
Abstract
Accumulating evidence indicates a significant astrocytic involvement in cerebral ischemia neuropathology, but little is known about the immediate astrocytic responses to ischemia insults in terms of electrophysiology and their pathologic implications. We show that astrocytes in acute rat hippocampal slices responded reversibly to more than 30 mins oxygen and glucose deprivation (OGD) treatment with depolarized membrane potentials (Vm) in whole-cell current clamp recording. This depolarization was multiphasic, showing an initial ~11 mins small-amplitude depolarization plateau, followed by a 6-mins accelerated depolarization, and then a second plateau. Oxygen and glucose deprivation-induced astrocyte Vm depolarization was only marginally inhibited, ~10%, by inhibition of ionotropic glutamate, γ-aminobutyric acid, purinergic receptors, and glutamate transporters presumed to be present on astrocytes in situ, suggesting increase in extracellular [K+] was primarily responsible for the astrocytic Vm change. The Vm depolarization was five-fold greater when glycolysis was inhibited by iodoacetate in a short 8 mins OGD treatment, suggesting glycolytic ATP is critical in maintaining extracellular K+ homeostasis in the early phase of OGD. Addition of oxidative metabolism inhibitors had much less effect. Cessation of OGD was always followed by a rapid and transient 9 mV astrocyte Vm hyperpolarization relative to the control Vm that was inhibited by ouabain, indicating a reactively enhanced Na+/K+-ATPase activity in post-OGD reperfusion. Altogether, hippocampal astrocytes appear to be electrophysiologically more resistant to acute ischemia insults as compared with neurons, and this should allow astrocytes to rescue endangered neurons in the face of acute ischemia insults via their various homeostatic functions.
Introduction
Cerebral ischemia, caused by the occlusion of blood supply to part of or the entire brain, is a major cause of brain damage. However, the precise cellular mechanisms underlying ischemia-induced neuronal damage over the course of ischemia and reperfusion still require extensive exploration. Meanwhile, although accumulating evidence indicates an astrocytic involvement in cerebral ischemia, their precise roles in this pathologic process are largely unclear and to some extent even controversial (Nedergaard and Dirnagl, 2005; Dienel and Hertz, 2005). For example, astrocytes have been found to be either protective or deleterious to ischemia-induced neuronal dysfunction that depends on the ischemia and reperfusion times (Swanson and Kauppinen, 2004). Therefore, a full understanding of astrocytic responses to ischemic insults and their underlying mechanisms is important for the future development of glial-oriented therapeutic strategies to combat the consequences of cerebral ischemia.
Previous sharp electrode intracellular recording studies showed that hippocampal astrocytes in situ responded to hypoxia with membrane potential (Vm) depolarization (Müller and Somjen, 2000; Leblond and Krnjevic, 1989). However, the mechanisms behind this Vm change were not examined. The in vivo glial Vm depolarization has been traditionally viewed as a passive response to the increased extracellular K+ on neuronal excitation (Orkand et al, 1966) because of the predominant K+ conductance expression by mature astrocytes (Kuffler et al, 1966; Picker et al, 1981). Therefore, if astrocytes could survive in early ischemic insults, they may potentially act as K+ electrodes to sense ischemia-induced extracellular K+ fluctuations. Accordingly, the ischemia-induced three phases of extracellular K+ accumulation in vivo described by Hansen (1985) should correspond to a multiphasic changes in astrocytic Vm.
However, ischemia is also associated with massive neurotransmitter release and astrocytes express a variety of neurotransmitter receptors (Verkhratsky and Kettenmann, 1996; Zhou and Kimelberg, 2004), whose activation during ischemia could mediate ion flux that contributes to the astrocyte Vm change. Thus, the relationship between increased [K+]o and neurotransmitter-mediated astrocyte Vm changes needs to be determined.
