
Abstract
Astrocytic glycogen is the sole glucose reserve of the brain. Both glycogen and glucose are necessary for basic neurophysiology and in turn for higher brain functions. In spite of low concentration, turnover and stimulation-induced degradation, any interference with normal glycogen metabolism in the brain severely affects neuronal excitability and disrupts memory formation. Here, I briefly discuss the glycogenolysis-induced glucose-sparing effect, which involves glucose phosphates as key allosteric effectors in the modulation of astrocytic and neuronal glucose uptake and phosphorylation. I further advance a novel and thus far unexplored effect of glycogenolysis that might be mediated by glucose phosphates.
Introduction
Glycogen metabolism in the brain is emerging as a fundamental process for maintenance of ion homeostasis, neuronal excitability and synaptic plasticity. 1 – 3 Pharmacological or genetic manipulations inducing dysfunctional brain glycogen metabolism have been shown to impair memory formation 4 – 7 as well as to increase susceptibility to epileptic seizures8,9 and cortical spreading depression. 10 Understanding the roles of glycogen in the brain is an important goal for neuroscience, especially considering that its metabolism is exquisitely sensitive to brain state (e.g. sleep-wake cycle) and changes in glycogen synthesis and degradation occur in response to neurotransmitters, hormones, extracellular K+ and available metabolic substrates.11,12
At the organ level, brain glycogen concentration is 10-fold lower than in muscle and 100-fold lower than in liver, 13 although at cellular level astrocytic glycogen level is about half of that in muscle (see below). In both humans and rodents, resting glycogen turnover in the brain is about 1–2% of cerebral metabolic rate of glucose (CMRglc). 14 – 18 There is evidence that glycogen consumed during 5–6 min of sensory stimulation is about 25–30% of the basal levels, and the rate of glycogenolysis approaches half of CMRglc by all cells during stimulation (see Table 5 in Dienel 19 ). These findings indicate that glycogenolysis can be substantial and readily detected during prolonged (i.e. tens of seconds to minutes) stimulation. However, the amount of glycogen degraded during brief (i.e. milliseconds to seconds) bursts of neuronal activity or certain types of sensory stimulation is probably only few percent of basal levels,16,20 which can be difficult to detect using available experimental procedures. 21 The quantitive relevance of glycogenolysis is apparent when the rapidity of breakdown is taken into account. Specifically, glycogen can be readily mobilized and the variable to take into account is not necessarily the amount of glycogen that is degraded but the rate of its degradation. 22 In particular, glycogenolysis yields glucose-1-phosphate (Glc-1-P), whose concentration can rise locally to substantial levels even after mobilization of relatively small amount of glycogen. Indeed, basal Glc-1-P concentration in brain is approximately 10 µmol/L, while glucose equivalents in astrocytic glycogen are ∼40 mmol/L (assuming a conservative estimate of 6 µmol/g of glycogen content in whole brain and an astrocytic volume fraction of 15%). As an illustration, mobilization of 2.5% of glycogen, something that can happen faster than concomitant CMRglc, 23 would yield 1.0 mmol/L of Glc-1-P, which means a 100-fold increase in Glc-1-P level. The actual increase in Glc-1-P concentration also depends on the velocity of its conversion to glucose-6-phospate (Glc-6-P) and glucose-1,6-bisphosphate (Glc-1,6-P2). Unfortunately, experimental measures of glycogenolysis-induced changes of glucose phosphates in the brain are not available. However, Glc-6-P has been reported to increase 2- to 4-fold after epinephrine-induced glycogen breakdown in skeletal muscle. 24 The fast release of glucose phosphates (whose basal concentration is very small) from the glycogen pool (whose concentration in terms of glucose equivalents is relatively high) would elicit powerful effects because the allosteric targets of glucose phosphates have very small half-maximal concentrations (see below). This argument is especially relevant considering that glycogenolysis occurs in peripheral astrocytic processes (i.e. filopodia and lamellipodia), which are characterized by high glycogen content 25 yet very narrow volume and restricted diffusion due to high cytosolic tortuosity.26,27
The critical importance of brain glycogen has been hitherto explained in terms of either the astrocyte-neuron lactate shuttle (ANLS)
28
or the glucose-sparing effect.
22
The former extends the hypothesis that circulating glucose is predominantly taken up by astrocytes and therein converted to lactate and exported to the extracellular space for neuronal uptake and utilization.
