
Abstract
Bcl-xL is a protein that blocks both apoptotic and necrotic cell death. The authors have previously shown that it is effective in maintaining mitochondrial membrane potential during glucose deprivation in cultured astrocytes. To further investigate the mechanism involved, the authors studied mitochondrial function and cytochrome c release. Oxygen consumption was monitored to assess oxidative respiration. State III respiration decreased significantly as early as 3 h after removal of glucose. At this time mitochondria hyperpolarize but cytochrome c is not yet released. Damage to the electron transport chain is not responsible for this change because uncoupled respiration was unchanged at this time point. At 5 h of glucose deprivation, when mitochondrial depolarization was observed, state IV respiration increased significantly, cytochrome c began to be released, and mitochondrial morphology changed from elongated to punctate. When Bcl-xL was overexpressed normal state III respiration and mitochondrial morphology were maintained and cytochrome c release was inhibited in the face of glucose deprivation stress.
Mitochondria play a central role in all kinds of cell death by controlling cellular energy metabolism, production of reactive oxygen species, and release of proapoptotic factors into the cytosol (Murphy et al., 1999; Beal, 2000; Ravagnan et al., 2002). The most prominent proapoptotic factor released from mitochondria is cytochrome c (Liu et al., 1996; Lim et al., 2002). One theory proposes that the opening of the mitochondrial permeability transition pore stimulates the release of proapoptotic factors (Zamzami et al., 1996; Lemasters et al., 1998; Marzo et al., 1998; Vande Velde et al., 2000). Because permeability transition-pore opening also results in inner membrane permeability to protons, it is associated with a decrease in mitochondrial transmembrane potential (ΔΨm) (Zoratti and Szabo, 1995). In many neuronal and nonneuronal apoptotic systems, however, cytochrome c release occurs before a loss of ΔΨm and proceeds in the presence of PTP inhibitors (Bossy-Wetzel et al., 1998; Krohn et al., 1999; Deshmukh et al., 2000). It has also been reported that mitochondrial membrane hyperpolarization (Vander Heiden et al., 1997; Scarlett et al., 2000) may precede cytochrome c release. The relation between the different states of mitochondrial respiration, ΔΨm, and cytochrome c release have not yet been described. Although we previously showed that Bcl-xL reduced glucose deprivation (GD)–induced cell death (Xu et al., 1999), to understand the mechanism of protection it is important to look at early cell responses to stress. Because GD is a slow injury we were able to correlate these important aspects of mitochondrial function as they change early in the injury process and determine the effect of Bcl-xL overexpression.
Bcl-xL is an antiapoptotic protein in the Bcl-2 family and localizes to intracellular membranes, including the outer mitochondrial membrane. Many studies have shown the protective effects of Bcl-xL against both apoptotic and necrotic cell death (Kane et al., 1995; Vander Heiden and Thompson, 1999; Xu et al., 1999; Gottlieb et al., 2000; Gross, 2001). Bcl-xL has been shown to inhibit release of cytochrome c and other mitochondrial proapoptotic proteins (Kim et al., 1997; Poppe et al., 2001). Despite investigations in many laboratories, the mechanism of Bcl-xL protection remains unclear. Our recent study showed that Bcl-xL overexpression in astrocytes markedly delayed and reduced reactive oxygen species accumulation and blocked changes in mitochondrial membrane potential induced by GD (Ouyang et al., 2002). To further investigate the mechanism involved in the protective effect of Bcl-xL against the mitochondrial membrane potential changes, we examined the effect of Bcl-xL overexpression on cell respiration and cytochrome c localization early after GD in mouse astrocytes and correlated them with changes in mitochondrial membrane potential.
MATERIALS AND METHODS
All chemicals were obtained from Sigma (St. Louis, MO, U.S.A.) unless otherwise noted.
Primary astrocyte cultures were prepared from postnatal (days 1 to 3) Swiss Webster mice (Simonsen, Gilroy, CA, U.S.A.) as previously described (Ouyang et al., 2002). The cultures were infected with retroviral vectors encoding mouse Bcl-xL, or the Eschericia coli Lac-Z gene encoding β-galactosidase and selected with G418 as previously described (Papadopoulos et al., 1998; Xu et al., 1999). Cells were used for experiments after 25 d in culture.
