
Abstract
A central question in manganese neurotoxicity concerns mitochondrial dysfunction leading to cerebral energy failure. To obtain insight into the underlying mechanism(s), the authors investigated cell-specific pathways of [1–13C]glucose metabolism by high-resolution multinuclear NMR-spectroscopy. Five-day treatment of neurons with 100-μmol/L MnCl2 led to 50% and 70% decreases of ATP/ADP and phosphocreatine–creatine ratios, respectively. An impaired flux of [1-13C]glucose through pyruvate dehydrogenase, which was associated with Krebs cycle inhibition and hence depletion of [4–13C]glutamate, [2–13C]GABA, and [13C]glutathione, hindered the ability of neurons to compensate for mitochondrial dysfunction by oxidative glucose metabolism and further aggravated neuronal energy failure. Stimulated glycolysis and oxidative glucose metabolism protected astrocytes against energy failure and oxidative stress, leading to twofold increased de novo synthesis of [3–13C]lactate and fourfold elevated [4–13C]glutamate and [13C]glutathione levels. Manganese, however, inhibited the synthesis and release of glutamine. Comparative NMR data obtained from cocultures showed disturbed astrocytic function and a failure of astrocytes to provide neurons with substrates for energy and neurotransmitter metabolism, leading to deterioration of neuronal antioxidant capacity (decreased glutathione levels) and energy metabolism. The results suggest that, concomitant to impaired neuronal glucose oxidation, changes in astrocytic metabolism may cause a loss of intercellular homeostatic equilibrium, contributing to neuronal dysfunction in manganese neurotoxicity.
Manganese, required for the activity of many enzymes, is essential for normal physiologic functioning (Underwood, 1981). Chronic exposure to manganese, however, leads to neurotoxicity (manganism) and is characterized by neuropsychiatric symptomatology that often resembles Parkinson disease (Huang et al., 1989; Mena et al., 1967). Neuropathologic changes are manifested mainly in the globus pallidus and include neuronal loss, gliosis, and Alzheimer type II astrocytosis (Pentschew et al., 1963; Yamada et al., 1986). The primary basis for manganese neurotoxicity remains unclear, but changes of both astrocytic and neuronal integrity indicate that several biochemical events may be involved. The accumulation of manganese in mitochondria (Wedler et al., 1989) suggests the possibility of mitochondrial derangements. As mitochondrial function is tightly coupled to cellular energy production, compromised brain energy metabolism leading to decreased oxidative phosphorylation may be a central feature of manganese neurotoxicity.
Astrocytes sequester manganese (Aschner et al., 1992; Tholey et al., 1990; Wedler et al., 1989) and are considered as the likely initial targets of manganese neurotoxicity (Henriksson and Tjalve 2000; Spranger et al., 1998). The glial localization of pyruvate carboxylase (PC), the key anaplerotic enzyme in the brain (Shank et al., 1985), and glutamine synthetase (GS) (Norenberg and Martinez-Hernandez, 1979), suggest an important role of these cells in brain energy metabolism. In this context, changes in astrocyte metabolism may either modify primary neuronal injury or induce secondarily neuronal dysfunction. In contrast to other organs, brain energy metabolism depends almost exclusively on the oxidation of glucose (Gjedde and Marrett, 2001). Via tricarboxylic acid (TCA) cycle activity, glucose metabolism also leads to the biosynthesis of neurotransmitters such as glutamate, aspartate, and γ-aminobutyric acid (GABA). The metabolic activities of the energy-producing pathways are under strict coordinated control and involve glycolysis, the TCA cycle, oxidative phosphorylation, and the pentose–phosphate pathway. So far, however, no data are available from simultaneous studies to correlate the mechanisms that link manganese-induced mitochondrial dysfunction and energy failure with glucose metabolism. The use of stable isotope-labeled precursors is a common technique used to study biochemical pathways. In particular, 13C-labeled substrates and the high chemical specificity of 13C-NMR permits the identification of 13C label in specific positions of given compounds and allows for the simultaneous calculation of substrate concentrations and multiple inter- and intracellular metabolic fluxes in the intact brain and cell cultures in a comprehensive manner, which is not possible with classic studies using 14C-labeled isotopes (Bachelard and Badar-Goffer, 1993; Cruz and Cerdan, 1999; Gruetter et al., 1994).
In the present study, we applied multinuclear NMR spectroscopy and an in vitro approach using primary astrocytes, neurons, and their cocultures to analyze metabolic pathways coupled to oxidative phosphorylation by simultaneous determination of high-energy phosphates, cell-specific de novo synthesis of metabolites derived from anaerobic and oxidative [1–13C]glucose metabolism, and the involvement of intercellular trafficking after manganese treatment.
MATERIALS AND METHODS
Cell cultures
Primary cell cultures of cortical astrocytes, cortical neurons, and cocultures of both cell types were obtained as described previously (Booher and Sensenbrenner, 1972; Zwingmann et al., 2000). Astrocytes were prepared from the brains of 1- or 2-day-old Sprague-Dawley rats and cultured in Dulbecco modified Eagle medium (DMEM) containing 10% fetal calf serum for 3 weeks. Neurons were prepared from 16- to 18-day-old rat embryos according to Richter-Landsberg (1988) and cultured in basal medium Eagle containing 0.5% fetal calf serum (to avoid astrocytic proliferation). For cocultures, astrocytes were first cultivated in DMEM until reaching confluence; thereafter, neuronal cells were added and cultivated as described for neurons. Approximately 107 to 108 cells were used for each NMR experiment. In cocultures, the ratio of astrocytes to neurons was found to be 4:6. All cultures were kept in an incubator with a humidified atmosphere of 5% CO2 at 37°C. Astroglial and neuronal cultures were characterized by GFAP immunoreactivity and antineurofilament antibodies, respectively. The purity of the individual cells was found to be >95% for primary astrocytes and >90% for primary neurons.
Incubation and preparation of cell extracts
Confluent cell cultures were incubated every 24 h with fresh media containing 100-μmol/L MnCl2 on 5 consecutive days. During the last 12 h, control cultures and manganese-treated cells were incubated for 12 h with DMEM containing 5-mmol/L [1–13C]glucose (Cambridge Isotope Laboratories, Andover, MA, U.S.A.). After removal of the incubation media (lyophilized for subsequent NMR measurements), the cells were extracted with perchloric acid (PCA) as described previously (Zwingmann et al., 2000). Because of the paramagnetic effect of manganese on T1 relaxation times, di- and trivalent cations were complexed by incubation of the supernatant with iminodiacetic acid (Chelex 100; Sigma, St. Louis, MO, U.S.A.) for 20 minutes. After removing of the chelating resin by filtration, the supernatants were lyophilized.
Biochemical analysis
The enzymatic assays (determination of lactate concentrations) were performed according to the supplier's instructions (Roche Molecular Biochemicals, Mannheim, Germany) with measurement of the UV/VIS absorption of NADH at 340 nm. The protein content was quantified according to the Biuret reaction (Goa, 1953) using bovine serum albumin as a standard and VIS absorption at 540 nm. All biochemical assays were adapted for use in 96-well microtiter ELISA plates. Samples were measured in four concentrations in the plate, each with four replicates.
Nuclear magnetic resonance spectroscopy
The lyophilized samples were redissolved in 0.5 to 0.6 mL deuterium oxide (D2O; Merck, Darmstadt, Germany) and centrifuged. Before the NMR analysis, the samples were neutralized with DCl and NaOH to allow unique chemical shift assignments and to prevent glutamine-carbamate formation in the media.
1H-, 13C- and 31P-NMR spectra were recorded on Bruker spectrometers DRX600 or AMX360/AM 360 (operating for 1H-NMR at frequencies of 600 MHz or 360 MHz, for 13C-NMR at 150.9 MHz or 90.5 MHz, and for 31P-NMR at 145.7 MHz). 1H-NMR spectra were recorded with a 5-mm H,C,N inverse triple-resonance probe using 200 to 400 accumulations, a flip angle of 40°, a repetition time of 15 seconds, a spectral width of 7,183 Hz (DRX 600) or 3,623 Hz (AM/AMX 360), a data size of 16 K, and zero filling to 32 K. Chemical shifts were referenced to lactate (1.33 ppm). 13C-NMR spectra were recorded with a 5-mm 1H/13C dual probe using 15,000 to 20,000 accumulations, a repetition time of 2.5 seconds, a flip angle of 27°, composite pulse decoupling with WALTZ-16, a spectral width of 47,619 Hz (DRX 600) or 20,833 Hz (AM/AMX 360), a data size of 32 K (16 K), and zero filling to 64 K (32 K). Chemical shifts were referenced to lactate (21.3 ppm). 31P-NMR spectra were recorded with a 5-mm HX probe using 5,000 to 8,000 accumulations, a flip angle of 80°, a repetition time of 3.5 seconds, a spectral width of 5,155 Hz, composite pulse decoupling with WALTZ-16, a data size of 16 K, and zero filling to 32 K. The resonance of phosphocreatine (−2.33 ppm) was used as internal shift reference.
Quantification of metabolite concentrations
The concentrations of lactate were determined enzymatically (see Biochemical analysis). The pool sizes of other metabolites were determined from fully relaxed 1H-NMR spectra using the known lactate concentration as internal standard or using (trimethylsilyl)propionic-2,2,3,3,d4-acid (TSP) as external standard. 31P-NMR spectra were analyzed with reference to the phosphocreatine concentrations, calculated from 1H-NMR spectra (nmol/mg protein for cell extracts, μmol/mg protein for media).
Calculation of fractional 13C-enrichments and amounts of 13C-labeled metabolites
The 13C-enrichments in C-3 of lactate were determined by the heteronuclear spin-coupling pattern in 1H-NMR spectra as follows:
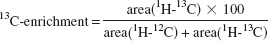
where the sum (area [1H − 12C] + area [1H − 13C]) is equivalent to the pool size of lactate. The values were corrected for 1.1% natural abundance 13C. 13C-enrichments in individual carbons of amino acids are derived from 13C-NMR spectra using the known 13C-enrichment in lactate as described previously (Zwingmann et al., 2000):
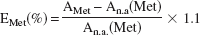
where AMet represents 13C carbon peak area of the metabolite, An.a. its natural abundance signal intensity, and 1.1 is the percentage factor of the 13C-isotope. The 13C signal intensities were corrected for nuclear Overhauser enhancement effects. The absolute amount of 13C in specified carbon positions (nmol/mg protein) is the product of the pool size times the fractional 13C-enrichment. Due to overlapping signals, glutathione and citrate cannot be integrated reliably by 1H-NMR, and changes in 13C-label incorporation were deduced from relative signal intensities in 13C-NMR spectra (normalized by the corresponding signal areas obtained after incubation of cells with unlabeled glucose).