Removal of glucose and oxygen from the perfusion solution for acutely prepared brain slices is a commonly used in vitro ischemia model (Martin et al, 1994; Allen et al, 2005; Chao et al, 2006). In this study, we applied glucose and oxygen-deficient artificial cerebrospinal fluid (aCSF) (oxygen and glucose deprivation (OGD) solution) to acute rat hippocampus slices and examined the OGD-induced astrocyte Vm changes by patch-clamp recording. The results show that astrocytes responded to OGD with three phases of Vm depolarization and a rapid hyperpolarization in the post-OGD recovery period. The results also show that ATP from glycolysis is the major energy source in maintaining the early slow and small-amplitude phase of Vm depolarization during OGD treatment.
Materials and methods
Hippocampal Slice Preparation
Acute hippocampal slices were prepared from 3- to 4-week-old rats. The procedure was performed in accordance with a protocol approved by the Wadsworth Center, New York State Department of Health Institutional Animal Care and Use Committee. Animals were anesthetized before decapitation, and their brains were removed from the skull and placed in an ice-cold, oxygenated (5% CO2–95% O2, pH 7.35) slice preparation solution containing (in mmol/L) 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 10 MgCl2, 10 glucose, 0.5 CaCl2, and 240 sucrose. Final osmolarity was 350±2 mosM. Coronal slices of 300 μm thickness were cut with a Vibratome 1500 (Ted Pella Inc., Redding, CA, USA) and transferred to a nylon-basket slice holder in aCSF containing (in mmol/L) 125 NaCl, 25 NaHCO3, 10 glucose, 3.5 KCl, 1.25 NaH2PO4, 2.0 CaCl2, and 1.0 MgCl2 (osmolarity, 295±5 mosM) at 20 to 22°C. The slices were allowed to recover in aCSF with continuous oxygenation for at least 60 mins before recording.
Electrophysiology
For in situ whole-cell recording, individual hippocampal slice was gently transferred into the recording chamber that was constantly perfused with oxygenated aCSF (2.5 mL/ min). The volume of the solution in the chamber was 250 μL during perfusion. Astrocytes in the CA1 region were initially identified based on their locations, morphology, and size—confirmed by their characteristic electrophysiologic features (see Results). Whole-cell membrane currents were amplified by a MultiClamp 700A amplifier, sampled by a Digidata 1322A Interface, and data acquisition was controlled by pCLAMP 9.0 software (all from Axon Instruments, Union City, CA, USA) installed on a Dell personal computer. Low-resistance patch pipettes were fabricated from borosilicate capillaries (OD (outer diameter): 1.5 mm; Warner Instrument Corporation, Hamden, CT, USA) using a Flaming/Brown Micropipette Puller (Model P-87, Sutter Instrument Co., Novato, CA, USA). When filled with K-gluconate-based electrode solution (see below), the electrode resistance was within 3 to 5 MΩ. Membrane potential was read in ‘I=0’ mode when recording was performed in voltage clamp mode or continuously monitored in current clamp mode. Membrane capacitance (Cm) and access resistance (Ra) were measured by using the ‘Membrane test’ protocol built into the pCLAMP 9.0. Patch pipettes were filled with a solution containing (mmol/L) 3 KCl, 137 K-gluconate, 0.5 CaCl2, 1 MgCl2, 5 EGTA (ethylene glycol bis(β-aminoethylether)-N,N,N',N',-tetraacetic acid), 10 HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 3 Mg-ATP, and 0.3 Na-GTP (pH was adjusted to 7.25 to 7.27 at 20°C with KOH, 280±5 mosM). In some experiments, ATP and GTP were omitted from pipette solution to study astrocyte Vm response to OGD in this ATP/GTP-free condition. All the experiments were conducted at room temperature (22 to 24°C).
Oxygen and Glucose Deprivation Procedure
Glucose deprivation was achieved by substituting D-glucose with equimolar sucrose in the standard aCSF (see above). Oxygen deprivation was achieved by bubbling D-glucose-deficient aCSF with 95% N2 and 5% CO2 for 30 mins before each experiment and continuing throughout the OGD treatment, while maintaining the same pH at 7.3 to 7.4 as in the control normal aCSF solution.