Tetramethylrhodamine ethylester, a potentiometric fluorescent dye that incorporates into mitochondria in a ΔΩm-dependent manner, was purchased from Molecular Probes (Eugene, OR, U.S.A.) and used to determine the time course of changes in ΔΩm as previously described (Reichert et al., 2001; Ouyang et al., 2002).
Cell respiration was measured following the method of Moreadith and Fiskum (1984) and Fiskum et al. (2000) with some modification (Ouyang et al., 1997a,b). Astrocytes, either control uninfected, Lac-Z expressing, or Bcl-xL overexpressing, from one well of a 6-well plate, were “permeabilized” by adding 0.01% (volume to volume) digitonin. The respiration medium contained 130 mmol/L KCl, 2 mmol/L KH2PO4, 1 mmol/L MgCl2, 5 mmol/L HEPES, 5 mmol/L malate, and 5 mmol/L glutamate. State III respiration was initiated by adding 0.4 mmol/L ADP. Oligomycin (2 μg/mL) was used to initiate state IV respiration, and the uncoupled rate was determined by addition of 0.1 μmol/L carbonylcyanide m-chlorophenylhydrazone. The oxygen consumption rate was measured using an oxygen electrode (Microelectrodes, Bedford, NH, U.S.A.) connected to a digital multimeter, recorded using ScopeView software (RadioShack, Fort Worth, TX, U.S.A.), and normalized to the protein content.
Fluorescence immunocytochemistry was performed on cell cultures on coverslips similar as previously described (Ouyang et al., 1999). The coverslips were washed twice in phosphate-buffered saline (PBS) for 5 minutes at room temperature and then in PBS containing 0.2% Triton-X100 (TX100) for 30 minutes. Nonspecific binding sites were blocked in 3% bovine serum albumin in PBS/0.2% TX100 for 30 minutes. Antibody for cytochrome c was from Pharmingen (Catalog No. 556432; San Diego, CA, U.S.A.). The cells were incubated with cytochrome cprimary antibody (1:500 dilution in PBS/0.1% TX100 and 1% bovine serum albumin) overnight at 4°C, then washed in PBS containing 0.1% TX100, 3 times at room temperature. The primary antibody was visualized with fluorescein-labeled anti-mouse secondary antibody, and propidium iodide was used to stain nuclei. Coverslips were washed several times in PBS/0.1% TX100, mounted on glass slides using Fluoromount-G (Southern Biotechnology Associates, Birmingham, AL, U.S.A.) and observed with an epifluorescence microscope (Nikon Diaphot; Nikon Corporation, Tokyo, Japan).
Statistics
All data reported represent at least 3 independent experiments. Data reported are means ± SD. Statistical difference was determined using ANOVA followed by Scheffé test with P < 0.05 considered significant, using StatView software (SAS Institute, Cary, NC, U.S.A.).
RESULTS AND DISCUSSION
The oxygen consumption rate was measured using an oxygen electrode, and typical response curves are shown in Fig. 1A. Rates of state III (phosphorylation) respiration are measured in the presence of oxidizable substrates and ADP. State III reflects the maximal rate of coupled respiration, that is, when electron transport is coupled to ATP synthesis. State IV (resting) respiration can be measured in the absence of ADP or in the presence of the ATP synthase inhibitor oligomycin and reflects the rate of leakage of protons back across the inner mitochondrial membrane into the matrix. Uncoupled respiration can be measured by addition of an uncoupler such as carbonylcyanide m-chlorophenylhydrazone and reflects the maximal rate at which substrate oxidation and electron flow through the chain to O2 can take place in the absence of the rate restriction imposed by the synthase or the adenine nucleotide translocator (Murphy et al., 1999). Acceptor control ratio (ACR, state III/state IV) and respiratory control ratio (RCR, uncoupled rate/state IV rate) are expressions of the capacity to perform oxidative phosphorylation (Myers et al., 1995).