13C-isotopomer analysis
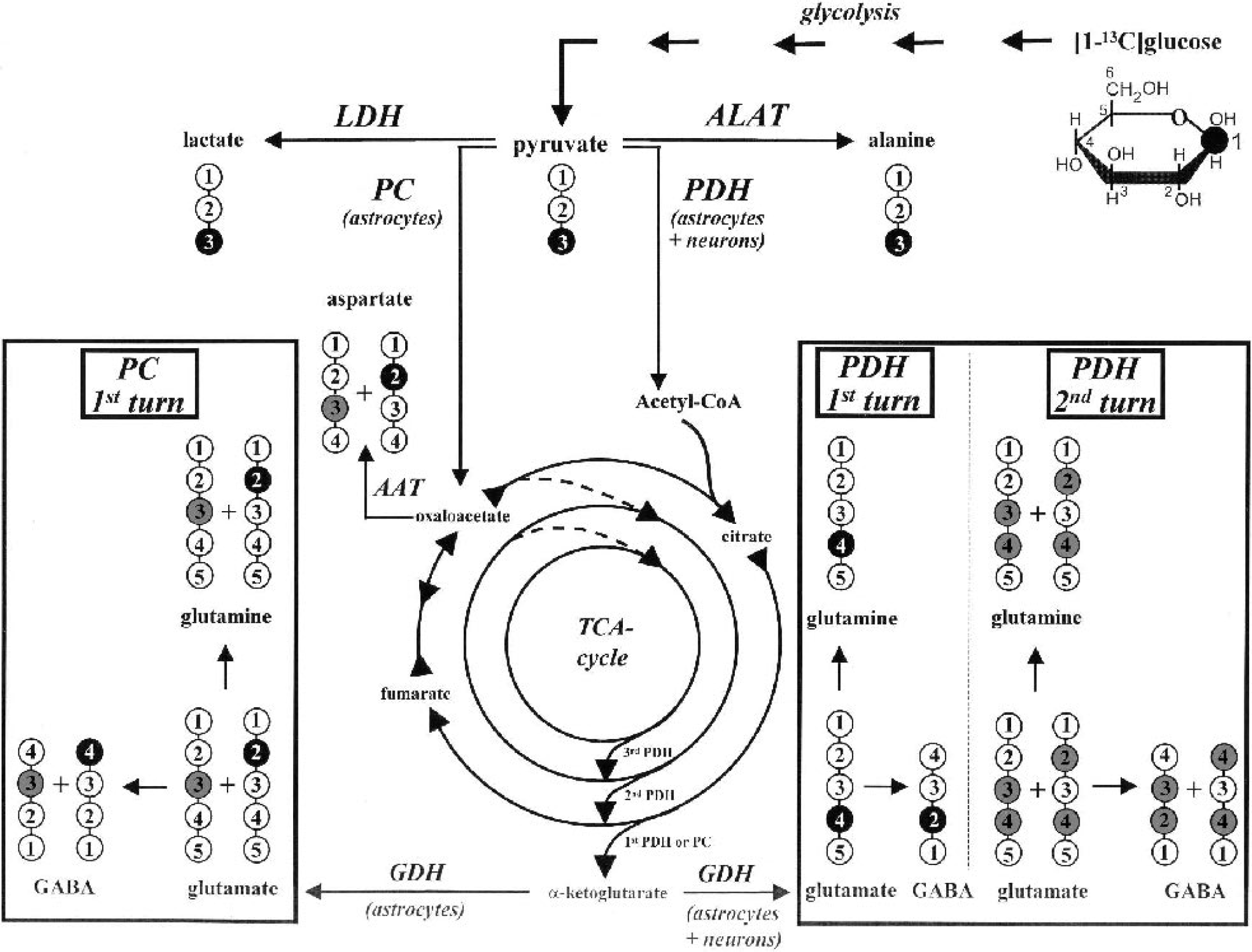
Metabolic fate of 13C-label from [1–13C]glucose. Label distribution in glycolytic and tricarboxylic acid (TCA) cycle intermediates during metabolism of [1–13C]glucose. For label distribution in TCA cycle intermediates, only one TCA cycle turn from pyruvate to malate is considered; for glutamine, glutamate, and GABA, the label distribution after two TCA cycle turns is presented. One 13C-label position in lactate and alanine synthesized via lactate dehydrogenase (LDH) and alanine aminotransferase (ALAT), respectively, and in TCA cycle-related metabolites synthesized through pyruvate carboxylase (PC) or pyruvate dehydrogenase (PDH) during the first TCA cycle turn. Gray circles indicate positions of the 13C-label due to equilibration of oxaloacetate with fumarate and the 13C-label within the second TCA cycle turn via PDH. GABA, γ-aminobutyric acid; AAT, aspartate aminotransferase.
Statistical analysis
The NMR studies were carried out on four to six independent experiments. Data are expressed as mean ± SD values. Data from all experimental groups were analyzed and compared using ANOVA and post hoc Tukey test. Differences were considered significant when P < 0.05.
RESULTS
Cellular energy state
Analysis of 31P-NMR spectra of PCA extracts (Fig. 2) depicting high-energy phosphates such as nucleoside diand triphosphates (NDPs and NTPs), in particular ADP, ATP, and PCr, showed that 5-d treatment of astrocytes with 100-μmol/L MnCl2 produced no change in NTP and PCr concentrations (Table 1). In cultured neurons, on the other hand, NTP/NDP and PCr/creatine ratios were 51% and 28% lower compared with control values, respectively (P < 0.001; creatine was calculated from 1H-NMR spectra), indicating a decreased energy state. When astrocytes were cocultured with neurons (astrocytes:neurons = 4:6), PCr was lowered to 32% (P < 0.01), but NTP levels were unchanged. Considering the relative concentrations of PCr in astrocytes and neurons, the theoretical intermediate of PCr in the two cell types under control conditions and after manganese treatment (11.45 vs. 10.67 nmol/mg protein) showed a higher decrease of PCr in manganese-treated cocultures compared with neuron cultures (P < 0.001).
Effect of manganese on the concentration of high-energy phosphates
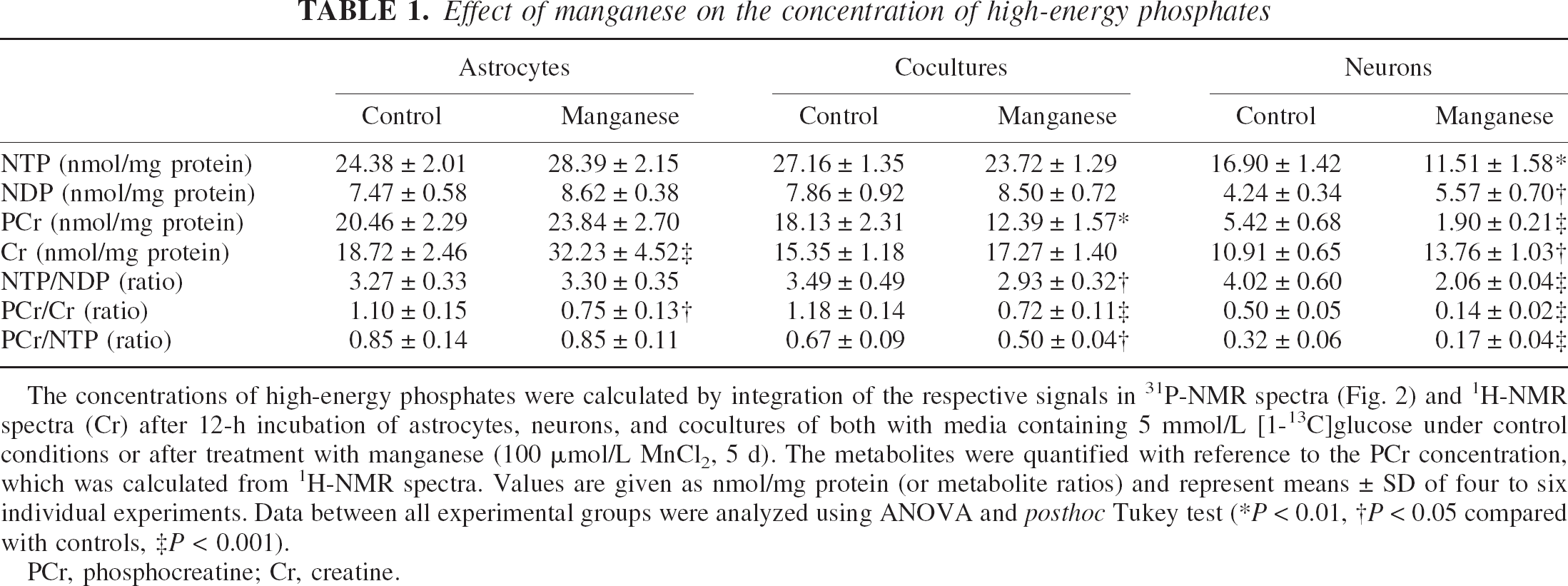
The concentrations of high-energy phosphates were calculated by integration of the respective signals in 31P-NMR spectra (Fig. 2) and 1H-NMR spectra (Cr) after 12-h incubation of astrocytes, neurons, and cocultures of both with media containing 5 mmol/L [1-13C]glucose under control conditions or after treatment with manganese (100 μmol/L MnCl2, 5 d). The metabolites were quantified with reference to the PCr concentration, which was calculated from 1H-NMR spectra. Values are given as nmol/mg protein (or metabolite ratios) and represent means ± SD of four to six individual experiments. Data between all experimental groups were analyzed using ANOVA and posthoc Tukey test (*P < 0.01, †P < 0.05 compared with controls, ‡P < 0.001).
PCr, phosphocreatine; Cr, creatine.