Solutions and Reagents
Data Analysis
Data are presented as means±s.e.m., unless indicated otherwise. Student's t-test was performed to assess the statistical significance of the difference in Vm changes before and after treatment in the same experimental group. One-way analysis of variance (ANOVA) test was performed to determine the statistical significance of the difference in Vm changes between two experimental groups. Differences were considered significant at P < 0.05.
Results
Identification of Mature Astrocytes in Acute Hippocampal Slices
To examine early astrocyte responses to ischemia insults close to the in vivo conditions, freshly prepared hippocampal slices were used, as the cytoarchitecture and local neuronal circuits remain intact. Astrocytes were initially identified according to their characteristic soma shapes and locations in CA1 region visualized through an infrared differential interference contrast (IR-DIC) video microscopy (Figure 1A), as described before (Zhou et al, 2006). Astrocytes were small in soma size (≤10μm), appearing either as round or irregular shapes, with one or two visible primary processes extending from the soma. Their identity was confirmed further by their distinctive electrophysiologic characteristics shown in whole-cell voltage-clamp recording. As we described before, mature astrocytes predominantly express linear I to V conductance that can be evoked by a series of de- and hyperpolarization voltage steps (Figures 1B to 1C) (Zhou et al, 2006). Such a linear whole-cell I to V relationship is predominately due to the expression of leak K+ channels, as the reversal potential of the whole-cell currents (Vm = −75 mV) is much closer to the equilibrium potential of K+ (EK= −96 mV) than to any other ion/ anion in the recording solutions (Figure 1C). The cells were also characterized by their low membrane resistances (Rm), typically less than 10MΩ. As indicated before, after the third postnatal weeks, this type of astrocytes appear as the only L-glutamate/L-aspartate transporter (GLAST) (+) astrocytes in rat hippocampal CA1 region (Zhou et al, 2006). Therefore, all the recordings were obtained from rats older than 21 postnatal weeks, and only astrocytes showing these characteristics were selected for study. Accordingly, only mature astrocytes from the rat hippocampal CA1 region were studied.
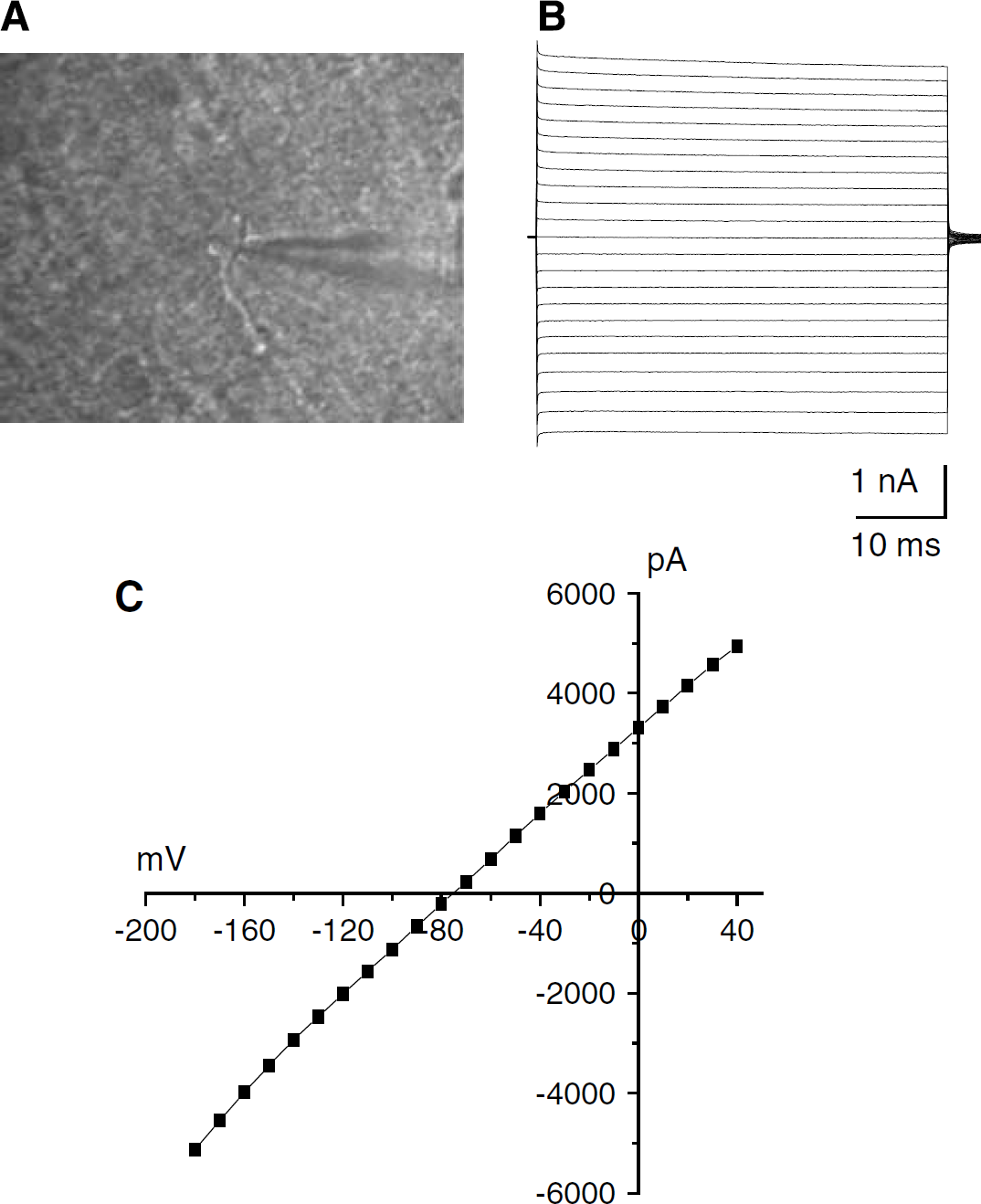
Identification of mature astrocytes in rat hippocampal CA1 region. (