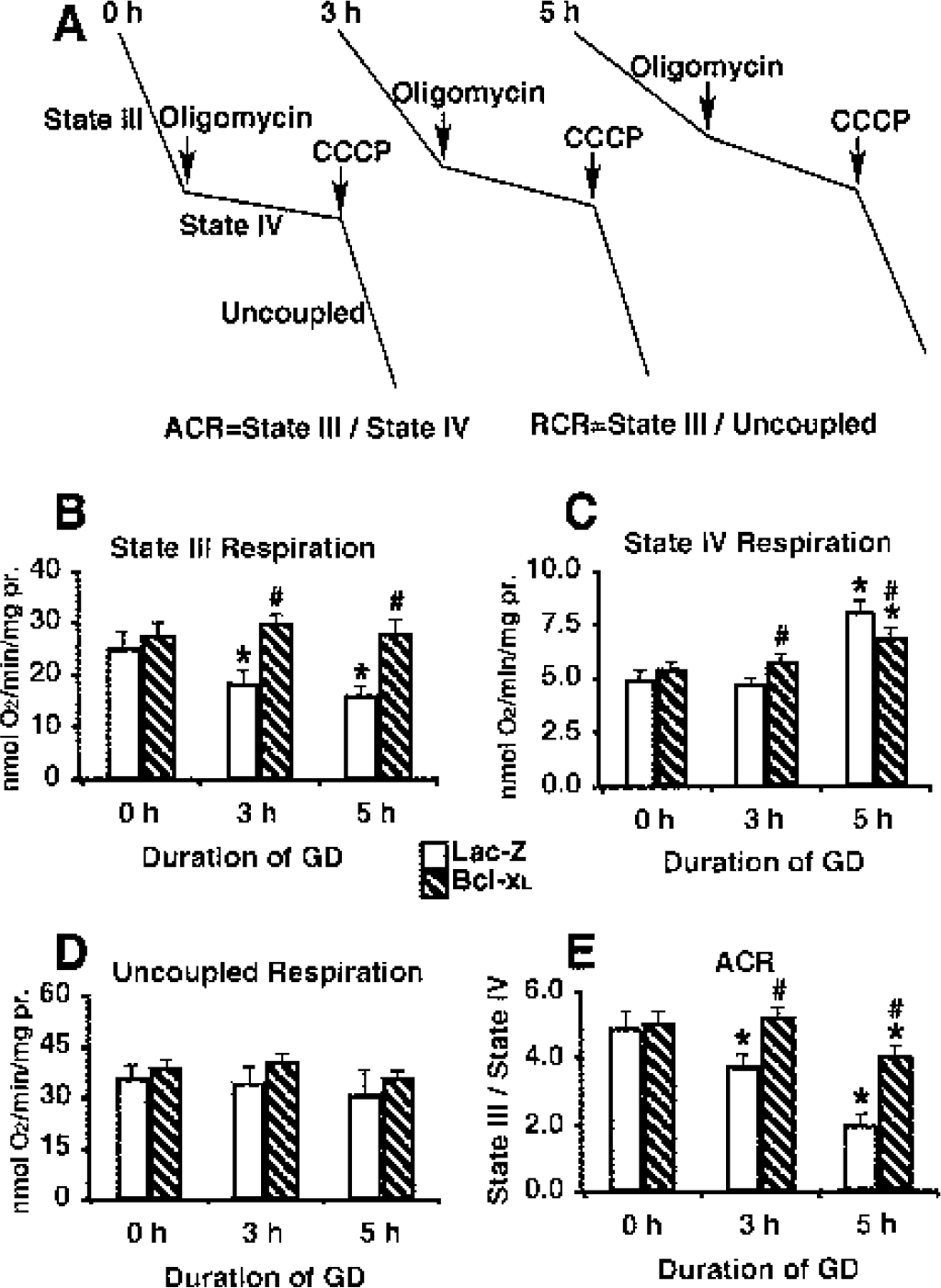
Mitochondrial respiration after glucose deprivation (GD) exposure in mouse astrocytes.