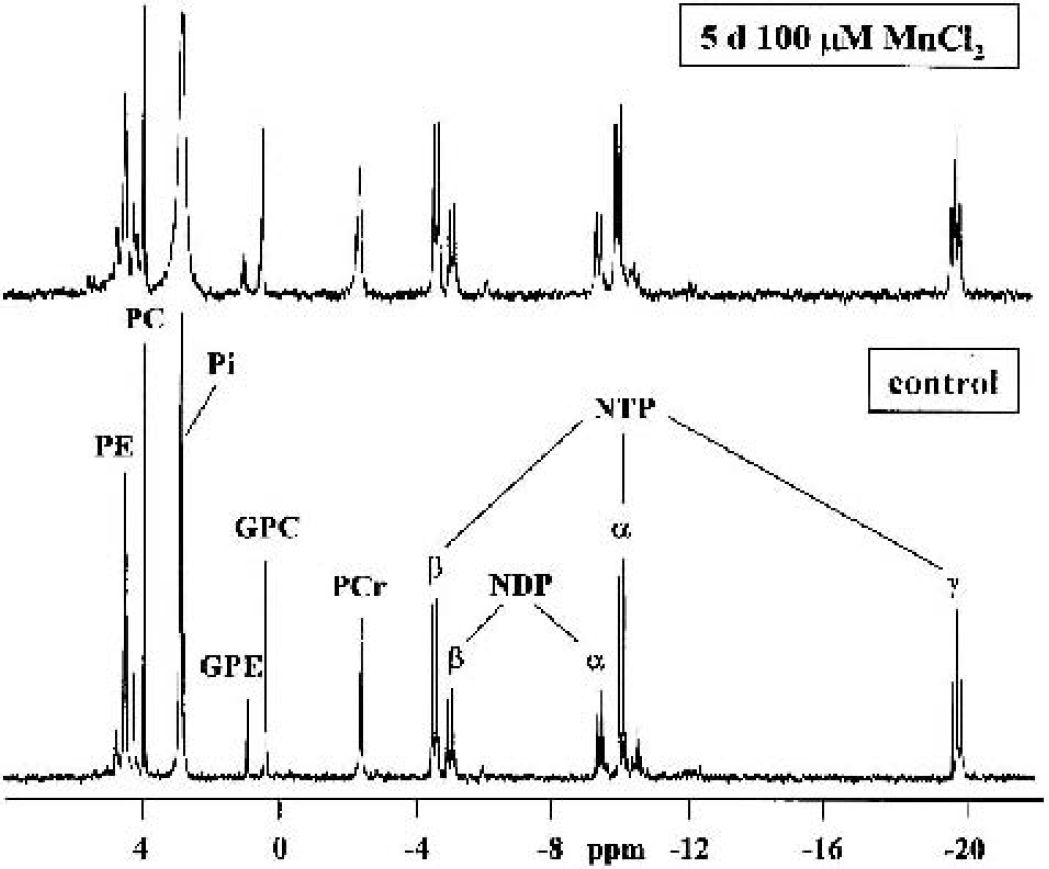
31P-NMR spectra of perchloric acid extracts of manganese-treated (100-μmol/L MnCl2, 5 d) or control astrocytes. NTP/NDP, nucleoside triphosphate/diphosphate; PCr, phosphocreatine; Pi, inorganic phosphate; (G)PE, glycero)phosphoethanolamine; (G)PC, (glycero)-phosphocholine.
Glycolytic metabolic flux of glucose
As a measure of the glycolytic flux, the consumption of [1–13C]glucose ([13C]Glc) and the de novo synthesis of [3–13C]lactate ([13C]Lac) were calculated from 1H- and 13C-NMR spectra of media (Fig. 3) and PCA extracts (Tables 2 and 3).
Effect of manganese on extracellular [1-13C]glucose and [3-13C]lactate

Extracellular concentrations of [1-13C]glucose and [3-13C]lactate, synthesized de novo from [1-13C]glucose, were calculated from 1H and 13C-NMR spectra of lyophilized media before and after 12-h incubation of cell cultures with media containing 5 mmol/L [1-13C]glucose under control conditions or following treatment with manganese (100 μmol/L MnCl2, 5 d); no unlabeled glucose was detected in 1H-NMR spectra. Values are given as μmol/mg protein or percentage of control after manganese treatment (in media based on the amounts of [1-13C]glucose/[3-13C]lactate, taken up from/released into the media during 12 h of incubation time) and represent means ± SD of four to six individual experiments. Data between all experimental groups were analyzed using ANOVA and post hoc Tukey test (*P < 0.001; †P < 0.05 compared with controls; for differences between individual cell types see text).
Effect of manganese on intracellular [1-13C]glucose and [3-13C]lactate
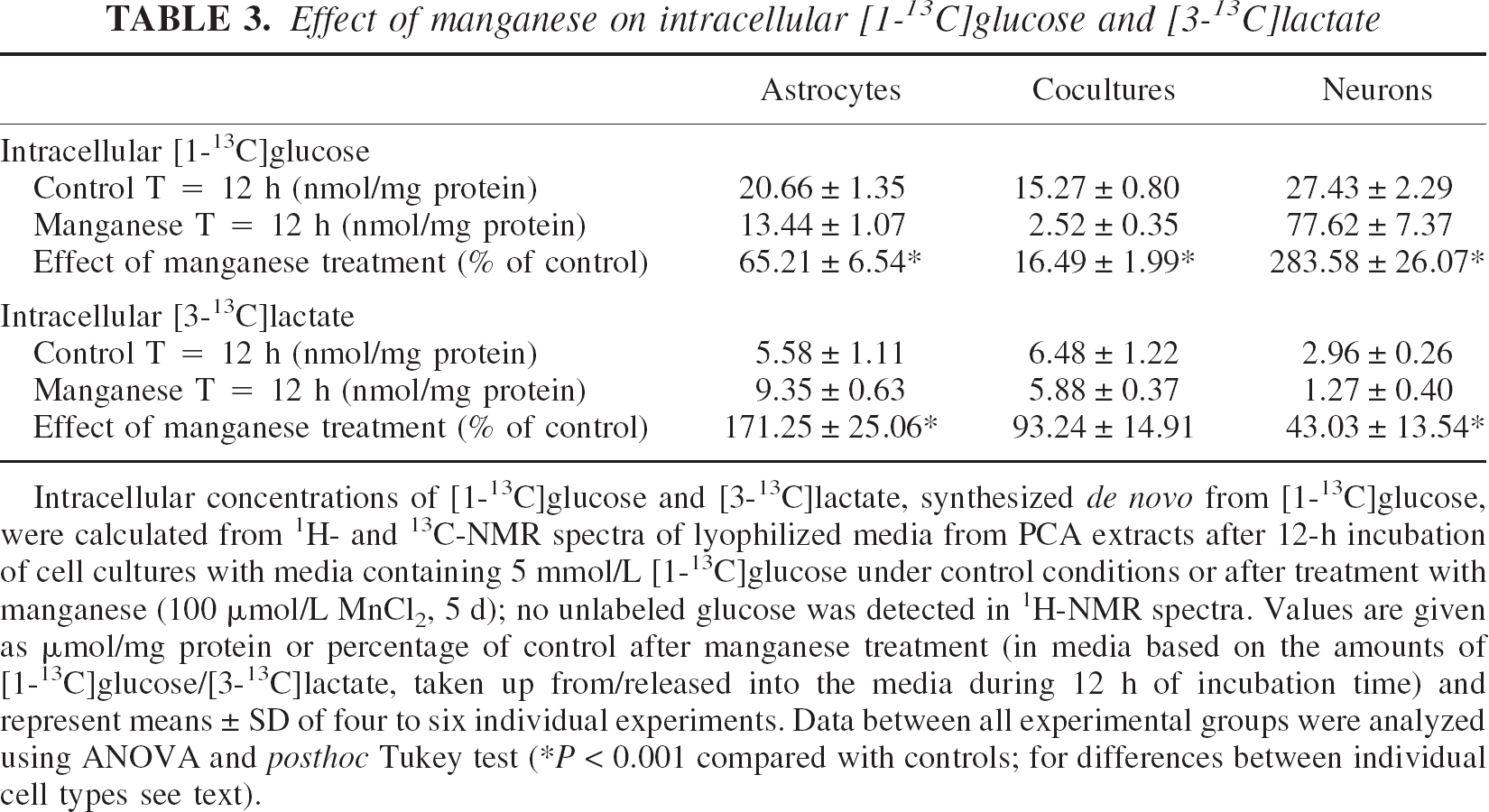
Intracellular concentrations of [1-13C]glucose and [3-13C]lactate, synthesized de novo from [1-13C]glucose, were calculated from 1H- and 13C-NMR spectra of lyophilized media from PCA extracts after 12-h incubation of cell cultures with media containing 5 mmol/L [1-13C]glucose under control conditions or after treatment with manganese (100 μmol/L MnCl2, 5 d); no unlabeled glucose was detected in 1H-NMR spectra. Values are given as μmol/mg protein or percentage of control after manganese treatment (in media based on the amounts of [1-13C]glucose/[3-13C]lactate, taken up from/released into the media during 12 h of incubation time) and represent means ± SD of four to six individual experiments. Data between all experimental groups were analyzed using ANOVA and posthoc Tukey test (*P < 0.001 compared with controls; for differences between individual cell types see text).
13C-NMR spectra of lyophilized incubation media before and after incubation of astrocytes and neurons with media containing 5-mmol/L [1–13C]glucose. The spectra were minimized in size to show the 13C-signals of glucose and lactate (the major metabolite labeled with 13C).
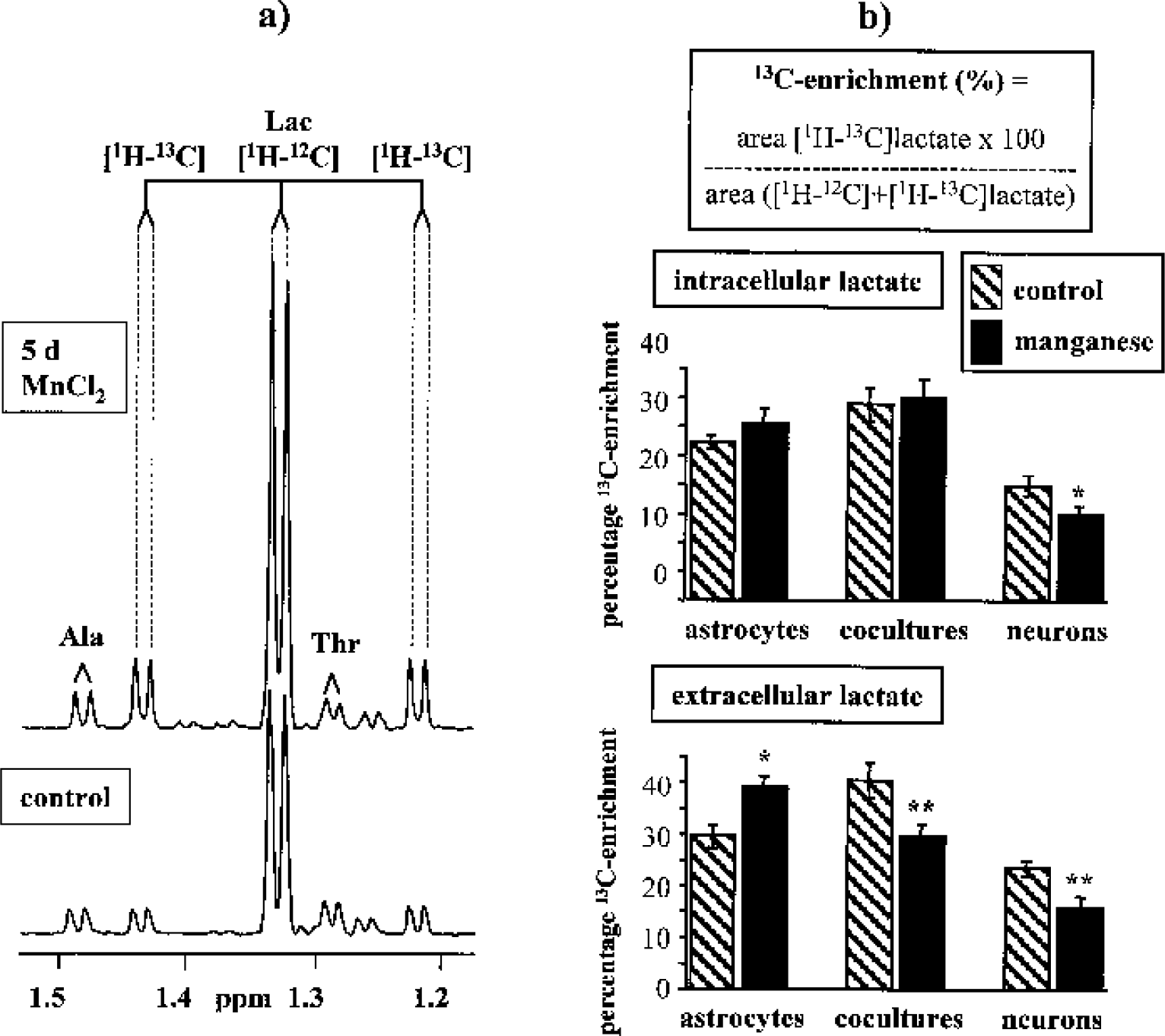
Percentage 13C-enrichment in intra- and extracellular lactate.