Oxygen and Glucose Deprivation Induces a Multiphase Astrocyte Membrane Potential Change
Since we wanted to study the real time course of astrocyte Vm response to early ischemia-like insults, ischemic solutions were prepared by simply substituting glucose by sucrose and oxygen by bubbling with nitrogen in normal aCSF (Allen et al, 2005), as described in the Materials and methods. Although alternative ischemic solution containing high K+ low Ca2+ acidic pH has also been used (Rytter et al, 2003), these mimic the consequences of ischemic condition, so it was not used as it would not serve the purpose of this study.
To monitor continuously the changes in astrocytic Vm in response to acute OGD insults, the current clamp recording mode was applied. A pair of ±500 pA/5 ms current pulses, separated from each other by 50 ms, was delivered to the recorded cell every 20 secs to monitor the changes in whole-cell access resistance (Ra). In all the accepted recordings, the Ra values were less than 15 MΩ, and the variation of Ra in each recorded cell was less than ~ ±15% throughout the experiment. The average value of membrane capacitance (Cm) was 597±76 pF (n = 27).
As shown in Figure 2A (left panel), when the recording chamber was continually perfused with OGD solution for 30 mins, the astrocyte Vm showed a multiphasic, rather than linear, depolarization. This was characterized by an initial small-amplitude depolarization phase, followed by an accelerating depolarizing period, and a second plateau. On returning to normal media, the Vm repolarized rapidly with a transient hyperpolarization relative to the initial control Vm before slowly repolarizing back to the pre-OGD level. Quantitatively, as shown in Figure 2B, the first Vm depolarization started immediately and reached a plateau that lasted for approximately 7 mins after the start of 4 mins OGD treatment. At the end of this phase, the Vm depolarization amounted to an average amplitude of 3.8±1.6 mV (n = 7). The accelerated phase appeared after approximately 11 mins of OGD application and reached a steady-state plateau at approximately 17 mins with an additional 6.0±1.8 mV (n = 7) membrane depolarization. The Vm remained stable in the third phase (17 to 30 mins). On OGD withdrawal, this depolarized Vm returned rapidly to the resting Vm level in approximately 4 mins and was followed by a transient further 3.4±1.6 mV hyperpolarization (n = 7) below the initial pre-OGD level.