State III respiration decreased significantly as early as 3 h after removal of glucose (Figs. 1A and 1B). At this time point state IV respiration (Figs. 1A and 1C) and uncoupled respiration (Figs. 1A and 1D) did not change. This is a functional change and it is observed at the time at which mitochondria hyperpolarized (Ouyang et al., 2002). At 5 h GD state IV respiration increased significantly (Figs. 1A and 1C) and state III respiration declined further compared to 3 h (Fig. 1B). In contrast, uncoupled respiration did not change much compared with 0 h GD (Fig. 1D). Because state IV respiration reflects mitochondrial membrane leakage, at 5 h GD the membrane is very leaky. This is the time point at which mitochondria underwent depolarization (Ouyang et al., 2002). It is clear from the preceding results that uncoupled respiration did not change at 3 h or 5 h GD (Fig. 1D), when state III (Fig. 1 B) and state IV (Fig. 1 C) did. Because uncoupled respiration reflects the integrity of the electron transport chain, the electron transport chain was not damaged at 3 h or 5 h GD. The significant decrease in oxidative phosphorylation capacity seen at 3 h in Lac-Z control cells (Fig. 1E) was due to the decrease of state III respiration at 3 h, whereas the further decrease at 5 h reflected both the further decline of state III respiration and the increase in state IV respiration at 5 h. Bcl-xL overexpression prevented the decrease in state III respiration at 3h and 5 h and moderated the increase in state IV respiration at 5 h GD. As shown in Fig. 1C, Bcl-x overexpressing cells initially had slightly higher state IV respiration at 0 h GD compared with the control group, and the tendency became apparent at 3 h GD. This agreed with the slight depolarization at 3 h GD in the Bcl-x group (Ouyang et al., 2002). At 5 h GD, the mitochondrial membrane became very leaky as seen in a significant increase in state IV respiration (Fig. 1C) and marked depolarization (Ouyang et al., 2002) in control cells. Bcl-xL overexpressing cells, however, maintained a slightly increased state IV respiration (Fig. 1C) and moderately depolarized ΔΩm (Ouyang et al., 2002) at 5 h GD. This was associated with both maintained mitochondrial morphology and absence of cytochrome c release (see below). To the best of our knowledge this is the first report of the effect of Bcl-xL on the different respiratory states of astrocyte mitochondria in response to GD.
Figure 2 shows immunostaining for cytochrome c in astrocytes after 3 h, 5 h, 8 h, or 24 h of GD. There was no obvious release of cytochrome c from mitochondria at 3 h GD (Fig. 2B) when compared with time 0 h (Fig. 2A). At 5 h GD, however, about 1/4 of the astrocytes showed an evenly distributed immunostaining pattern that reflects release of cytochrome c from mitochondria to the cytosol (Fig. 2C, arrow). At 8 h GD, whereas some cells still showed a normal mitochondrial pattern (Fig. 2D, upper cell), some cells showed cytochrome c release (Fig. 2C), and some cells showed a special pattern with a dark ring around the nucleus (indicating depletion of cytochrome c) and the shape of the mitochondria changed from elongate to round or punctate (Fig. 2 D, arrow head). By 24 h GD, almost all of the cells had changed to this pattern (Fig. 2 E, arrow heads). Bcl-xL overexpression prevented the loss of cytochrome c from mitochondria and the change in morphology (Fig. 2F). Cytochrome c release occurred at the time point when state IV respiration changed and after state III respiration had already decreased.