Concentrations of TCA cycle–related metabolites and organic osmolytes
The concentrations of amino acids were calculated from the 2.2- to 3.6-ppm region of 1H-NMR spectra (Table 4). Glutamine declined to 45% in cocultures of astrocytes and neurons compared with nontreated cells (P < 0.001), whereas no changes were observed in pure astrocytes or neurons. The concentration of glutamate increased in manganese-treated astrocytes and cocultures (5 d, 100 μmol/L) to 180% and 170% of control, respectively (P < 0.01; not statistically significant between astrocytes and cocultures). Whereas manganese increased aspartate in astrocytes to 125% (P < 0.05), it remained unchanged in cocultures. Manganese-treated neurons manifested 30% to 35% decreased intracellular levels of glutamate and aspartate (P < 0.05). After 5-d treatment with manganese, a generalized increase of the TCA-cycle intermediate succinate to 140% to 185% of control levels (P < 0.01) was observed in the order cocultures>neurons>astrocytes (P < 0.001 neurons and cocultures vs. astrocytes). In addition, the concentrations of organic osmolytes were calculated (3.4 to 4.05 ppm). Taurine levels were increased in astrocytes to 250% of control (P < 0.001), but decreased in neurons (P < 0.05) and, in particular, in cocultures to 60% of control (P < 0.001; P < 0.01 cocultures vs. neurons). Hypotaurine, the direct precursor for taurine, and myo-inositol, an astrocytic marker molecule, were unchanged in pure astrocytes after 5-d manganese treatment, but decreased to 25% and 55% of control levels (P < 0.001) in cocultures.
Effect of manganese on the concentration of tricarboxylic acid cycle–related metabolites and organic osmolytes
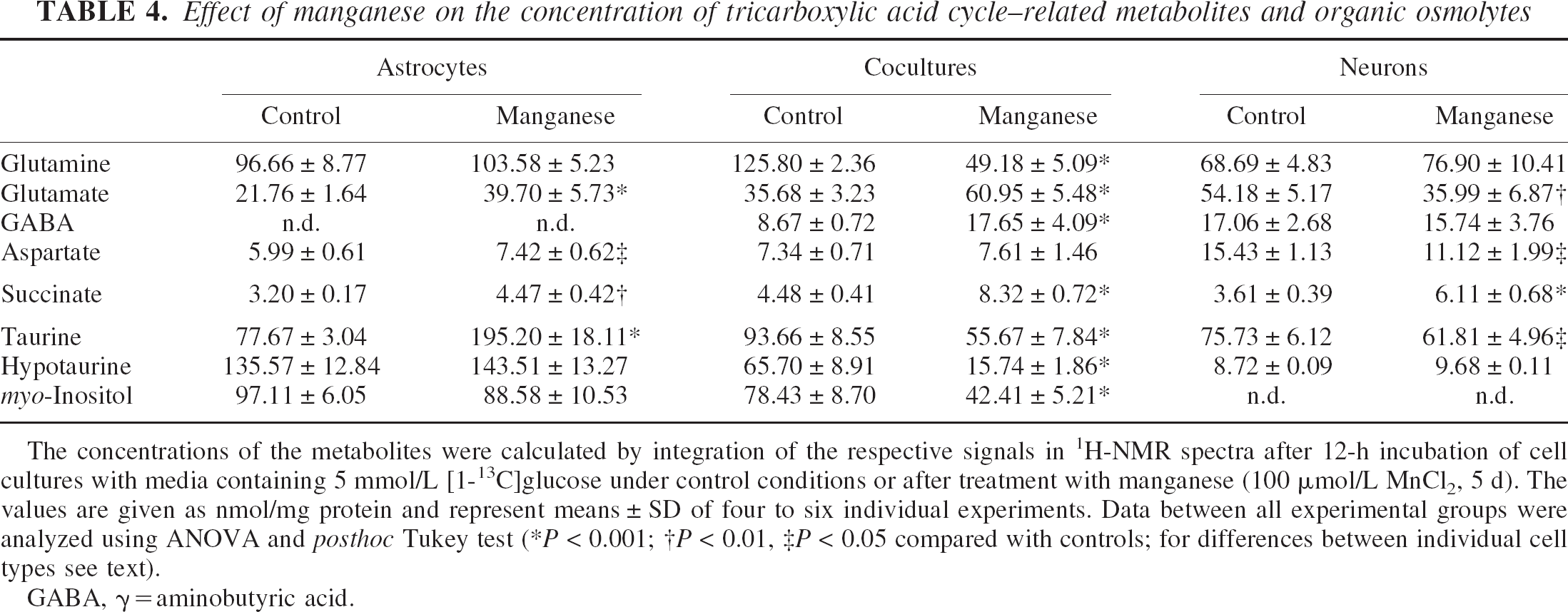
The concentrations of the metabolites were calculated by integration of the respective signals in 1H-NMR spectra after 12-h incubation of cell cultures with media containing 5 mmol/L [1-13C]glucose under control conditions or after treatment with manganese (100 μmol/L MnCl2, 5 d). The values are given as nmol/mg protein and represent means ± SD of four to six individual experiments. Data between all experimental groups were analyzed using ANOVA and posthoc Tukey test (*P < 0.001; †P < 0.01, ‡P < 0.05 compared with controls; for differences between individual cell types see text).
GABA, γ = aminobutyric acid.
Cell-specific [1–13C]glucose metabolism
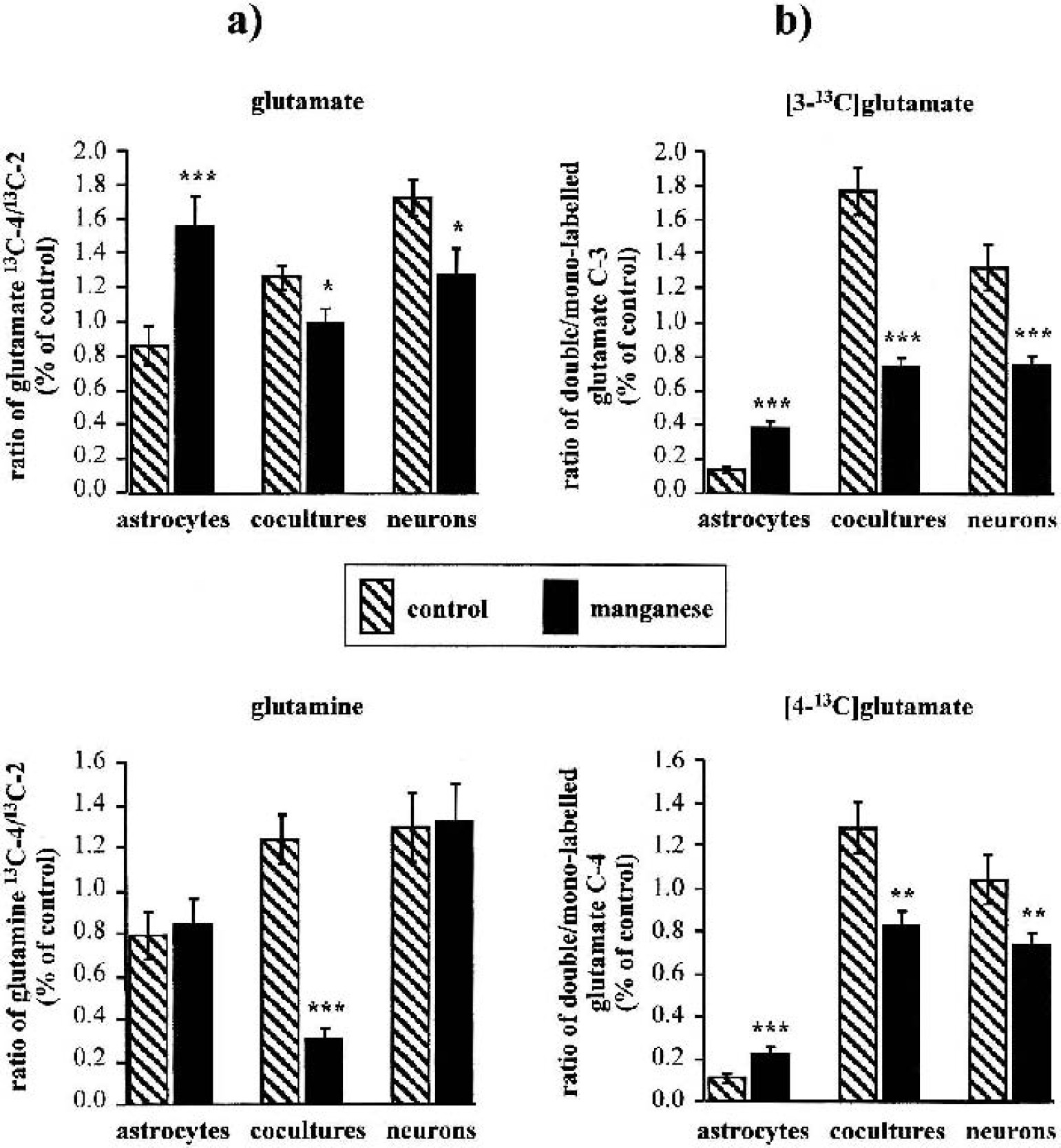
Figure 5 shows representative 13C-NMR spectra obtained from extracts of cocultures of astrocytes and neurons after 12-h incubation with 5-mmol/L [1–13C]glucose. Whereas 13C-labeled glutamate was hardly detected in astrocytes under control conditions, 5-d exposure of astrocytes to 100-μmol/L manganese led to increased synthesis of the isotopomer [4–13C]glutamate, formed in the PDH pathway, reflected by an increased total amount and 13C-enrichment to 400% and 220% of control, respectively (Tables 5 and 6). Consistent with unchanged glutamine concentrations (Table 4), the increased glutamate synthesis was not accompanied by subsequent incorporation of 13C-label into glutamine. When astrocytes were cocultured with neurons, manganese exposure resulted not only in stimulated PDH-mediated synthesis of [4–13C]glutamate, but in particular in increased flux through PC leading to increased [2–13C]glutamate and [3–13C]aspartate synthesis (P < 0.001). Despite stimulated glutamate synthesis in cocultures, however, manganese exposure led to decreased [13C]glutamine levels to 7% (C-4) and 26% (C-2) of control values (P < 0.001) (Table 5). A similar observation was made from 13C-NMR spectra of the incubation media. In control cells, the release of 13C-labeled glutamine within 12-h incubation with [1–13C]glucose was in the following order: cocultures (357.84 ± 48.73 nmol/mg protein), astrocytes (165.70 ± 15.92 nmol/mg protein), and neurons (which did not release 13C-labeled glutamine) (P > 0.001 between the individual cell types). After exposure to manganese, however, 13C-labeled glutamine was not detected in media of cocultures (data not shown). In contrast to astrocytes and cocultures, a decrease of mitochondrial [13C]glucose metabolism in manganese-treated neurons was associated with a 55% decrease in 13C-enrichment in [4–13C]glutamate, synthesized via PDH (P < 0.001), and reduced 13C-label incorporation from [13C]Glc into aspartate to 40% of control values (P < 0.001). The 13C-isotopomer pattern of GABA is interesting in that for neurons, the labeling pattern of GABA resembled that for 13C-labeled glutamate (i.e., manganese exposure selectively decreased de novo synthesis of [2–13C]GABA compared with [4–13C]GABA) (P < 0.01), which is metabolically derived from [4–13C]glutamate (Fig. 1). In manganese-treated cocultures, the 13C-enrichment and the absolute amount of 13C in [2–13C]GABA increased dramatically to almost 200% and 400% of control values, respectively.