29
The latter implicates glucose phosphates in the modulation of cellular glucose utilization and is the topic of the following sections. I will briefly discuss how glycogenolysis-derived glucose phosphates can constitute an allosteric signaling pathway mediating a number of immediate effects that can be of critical importance for brain function (Figure 1). On the contrary, rapid production of lactate is unlikely to exert any immediate metabolic effect, as during enhanced glycogenolysis the oxygen–glucose index falls substantially.
30
Thus, any significant oxidation of glycogen-derived lactate must take place in a later stage after glycogenolysis has superseded. The rapid increase in tissue lactate can nevertheless be influential due to its non-metabolic effects (e.g. receptor- and/or redox state-mediated signaling).
31
Readers interested in ANLS hypothesis in the context of brain glycogen metabolism are referred to recently published reviews.1,28,32,33
Schematics of the proposed glucose phosphates-mediated allosteric signaling pathway associated with astrocytic glycogenolysis. Glycogen degradation in astrocytes can be triggered by (i) adenylate kinase (AK)-mediated production of AMP after ATP hydrolysis by Na+/K+-activated adenosine triphosphatase (NKA) and/or (ii) monoamine-initiated G-protein coupled receptors (GPCRs)-mediated second-messenger signaling cascades, either the cAMP/protein kinase A (PKA) pathway or the phospholipase C (PLC)/Ca2+ pathway (for a detailed description, see DiNuzzo et al.
84
). Since glycogenolysis is faster than uptake and phosphorylation of glucose, the intracellular concentration of Glc-1-P increases. Glc-1-P enhances active K+ uptake by NKA. Glc-1-P is isomerized by phoshoglucomutase (PGM) to Glc-6-P and also converted to Glc-1,6-P2 by Glc-1,6-P2 synthase (not shown). Glc-6-P inhibits glucose phosphorylation by hexokinase (HK). Besides being a cofactor in the PGM-catalyzed reaction, Glc-1,6-P2 further inhibits HK and stimulates phosphofructokinase (PFK), thereby favoring glycolytic processing of glycogen-derived Glc-6-P and mitochondrial metabolism of downstream pyruvate (not shown) as well as oxidative phosphorylation. The decrease in substrate flow through HK entails a reduction of extracellular glucose uptake by astrocytes and glucose-sparing for neuronal utilization (dashed arrow). Overall, the effects of glycogenolysis are net changes in astrocytic uptake of potassium (increased) and glucose (decreased).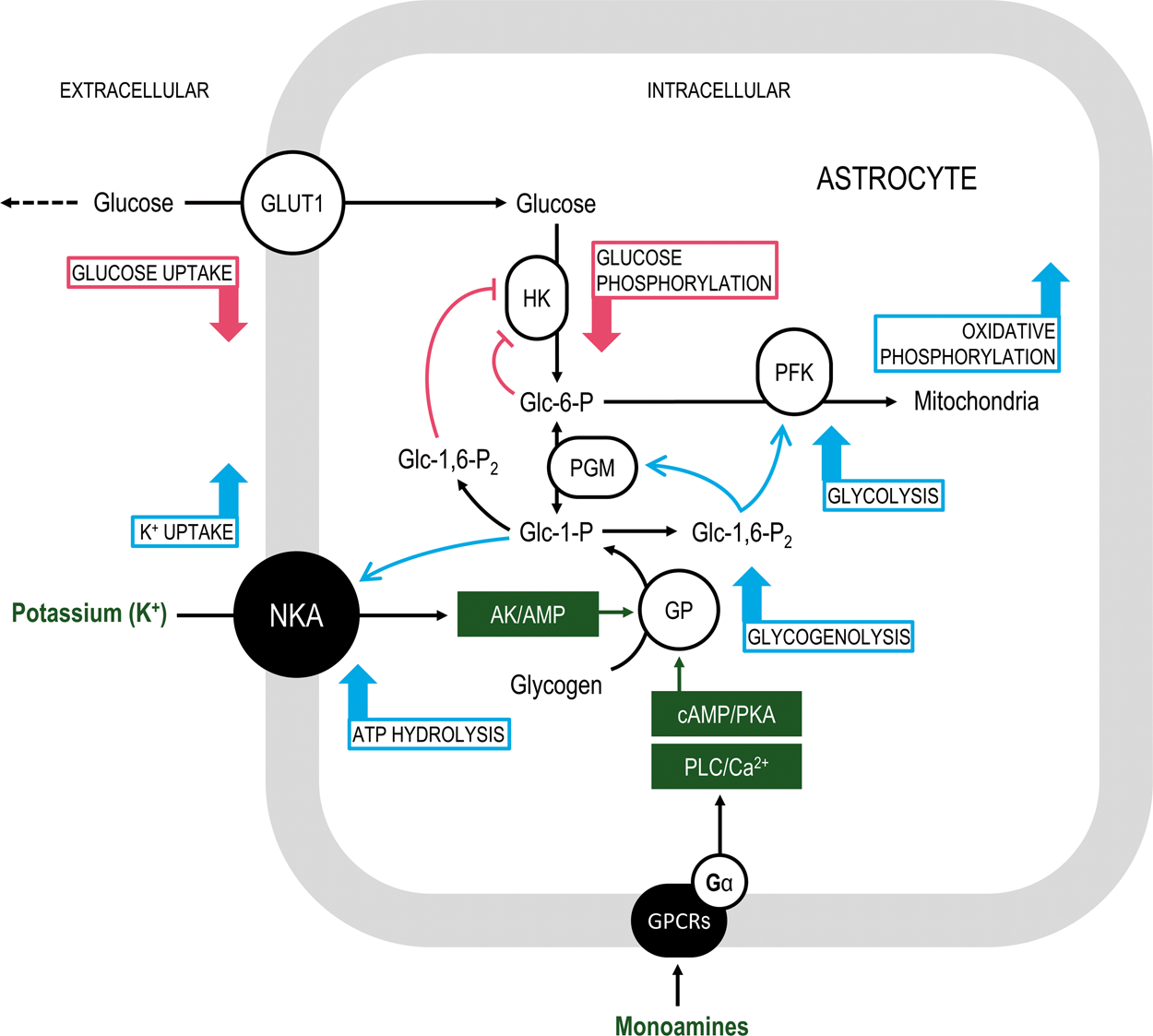