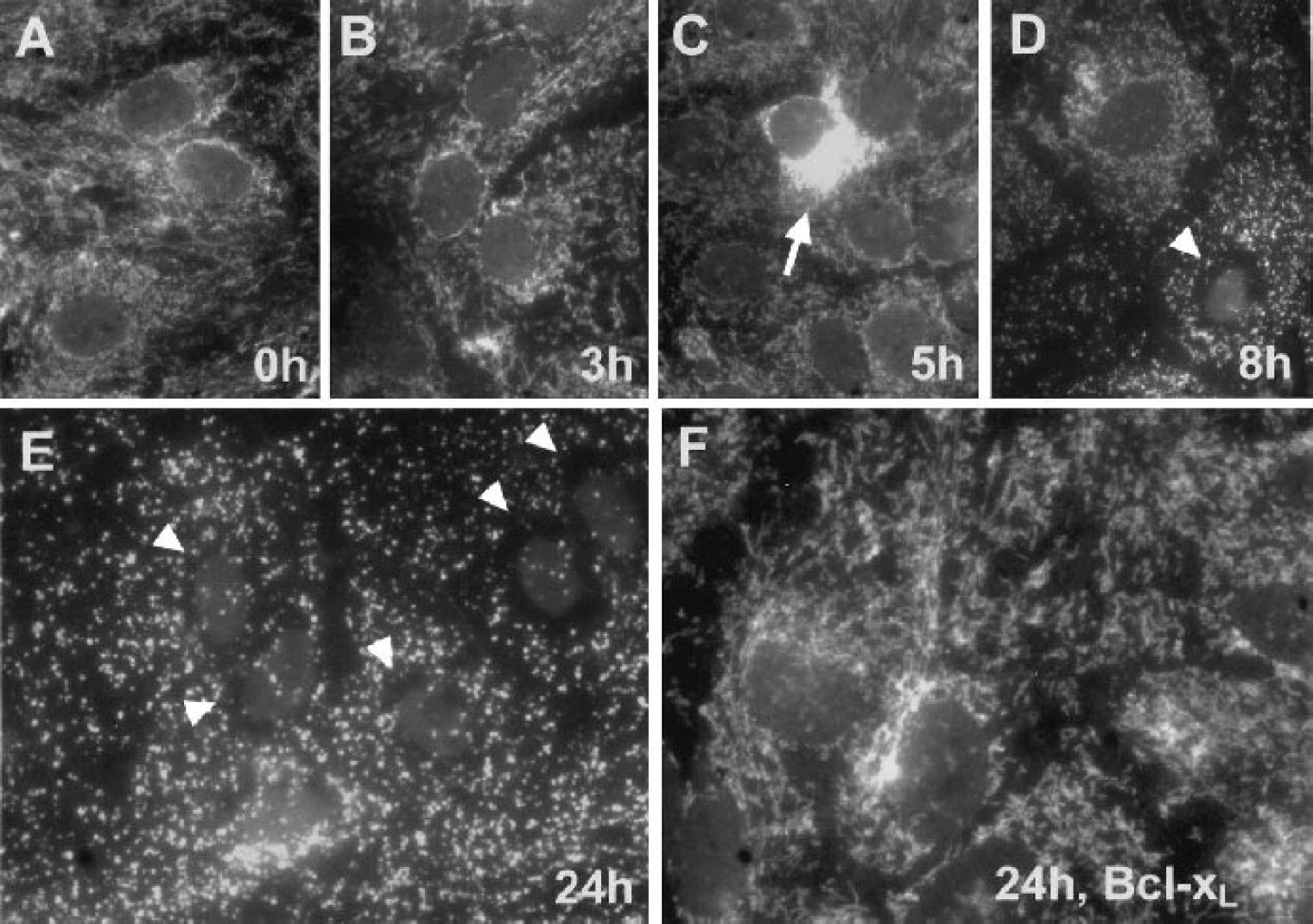
Photomicrographs of astrocytes expressing Lac-Z immunostained for cytochrome c after 0 h
A very strong case has been made for the role of mitochondria in inducing apoptosis and controlling cell death commitment (Reed and Kroemer, 2000) and for the role of Bcl-2 (Harris and Thompson, 2000). It is now known that mitochondrial release of cytochrome c is a crucial event in caspase activation and that antiapoptotic Bcl-2 proteins, including Bcl-2 and Bcl-xL, can prevent this release whereas proapoptotic family member can promote release. Several hypotheses have been proposed to explain the effect of the Bcl-2 family on mitochondria. These include regulating the voltage dependent anion channel on the outer membrane of mitochondria (Tsujimoto and Shimizu, 2000), the adenine nucleotide translocator on the inner mitochondrial membrane (Vieira et al., 2000) and the F0F1-ATPase/H+ pump (Matsuyama and Reed, 2000). Our study shows the ability of Bcl-xL to inhibit reduction in state III respiration and increase in state IV respiration triggered by the stress of GD, as well as inhibiting cytochrome c release and change in mitochondrial morphology.