Effect of manganese on 13C-label incorporation from [1-13C]glucose into tricarboxylic cycle–related amino acids: total amounts of 13C
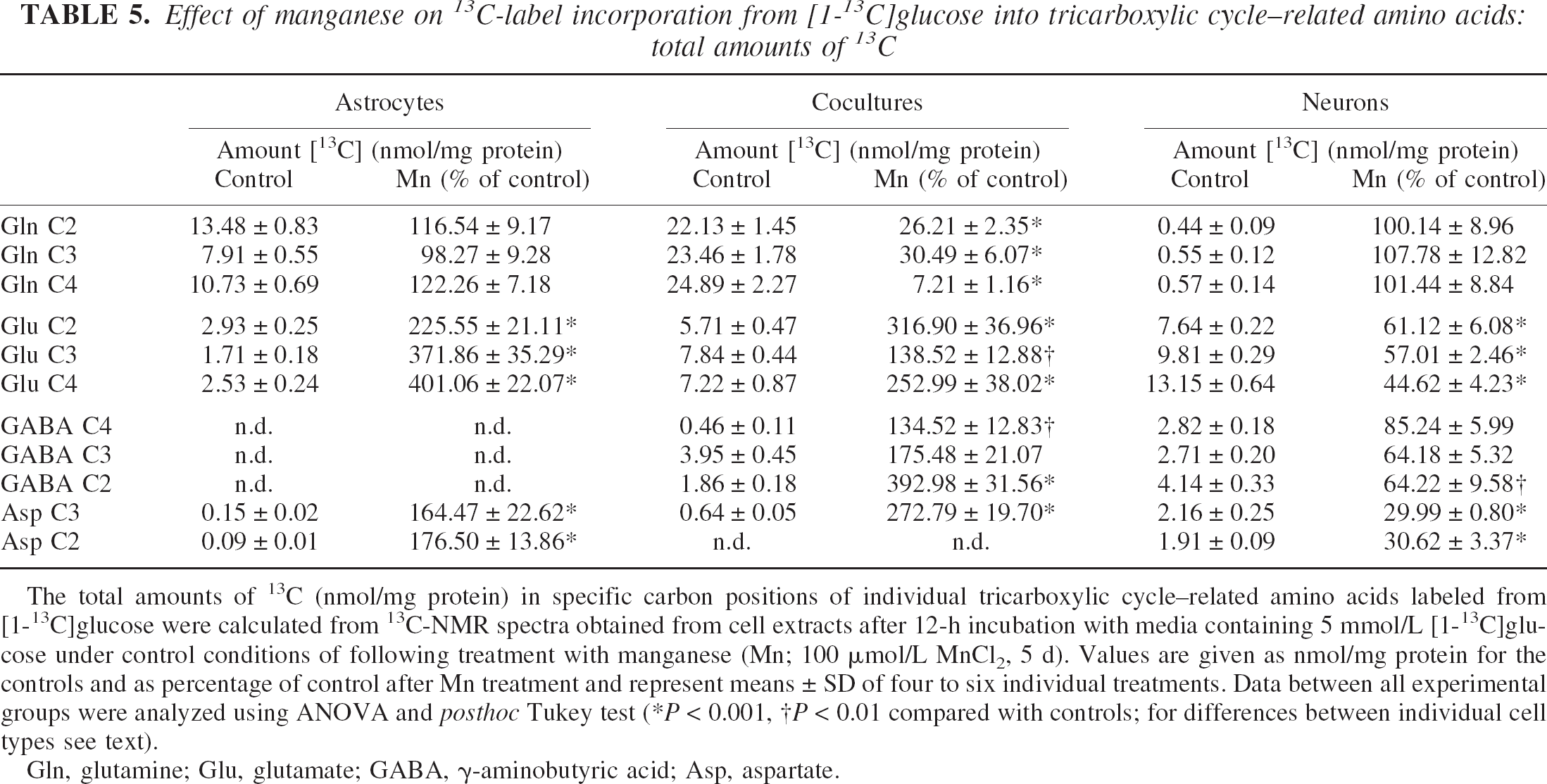
The total amounts of 13C (nmol/mg protein) in specific carbon positions of individual tricarboxylic cycle–related amino acids labeled from [1-13C]glucose were calculated from 13C-NMR spectra obtained from cell extracts after 12-h incubation with media containing 5 mmol/L [1-13C]glucose under control conditions of following treatment with manganese (Mn; 100 μmol/L MnCl2, 5 d). Values are given as nmol/mg protein for the controls and as percentage of control after Mn treatment and represent means ± SD of four to six individual treatments. Data between all experimental groups were analyzed using ANOVA and posthoc Tukey test (*P < 0.001, †P < 0.01 compared with controls; for differences between individual cell types see text).
Gln, glutamine; Glu, glutamate; GABA, γ-aminobutyric acid; Asp, aspartate.
Effect of manganese on 13C-label incorporation from [1-13C]glucose into tricarboxylic cycle–related amino acids: percentage of 13C-enrichments
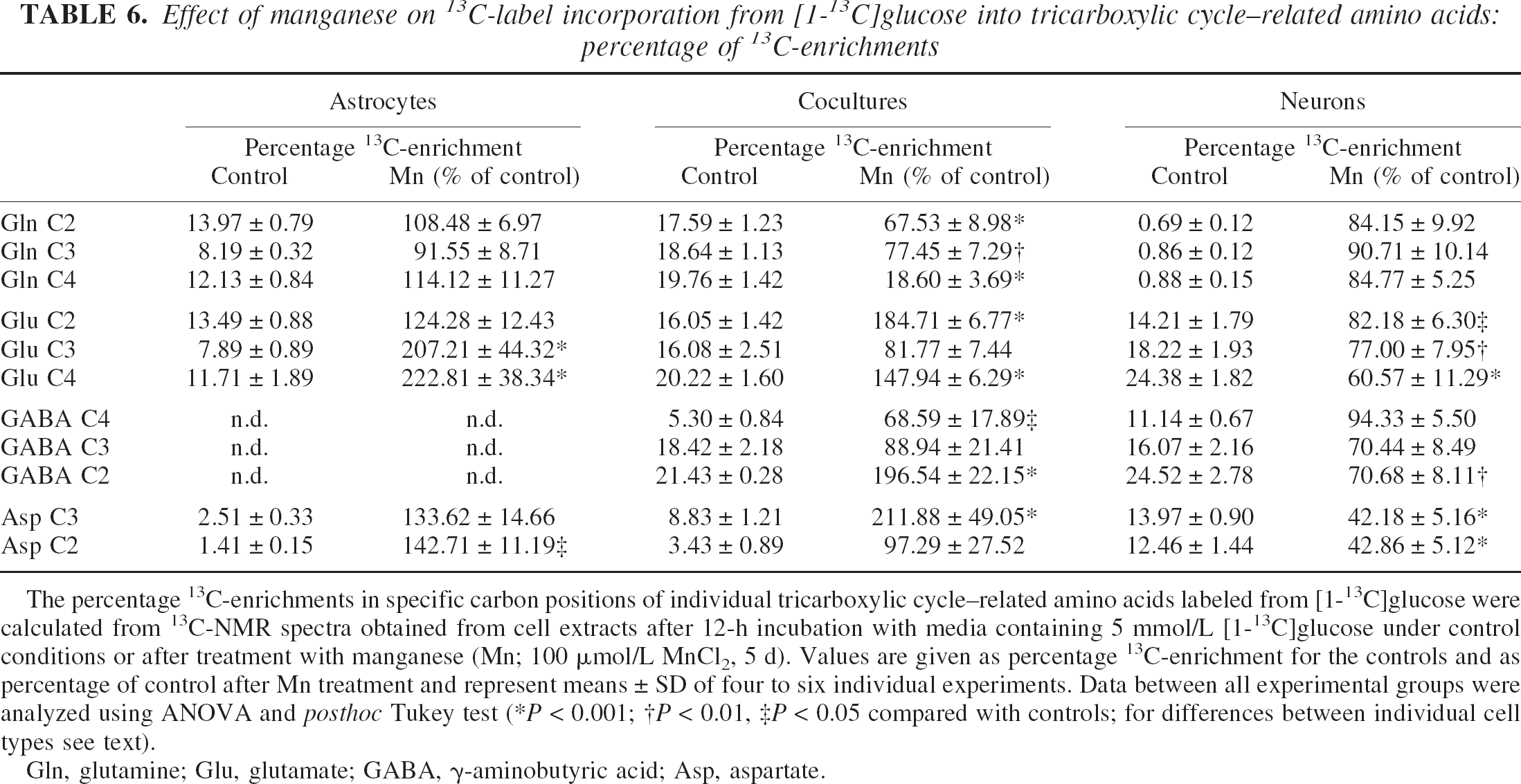
The percentage 13C-enrichments in specific carbon positions of individual tricarboxylic cycle–related amino acids labeled from [1-13C]glucose were calculated from 13C-NMR spectra obtained from cell extracts after 12-h incubation with media containing 5 mmol/L [1-13C]glucose under control conditions or after treatment with manganese (Mn; 100 μmol/L MnCl2, 5 d). Values are given as percentage 13C-enrichment for the controls and as percentage of control after Mn treatment and represent means ± SD of four to six individual experiments. Data between all experimental groups were analyzed using ANOVA and posthoc Tukey test (*P < 0.001; †P < 0.01, ‡P < 0.05 compared with controls; for differences between individual cell types see text).
Gln, glutamine; Glu, glutamate; GABA, γ-aminobutyric acid; Asp, aspartate.