Glucose-6-phosphate
An important difference between glycolysis and glycogenolysis lies in the way of generating Glc-6-P, respectively (i) phosphorylation by hexokinase (HK) or (ii) glycogen degradation by glycogen phosphorylase (GP) to Glc-1-P followed by the action of phosphoglucomutase (PGM, the enzyme interconverting Glc-6-P and Glc-1-P). Although glycolysis and glycogenolysis are differently regulated, once Glc-6-P is generated both pathways share the same downstream reactions. Glc-6-P inhibits HK with an IC50 of 17 µmol/L. 34 Since basal Glc-6-P concentration in the brain is about 100 µmol/L, HK normally operates at ∼15% of its maximal catalytic capacity. 35 With these numbers, doubling of Glc-6-P concentration translates to halving of HK-catalyzed reaction rate (∼[1+Glc-6-P/IC50]−1), which has been theoretically shown to substantially reduce substrate flow through HK and hence decrease astrocytic glucose utilization. 22 Notably, such glucose-sparing effect by glycogenolysis has been seemingly reported 60 years ago in rat diaphragm, where it was observed that “epinephrine […] leads to an intracellular accumulation of free glucose and to an increase in the glucose-6-phosphate concentration of muscle, the latter arising from a breakdown of muscle glycogen.” 36 In rat gastrocnemius, it was then observed that “epinephrine inhibits insulin-stimulated glucose uptake in skeletal muscle by inhibiting glucose phosphorylation,” which prompted the hypothesis “that the inhibition of glucose phosphorylation is due to the stimulation of glycogenolysis, which leads to the accumulation of hexose phosphates, which inhibit hexokinase.” 37 Subsequent work in rat skeletal muscle demonstrated that the “ineffectiveness of epinephrine to suppress insulin-stimulated glucose uptake occurred in conjunction with its inability to increase the intracellular concentration of glucose-6-phosphate, […] which inhibits hexokinase activity and glucose phosphorylation.” 38 Together, these studies link the control of glucose uptake to glycogenolysis through the ability of glycogen breakdown to increase intracellular Glc-6-P concentration and hence inhibit glucose phosphorylation at the HK-catalyzed reaction step.
