suggesting that postconditioning might protect against reperfusion injury by blocking free radicals generation. In addition, postconditioning blocked TUNEL-positive cells in the penumbra, suggesting that postconditioning might reduce infarct size through inhibiting apoptosis after stroke. The protective effects of postconditioning against myocardial ischemia are likely to have some common mechanisms to preconditioning. For instance, ROS products are reduced by both preconditioning and postconditioning after stroke; both pre- and postconditioning in myocardial ischemia also reduce ROS production (Hausenloy et al, 2005). In addition, both attenuate the disruption of the protective Akt and Erk signal pathways in myocardial ischemia (Hausenloy et al, 2005). The protective mechanisms underlying preconditioning against cerebral ischemia have been extensively studied. In both global and focal ischemia, protective effects of preconditioning have been linked with adenosine activity depending on potassium ATP channels (Reshef et al, 2000), overexpression of heat shock proteins (Chen et al, 1996), expression of early genes (Belayev et al, 1996), new protein synthesis (Puisieux et al, 2004), inhibition of inflammation (Rosenzweig et al, 2004), enhanced expression of antiapoptotic proteins (Nakatsuka et al, 2000), and activation of Akt pathways (Yano et al, 2001; Hashiguchi et al, 2004; Yin et al, 2005). Importantly, as observed in postconditioning, preconditioning has also been shown to inhibit activities of ROS (Glantz et al, 2005; Perez-Pinzon et al, 2005). Whether the mechanism underlying pre- and post-conditioning in the brain is similar with regard to other mechanisms needs further study.
There are some limitations to the current study. First, postconditioning was performed after partial reperfusion. Whether such a protective phenomenon is applicable to other ischemia/reperfusion models is not clear. Second, robust protection was only detected after relatively mild ischemia, which may limit postconditioning's translation into clinical stroke. Third, despite the observation that postconditioning reduced free radical generation and TUNEL-positive cells, it is not known if these effects were the cause or simply the result of neuroprotection. Thus, further research regarding the mechanisms underlying postconditioning's protective effects is needed. In addition, infarct size was measured at 2 days after stroke; whether postconditioning merely delays ischemic damage needs further study. Nevertheless, because postconditioning, by definition, is performed after the insult. It may be applicable in certain clinical settings for stroke patients or patients subjected to surgery and endovascular therapy associated with blood vessel occlusion and revascularization. This might include patients undergoing angioplasty and stenting of intracranial arteries, temporary proximal arterial occlusion during intracranial aneurysm surgery, or carotid endarterectomy as well as carotid artery stenting. It will be critical to define the precise temporal characteristics of postconditioning in the brain, including the interval when postconditioning must be applied, the number of cycles of re-occlusion used, and the duration of re-occlusion.
In conclusion, we demonstrate for the first time that ischemic postconditioning reduces infarct size after focal stroke as a function of stroke severity, probably by reducing apoptosis and free radical products.
Footnotes
Acknowledgements
The authors thank Guohua Sun, Xuwen Gao, and Hanfeng Zhang for technical assistance, and Elizabeth Hoyte for preparing the figures. This study was supported by NINDS Grants R01 NS27292 (GKS) and P01 NS37520 (GKS and RMS).