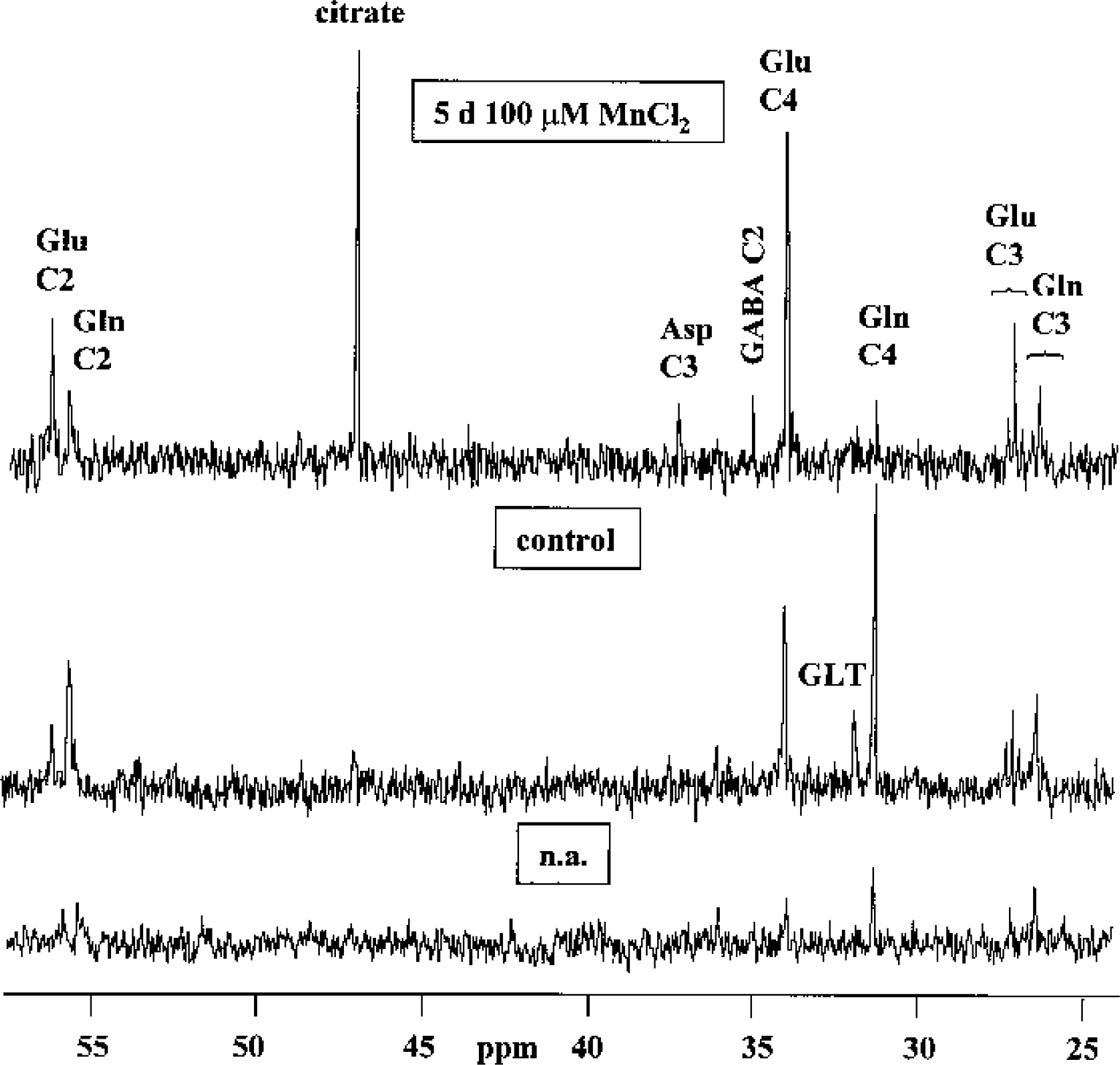
13C-NMR spectra of perchloric acid extracts obtained from cocultures of astrocytes and neurons after 12-h incubation with 5-mmol/L [1–13C]glucose under control conditions and after 5-d treatment with 100-μmol/L MnCl2. The bottom spectrum was obtained from cocultures incubated with a medium containing [12C]glucose and shows resonances resulting from natural abundance 13C (n.a.) in the metabolites. Asp, aspartate; Gln, glutamine; Glu, glutamate; GLT, glutathione; GABA, γ-aminobutyric acid.
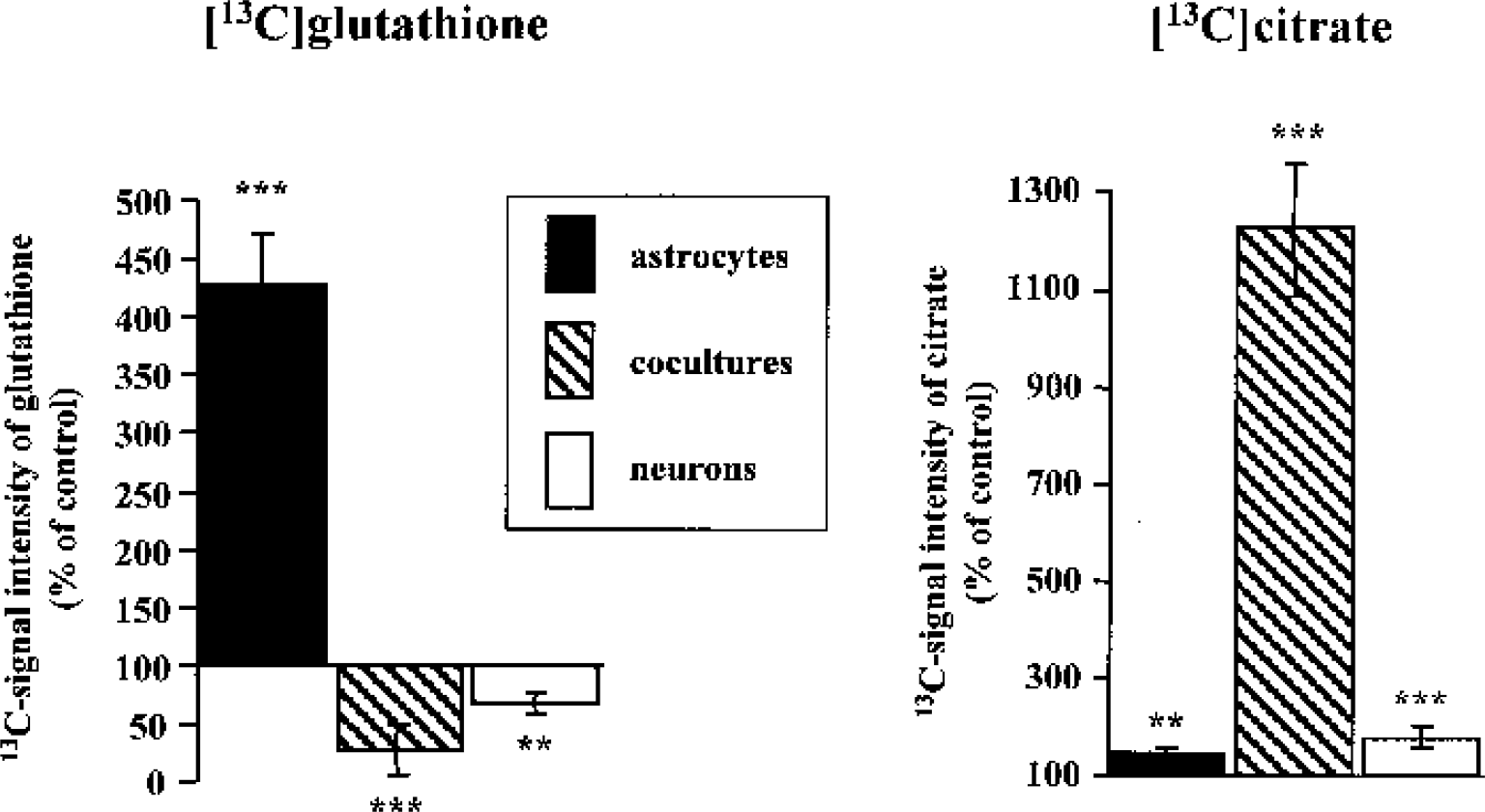
Effect of manganese on synthesis of glutathione and citrate. The 13C-signal intensities in glutathione and citrate, labeled from [1–13C]glucose, were calculated by integration of the respective signals in 13C-NMR spectra after 12-h incubation of astrocytes, neurons, and cocultures of both with media containing 5-mmol/L [1–13C]glucose under control conditions or after treatment with manganese (100-μmol/L MnCl2, 5 d). Values are given as percentage of control and represent means ± SD of four to six individual experiments. Data between all experimental groups were analyzed using ANOVA and post hoc Tukey test (***P < 0.001; **P < 0.01 compared with controls; see text for differences between individual cell types).
Relative contribution of pyruvate dehydrogenase (PDH) and pyruvate carboxylase (PC) 13C-enrichment in acetyl-CoA and tricarboxylic acid (TCA) cycling ratio.
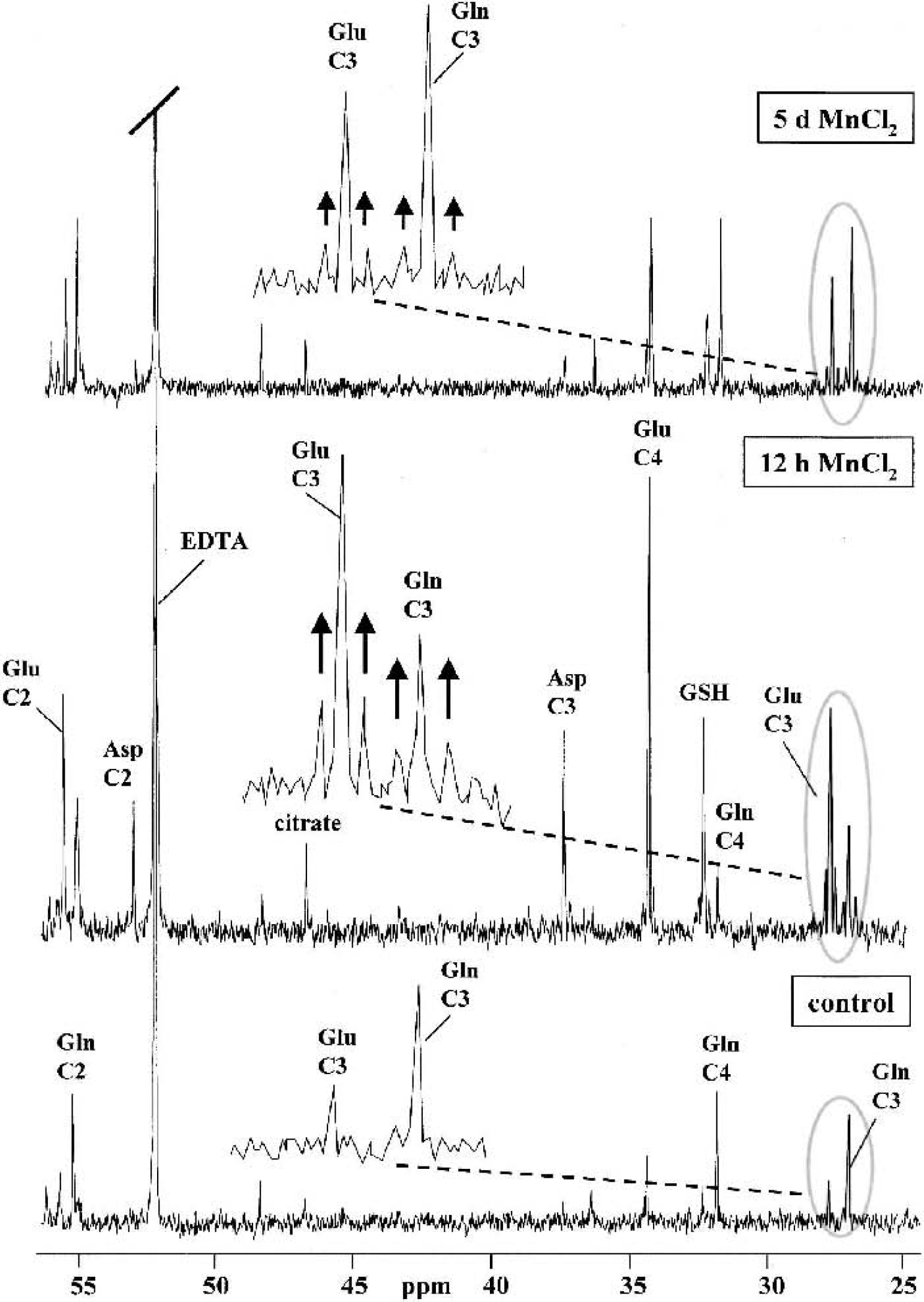
13C-NMR spectra of perchloric acid extracts obtained from astrocyte cultures after 12-h incubation with 5-mmol/L [1–13C]glucose under control conditions and after 5-d or 12-h treatment with 100-μmol/L MnCl2. For peak assignments see Fig. 6 The multiple pattern of [3–13C]glutamate and [3–13C]glutamine is shown in the expanded plots of the respective chemical shift region (the experimental spectra are smoothed by an exponential multiplication corresponding to a line broadening of 0.5 Hz).