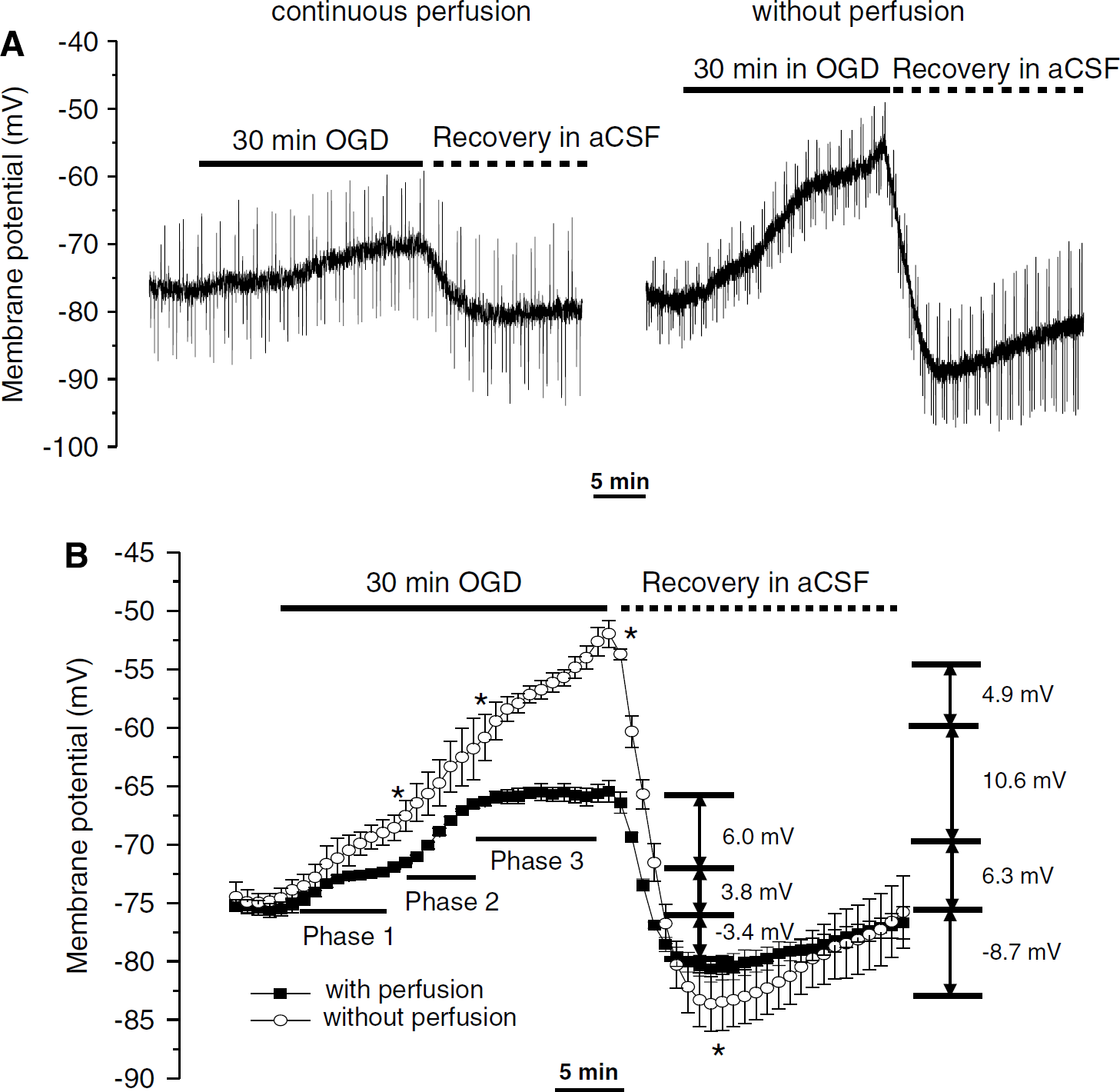
Oxygen and glucose deprivation-induced astrocyte membrane potential (Vm) changes. (
We were concerned that the continuing perfusion experimental condition might be too far from the in vivo ischemia conditions occurring in the enclosed brain, as OGD-induced increases in extracellular K+ neurotransmitters would be continuously washed away from the extracellular space. Therefore, we next measured the astrocyte Vm response to OGD with the perfusion halted after the recording chamber fluid was fully exchanged to the OGD solution. As a representative recording shows in Figure 2A (right panel), the mean astrocyte Vm response over 30 mins OGD under this nonperfusion condition showed a much stronger depolarization (Figure 2B). Specifically, the Vm depolarizations were 6.3±1.1, 10.6±1.6, and 4.9±1.2 mV (n = 7) in the three consecutive phases, respectively. The Vm hyperpolarization amounted to −8.7±2.3 mV (n = 7) in the post-OGD in aCSF. Although the trend of astrocyte Vm response was similar in both recording conditions, the amplitude of the phasic Vm depolarization measured in this closed condition and the hyperpolarization in aCSF after OGD withdrawal were significantly greater (P < 0.05).