The occurrence of glycogenolysis-induced glucose-sparing in the brain is corroborated by several experimental observations (reviewed in Dienel 19 ). Although it has not been directly measured, it presently represents the sole proposed scenario by which neurons can secure more glucose (i.e. in addition to their basal consumption rate) during brain stimulation. 39 – 41 Glucose is necessary to neurons for energizing the machinery of axonal vesicle transport and presynaptic vesicle recycling, 42 – 45 which agrees with the low mitochondrial density observed in presynaptic terminals. 46 Indeed, nearly 50% of presynaptic terminals lack mitochondria (reviewed in Dienel 19 ). Glycolytic energy provision in neurons is also critical for action potential waveform and transmitter release. 47 Furthermore, remodeling of dendritic spines during long-term potentiation requires glycogenolysis in astrocytes. 48 Accordingly, mitochondria are abundant in dendritic shafts but only occasionally or very rarely found in dendritic spines. 49 – 52 Such distribution is consistent with biophysical calculations that allocate most of the brain's energy budget to dendritic depolarizations.53,54 Thus, although some unused ATP might diffuse from nearby dendritic mitochondria to spine heads, it is conceivable that these structures are reliant on glycolysis. It should be noted that mitochondrial oxidative metabolism is tightly coupled with excitatory glutamatergic neurotransmission and represents the major pathway for ATP turnover in both neurons and astrocytes under a great variety of brain states (see Table 2 in DiNuzzo et al. 55 ). Nonetheless, aerobic glycolysis in the brain is substantially up-regulated during stimulation regardless of oxygen availability (for an extensive discussion on this topic, see Dienel and Cruz 33 ).
Glucose-1,6-bisphosphate
Glc-1,6-P2 and its synthetizing enzyme are abundant in the brain. 56 The brain is the tissue with highest capability of Glc-1,6-P2 synthesis, 57 with Glc-1-P as its predominant precursor (Km = 19 µmol/L). 58 In addition to inhibit HK 59 – 61 with an IC50 of 400 µmol/L, 62 it is well-known that Glc-1,6-P2 stimulates phosphofructokinase (PFK) and possibly pyruvate kinase. 63 – 66 Furthermore, Glc-1,6-P2 also acts as a co-enzyme and disinhibitor for PGM,61,67 although its basal concentration in the brain (>100 µmol/L) is already above that required to maintain PGM activity. 68 The net results of all these regulations are to switch-off glucose phosphorylation while boosting metabolism of glycogen-derived Glc-6-P. A similar toggle-like effect has been described in the rodent diaphragm. 69 In particular, it was suggested that Glc-1,6-P2 might sustain inhibition of glucose utilization after the transient increase of Glc-6-P had vanished due to its metabolism in the PFK-catalyzed reaction (after isomerization to fructose-6-phosphate). In addition, the increased availability of Glc-6-P eventually enhances its channeling into the pentose phosphate pathway, further stimulating PFK due to the ensuing increase in ribose-1,5-phosphate, 70 the synthesis of which is directly enhanced by Glc-1,6-P2 in the presence of Glc-6-P and ribose-1-phosphate (Glc-1,6-P2 is also a cofactor for the phosphopentomutase-catalyzed interconversion between ribose-5-phosphate and ribose-1-phosphate).71,72
Glucose-1-phosphate
Brain energy consumption is primarily driven by the action of Na+/K+-activated adenosine triphosphatase (NKA), which maintains far-from-equilibrium Na+ and K+ (and indirectly other ionic species) concentrations across cell membranes. 53 Neuronal activity is accompanied by increases in intracellular Na+, which mainly stimulates neuronal NKA (Km for Na+ is ∼25 mmol/L in neurons versus ∼10 mmol/L in astrocytes), as well as extracellular K+, a fair part of which is rapidly taken up by astrocytic NKA-mediated transport (Km for K+ is ∼6 mmol/L in astrocytes versus ∼2 mmol/L in neurons). 73 – 75 Neurons and astrocytes express several NKA isoforms that differ in subunit composition: neurons express low levels of α1β1 and high levels of α3β1, while astrocytes express the whole constellation α1β1, α1β2, α2β1, α2β2.76,77 Importantly, α1β1 and α2β1 are the only isoforms to be associated with the auxiliary protein FXYD7, which reduces the enzyme affinity for extracellular K+. 78 Low NKA affinity for extracellular K+ underlies the capacity of substantial upregulation of reaction rate and is a prerogative of active astrocytic K+ uptake. The FXYD7 association with α1β1 might reinforce the astrocyte-specific α2β1 isoform, which has an intrinsically low K+ affinity. Using computational flux balance analysis, it has been shown that NKA-mediated uptake of extracellular K+ by astrocytes accounts for the high rates of oxidative metabolism experimentally reported to occur in these cells. 