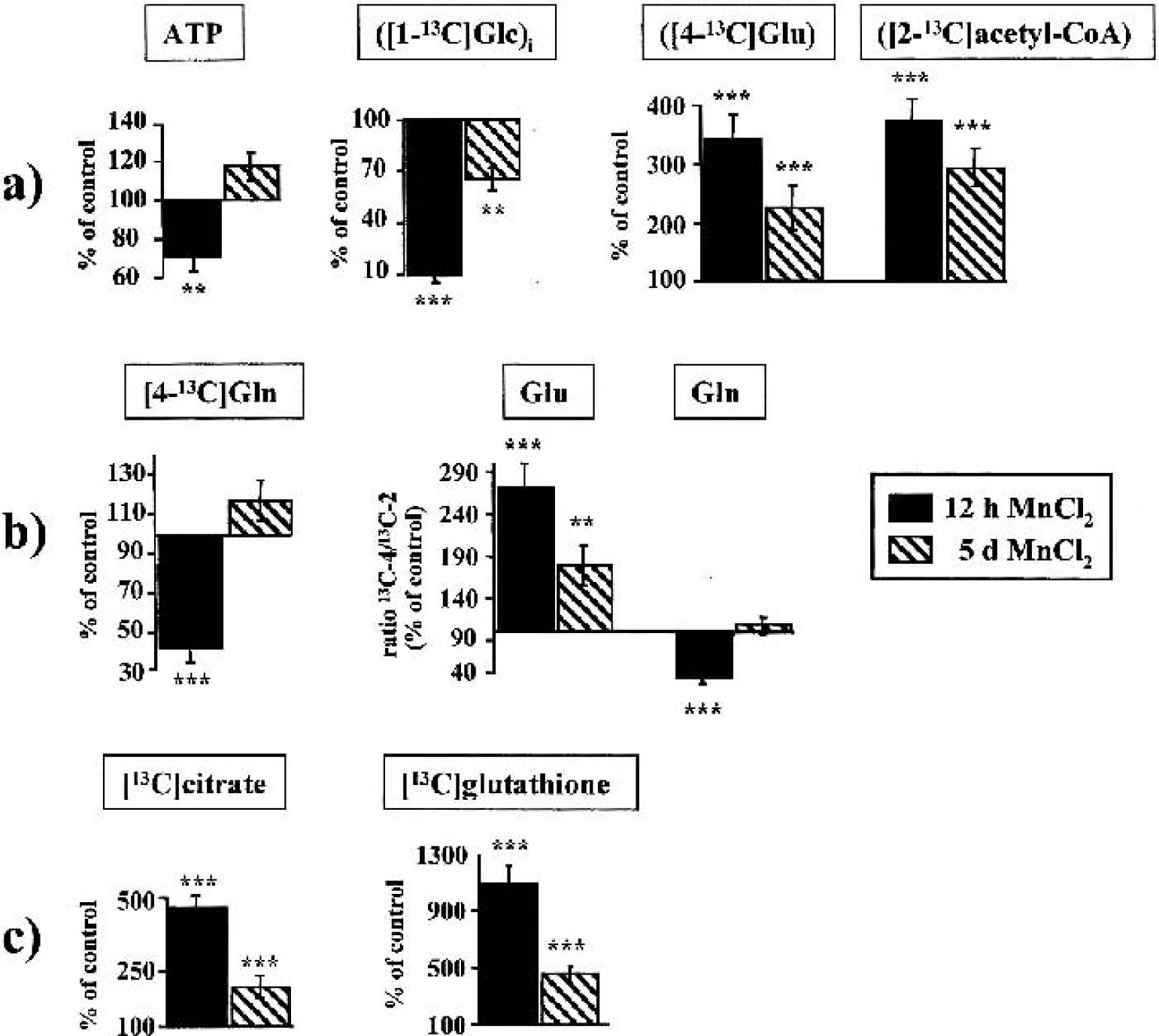
Short-term effects of manganese in neurons and astrocytes. The changes in 13C-labeled metabolites related to energy metabolism
DISCUSSION
Manganese neurotoxicity may involve mitochondrial dysfunction leading to decreased energy production. Previous studies, however, have provided little direct evidence for an altered brain energy metabolism and have not distinguished between the potentially different susceptibility of neural cells to manganese-induced metabolic derangements.
By means of multinuclear NMR investigations, allowing simultaneous monitoring of cellular energy state, [1–13C]glucose metabolism and intercellular metabolic trafficking, we have shown for the first time that manganese selectively impairs neuronal glucose oxidation leading to reduced antioxidant capacity and disruption of mitochondrial energy production. Astrocytes, in contrast, increased their oxidative and anaerobic glucose metabolism to protect against energy failure and oxidative stress but failed to evade other derangements, such as the distortion of glutamine synthesis, leading to a disturbed intercellular metabolic homeostasis and increased risk of manganese-induced neuronal energy failure.
A selective energy failure in cultured neurons was reflected by a rapid decrease of ATP and a dramatic depletion of PCr. Astrocytes were able to restore initially reduced ATP levels, consistent with previous results (Hazell et al., 1999). Interestingly, when neurons were cocultivated with astrocytes, the decrease of PCr was even higher than the theoretical intermediate between the individual cell types. Whereas neurons were not able to increase their glycolytic activity, astrocytes showed stimulated anaerobic metabolism of external [1–13C]glucose (with NAD+ regeneration by subsequent lactate synthesis) that enabled, at least in part, cytosolic ATP synthesis. Unlike [3–13C]lactate synthesis, changes in the total amount of lactate were not significant in manganese-treated astrocytes or neurons, as observed earlier (Hazell et al., 1999). The analysis of the percentage 13C-enrichments in [3–13C]lactate (i.e., the selective changes in de novo synthesized [3–13C]lactate derived from the pyruvate pool of glycolytic origin compared with exogenous substrates other than [1–13C]glucose), however, directly reflects cell-specific changes in glycolytic activity. Our results are consistent with increased lactate in brain tissue in parallel with an ATP depletion after striatal injection of MnCl2 (Brouillet et al., 1993), but give supplementary evidence for a cell-specific occurrence of both features. Nevertheless, brain energy production depends almost exclusively on mitochondrial glucose oxidation. Pyruvate dehydrogenase, transforming [1–13C]glucose into [2–13C]acetyl-CoA and hence into the TCA-cycle intermediate [4–13C]α-ketoglutarate, is active in both neurons and astrocytes and plays the dominant role in regulation of oxidation and mitochondrial energy production in the brain. Only in astrocytes can [1–13C]glucose be transformed into [2–13C]α-ketoglutarate via the astrocyte-specific PC, the main anaplerotic enzyme in the brain (Shank et al., 1985). 13C-NMR analysis showed a dramatic inhibition of neuronal oxidative glucose metabolism via PDH, decreased 13C-enrichment in acetyl-CoA entering the TCA cycle and reduced TCA cycle turnover, leading to deterioration of the neuronal energy state and impaired de novo synthesis of neurotransmitter amino acids (i.e., glutamate, aspartate, and GABA). In contrast, in astrocytes a strongly stimulated uptake and oxidation of [1–13C]glucose through PDH, which coincided with increased 13C-enrichment in acetyl-CoA and amplified 13C-cycling ratio within the TCA cycle, seems to be the most prominent mechanism to avoid manganese-induced cellular energy failure. Our results suggest that limited availability of external glucose for mitochondrial metabolism leads to an inability of neurons to antagonize an energy depletion by oxidative metabolism, and that limited flux through PDH further aggravates the energy failure.
Since astrocytes sequester manganese (Aschner et al., 1992; Tholey et al., 1990; Wedler et al., 1989), it has been suggested that these cells are likely initial targets of manganese toxicity (Henriksson and Tjalve, 2000; Spranger et al., 1998). The prevention of energy failure by stimulated glucose oxidation and glycolytic activity in manganese-treated astrocytes does not necessarily contradict this hypothesis. The accumulation of succinate and citrate in all cell types suggests an inhibition of intermediary metabolism at the level of succinate dehydrogenase (Hietanen et al., 1981) and of aconitase (Zheng et al., 1989). Similar to a direct effect of manganese on oxidative phosphorylation (Gavin et al., 1992) and complex I activity (Galvani et al., 1995), an inhibition of these TCA cycle enzymes ultimately leads to decreased availability of reduced equivalents for oxidative phosphorylation and hence to an energy deficit. Interestingly, accumulation of succinate and citrate as well as the decrease of [13C]acetyl-CoA entering the TCA cycle were highest when neurons were cocultivated with astrocytes. Therefore, manganese-induced changes in astrocytes may either modify a primary neuronal injury or induce secondarily neuronal dysfunction.