The multiphasic responses of astrocyte Vm to OGD resembles the dynamic accumulation of extracellular K+ in the ischemic rat cortex in vivo (Hansen, 1985), where the extracellular K+ accumulation also occurred in three phases with a time course and trends similar to what we found for astrocytic Vm. This coincidence suggests that astrocyte Vm might behave as a K+ electrode sensor in the early ischemic brain.
Activation of Astrocytic Ionotropic Receptors and Transporter Contributes only Marginally to Oxygen and Glucose Deprivation Induced Astrocyte Membrane Potential Depolarization
During cerebral ischemia, extracellular glutamate concentration can be elevated as high as ~44-fold above normal brain levels (Seki et al, 1999), through either Ca2+-dependent vesicular release (Drejer et al, 1985) or Ca2+-independent mechanisms, such as reversal of the glutamate transporter (Szatkowski et al, 1990) and activation of swelling-activated anion channels (Kimelberg et al, 1990; Feustel et al, 2004). Therefore, it is important to clarify whether ischemia-elevated glutamate acts on astrocytes or neuronal glutamate receptors contribute to the observed astrocyte Vm depolarization. However, ionotropic alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are hardly functionally detectable in rat astrocytes from rat hippocampus (Zhou and Kimelberg, 2001), and it is still unclear whether NMDA (N-methyl D-aspartate) receptors are also functionally expressed, as seems to be the case for mouse cortical astrocytes in slices (Lalo et al, 2006). We added inhibitors for AMPA receptors (30 μmol/L NBQX), NMDA receptors (50 μmol/L