55 It might seem therefore surprising that astrocytic glycogen is necessary for active K+ uptake (but see below). Direct experimental proof supporting the requirement of glycogenolysis for K+ uptake is currently limited to few studies.10,79,80 In cultured astrocytes, inhibition of glycogenolysis was found to abolish K+ uptake by these cells even in the presence of supraphysiological glucose concentration. 79 A first attempt to replicate these findings in brain slices failed, 81 most likely because the experimental design was sub-optimal (Leif Hertz, personal communication). In particular, (i) high glutamate, not K+, was used to increase neuronal activity, yet glutamate has been previously reported to have complex effect on glycogen, eventually increasing glycogen synthesis not breakdown, 82 and (ii) the experiment was performed 30 min after slicing, yet glycogen is known to be rapidly degraded during slice preparation and its recovery needs at least 3 h. 83 More recently, impairment of extracellular K+ clearance due to inhibition of glycogenolysis has been confirmed in vivo during whisker stimulation in mouse, with a suggested involvement of the astrocyte-specific α2 NKA catalytic subunit. 10 In order to explain these findings, it is indeed necessary to postulate the existence of a signal derived from glycogen and acting (as a signaling molecule, not metabolic fuel) on the sodium pump, as previously hypothesized.12,84 Glc-1-P can be identified as the glycogen-derived signal that is sensed by NKA.
Experimental support to this idea comes from studies performed by one single group that primarily investigated the role of galactose and other hexoses on brain ATPases activity.85,86 In particular, these authors found that Glc-1-P stimulated NKA from both rat brain homogenates and pure enzyme preparations from porcine cerebral cortex (Sigma A7510). 85 In particular, a 400% increase in enzyme activity was observed at 1.0 mmol/L Glc-1-P (the level potentially reached after degradation of 2.5% of astrocytic glycogen; see above) in pure enzyme preparations. The stimulation was thought to be direct and dependent on the phosphate, since glucose had no effect over a wide range of concentrations (up to 16 mmol/L). Moreover, the stimulation of NKA activity kept rising at increasing Glc-1-P levels, reaching + 3900% at 8 mmol/L in pure enzyme preparations (the increase was around 100% in rat brain homogenates). Although the above experiments have been performed using a 1-h incubation protocol, the relevant figures are consistent with a significant effect of Glc-1-P on NKA activity. For example, since the dose–response curve for the range of Glc-1-P concentrations used in the above-mentioned study was found to be linear, the reported effect translates in a 40% increase in NKA activity for a 100 µmol/L rise in Glc-1-P level, which in principle requires the degradation of only 0.25% of glycogen in astrocytes. As for the other glucose phosphates, the effect of Glc-1-P is most likely very local, i.e. occurring in volume- and diffusion-restricted subcellular astrocytic domains.
Concluding remarks
In the present paper, I argued that an allosteric signaling mechanism mediated by glucose phosphates might explain how glycogenolysis influences two fundamental processes underlying brain function, namely energy metabolism of glucose (HK effect) and ion homeostasis (NKA effect). In particular, the potential of Glc-1-P to stimulate NKA activity would be specific to astrocytic glycogen mobilization, because HK (which yields Glc-6-P) is much slower than active GP, which means that HK is unlikely to produce substantial rise in intracellular glucose phosphates. Importantly, glucose phosphates cannot leave the cells where they are generated. Indeed, glucose-6-phosphatase activity in brain is negligible87,88 and glucose phosphates cannot be dispersed among gap-junction coupled astrocytes (i.e. the astrocytic syncytium), as gap-junctions are not permeable to these compounds. 89 Thus, the rapid production of glucose phosphates in astrocytes following glycogenolysis could recapitulate the well-known intracellular Glc-6-P compartmentation, 90 – 95 which in the brain is consistent with the observation that within astrocytes carbons derived from glucose and glycogen do not mix. 96 The combined stimulation of NKA and glycolytic processing of glycogen-derived Glc-6-P might explain the apparently non-energetic coupling between NKA activity and ATP generated by glycolytic enzymes. 97 Finally, the potential effect of glycogenolysis on NKA activity provides a direct route by which monoaminergic signaling modulates neuronal excitability in the cerebral cortex and hippocampus. 11 The understanding of whether such mechanisms are functionally meaningful awaits unequivocal experimental confirmation. Submitting the above-mentioned concepts to experimental test will hopefully provide a route to understand why glycogen is so critical to brain function.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.