Besides neuronal glucose oxidation, neurons rely on the supply of substrates synthesized and released by astrocytes (Hertz et al., 1999). Several in vitro studies (Pellerin et al., 1998; Waagepetersen et al., 1998; Zwingmann et al., 2000) have proposed an astrocyte–neuron “lactate shuttle” for the maintenance of neuronal energy and neurotransmitter metabolism. The different effects of manganese on the percentage 13C-enrichments in intraand extracellular lactate are consistent with the presence of metabolically compartmentalized pools of cytosolic pyruvate in neurons and astrocytes (Bouzier et al., 1998; Cruz et al., 2001; Zwingmann et al., 2001), which may be modified under pathological conditions. Compared with pure astrocytes, however, manganese-induced stimulation of glycolytic flux was less after cocultivation with neurons, and in view of the decreased 13C-enrichment in extracellular lactate, astrocytic ability to regulate glycolytic synthesis of releasable lactate seems to be uncoupled from substrate requirements in manganese-treated neurons when cocultivated with astrocytes. Nevertheless, the normal rate of glycolysis in astrocytes does not exceed approximately 15 nmol glucose·min·−1mg−1 protein (Marrif and Juurlink 1999), a value almost reached in our manganese-treated cocultures, and neuronal oxidative metabolism cannot rise more rapidly than dictated by the subtype LDH5 isoenzyme prevailing in astrocytes (Gjedde and Marrett, 2001). Although the support of neuronal function with lactate as a sole energy substrate has been reported in several encephalopathies (Nemoto et al., 1974; Schurr et al., 1997), astrocyte-derived lactate may be insufficient to restore the energy deficit in neurons suffering from impaired oxidative metabolism of glucose.
Several positron emission tomography studies have shown that neurons regulate their oxidative metabolism using their own rather than astrocytic-generated pyruvate (Gjedde and Marrett, 2001). Since neurons lack anaplerotic activity, however, they cannot maintain de novo synthesis of neurotransmitter amino acids and permanently require new carbons from astrocytes. In contrast to lactate, glial PC activity and glutamine synthesis are significant in brain interplay between glutamatergic action and glucose metabolism (Gruetter et al., 2001). Manganese may play an important role in glial metabolism (Tholey et al., 1987) owing to the localization of PC and GS (Norenberg and Martinez-Hernandez, 1979) in these cells, with both metalloenzymes requiring manganese.
In astrocytes, manganese treatment resulted in abnormal stimulation of PDH versus PC activity and intracellular accumulation of de novo synthesized [4–13C]glutamate. From the manganese-induced accumulation of glutamate in these cells, one might expect an increased flux through GS, but glutamine synthesis remained unchanged and even decreased after brief exposure despite a more than fivefold elevation of [4–13C]glutamate. Despite cocultivation of astrocytes with neurons stimulating both synthesis and release of glutamine, which is consistent with increased GS messenger RNA and activity (Mearow et al., 1990; Wu et al., 1988), de novo synthesized glutamine was hardly detectable after manganese treatment. These findings are in disagreement with reports of increased GS expression in manganese-exposed rat brain (Zheng et al. 1999) and cultured astrocytes (Chen and Liao, 2002). The complexity of GS regulation provides several explanations for this paradoxical effect. For example, changes in the synthesis rate may not necessarily coincide with changes in protein expression. In addition, depending on the concentration, manganese may both activate and inhibit GS, and maximal GS activity occurs at a defined cation/ATP ratio (Tholey et al. 1987). Although the distortion of glutamine synthesis may conserve astrocytic energy consumption, this may have profound consequences for the normal metabolic balance between astrocytes and neurons. First, as glutamine synthesized in astrocytes serves as an important neuronal energy substrate and precursor of glutamate (Hertz et al., 1999), a limited supply of glutamine to neurons would additionally impair neuronal oxidative metabolism and deplete the neurotransmitter pool of glutamate as well. Second, the inability to convert glutamate to glutamine may represent one mechanism contributing to impaired reuptake of glutamate into astrocytes after manganese exposure (Hazell and Norenberg, 1997), making neuronal cells more prone to excitotoxicity. An impaired reuptake of neuronal glutamate is consistent with the relative greater decrease of glutamine metabolically derived from [4–13C]glutamate (PDH) compared with that derived from [2–13C]glutamate formed exclusively via astrocytic PC. Furthermore, the reduction in the total amount of [4–13C]glutamine was higher than the increase of [4–13C]glutamate (23.1 vs. 11.05 nmol/mg protein), further indicating impaired trafficking via the glutamine-glutamate cycle. Similarly, the accumulation of [2–13C]GABA in cocultures, which is metabolically derived from neuronal [4–13C]glutamate rather than from astrocytic glutamine, may be the result of impaired reuptake via the GABA shunt, an important pathway for the reentry of GABA into the TCA cycle (Hassel et al., 1998).
Several studies using various paradigms of oxidative stress have shown that reactive oxygen species can inactivate GS (Brand et al., 1999; Minana et al., 1997; Oliver et al., 1990; Schor, 1988), which would be consistent with manganese-induced oxidative stress involving formation of reactive oxygen species and free radicals (Chen and Liao, 2002; Spranger et al., 1998). The reduced label incorporation from [1–13C]glucose into the [4–13C]glutamate residue of neuronal glutathione, a major intracellular antioxidant (Dringen, 2000), after prolonged manganese exposure indicates a likely impairment of the neuronal antioxidant system in parallel with energy deficits. Astrocytes, in contrast, showed a rapid, more than 10-fold increase in the synthesis of glutathione from [1–13C]glucose, which likely reflects a protective response against formation of reactive oxygen species. The higher ratio of double- to mono-labeled 13C-4 in the glutamate residue of glutathione compared with glutamate revealed glutathione synthesis occurring consecutively to manganese-induced stimulation of PDH (i.e., after the carbon skeleton of glutamate has passed several TCA cycle turns). However, the initial dramatic stimulation of PDH activity and hence glutathione synthesis slowed down in astrocytes, and the even greater decrease of [2–13C]acetyl-CoA and [13C]glutathione in cocultures compared to pure neurons, together with the deterioration of the cellular energy state, suggests a failure of astrocytes to maintain energy metabolism and antioxidant defense mechanisms after sustained manganese exposure. Interestingly, manganese exerted a similar effect on the concentration of taurine in neurons and cocultures, which, like glutathione, is a free radical scavenger and can protect against excitotoxicity (Saransaari and Oja, 2000) and plays an important role as an organic osmolyte in astrocytes (Olson and Li, 2000). Interestingly, hypotaurine, the precursor for taurine, and myo-inositol, which both participate in cell volume regulation in astrocytes (Brand et al., 1993; Isaacks et al., 1999), were strongly decreased in these cells when cocultivated with neurons and may reflect effects of manganese on the control of ion and volume regulation in astrocytes in the presence of neurons.
In this study, a selective impairment of neuronal oxidative metabolism of glucose and astrocytic substrates was shown by 13C-isotopomer analysis of [1–13C]glucose metabolism, which labels TCA cycle metabolites in different carbon positions depending on the contribution of cell-specific and intercellular pathways. We have presented data indicating that manganese produces differential changes in glucose utilization in astrocytes and neurons that may play an important role in determining the relative vulnerability of neuronal cells in manganese neurotoxicity (Fig. 10). Inhibition of neuronal PDH impedes the ability of neurons to compensate for mitochondrial dysfunction by raising oxidative glucose metabolism and further aggravates neuronal energy failure. Although astrocytes maintain their normal energy state and antioxidant mechanisms by upregulation of glucose metabolism through increased glycolysis, PDH and Krebs cycle activity, astrocytic dysfunction in the presence of neurons (e.g., reduced glutamine synthesis) may cause a homeostatic dysequilibrium between both cell types, leading to further potentiation of the primary neuronal derangement and, conceivably, the eventual development of an excitotoxic state. These self-protective metabolic responses may also facilitate astrocyte survival and the development of reactive gliosis in the face of concomitant neuronal dysfunction. Our results provide a cell type-specific basis for some possible mechanisms associated with energy failure in manganese neurotoxicity. Future studies are necessary to characterize the sequence of metabolic events. Further NMR investigations using 13C-labeled substrates involved in energy and neurotransmitter metabolism as well as the cellular antioxidant defense system, combined with in vitro, ex vivo, and in vivo approaches, may provide additional information concerning the primary basis and significance of energy failure in manganese neurotoxicity.
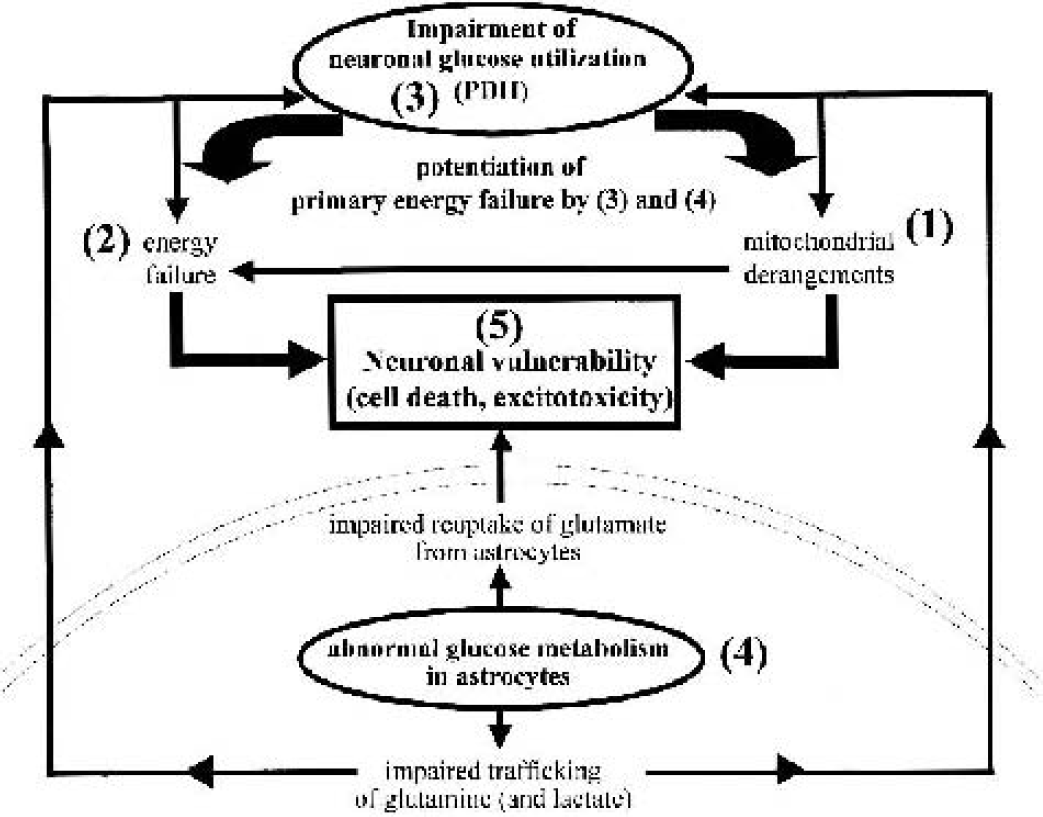
Relation between cell-specific glucose metabolism and neuronal vulnerability in manganese neurotoxicity. Mitochondrial dysfunction in neurons (