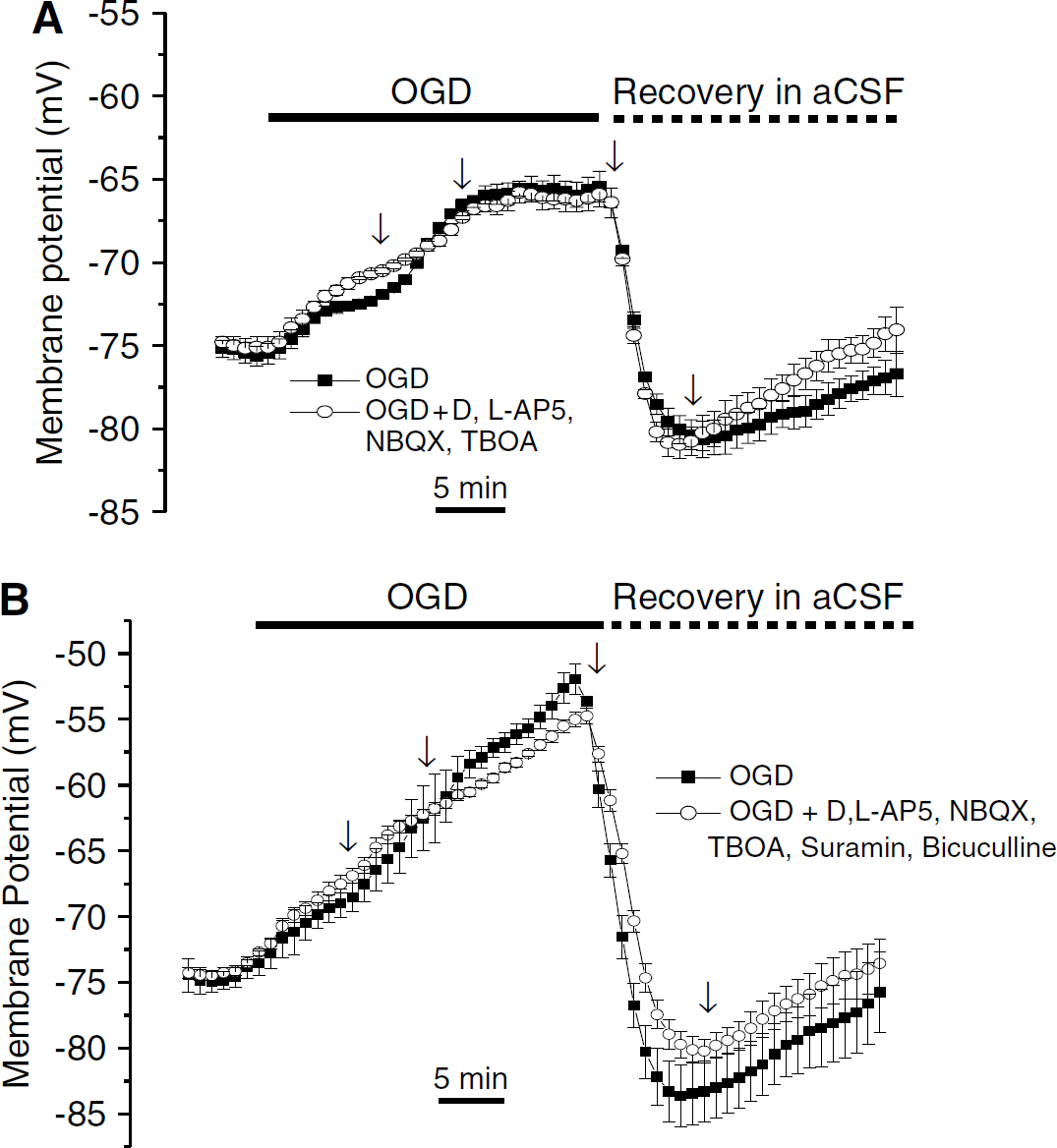
Inhibition of astrocytic ionotropic receptors and transporters produced a negligible effect on OGD-induced astrocyte Vm changes. The chamber was continuously perfused during OGD application in (
Hippocampal astrocytes in vivo also express ionotropic purinergic P2X receptors (Kukley et al, 2001) that are upregulated in OGD conditions in hippocampal slice cultures (Cavaliere et al, 2003; Rundén-Pran et al, 2005). Although the level of the endogenous P2X agonist, ATP, can decrease very quickly in the first few minutes of ischemia (Nicholls and Attwell, 1990), it is possible that ATP could be released as a coneurotransmitter (Pankratov et al, 2006) that modulates astrocyte Vm via P2X receptors in the early phase of OGD. We therefore tested for any potential contribution of P2X to the observed astrocyte Vm depolarization by studying the effect of the nonselective P2X receptor antagonist suramins (100 μmol/L) on the OGD-induced astrocyte Vm change. Suramin had no effect on either the amplitudes or the progression of the OGD-induced astrocyte Vm change, nor the post-OGD Vm hyperpolarization (n = 6, P > 0.05, data not shown).
Ischemia markedly increases the extracellular levels of most transmitters. It has been shown that increase in spontaneous γ-aminobutyric acid (GABA) neurotransmitter release occurs very early in ischemia/hypoxia insults (Globus et al, 1991; Fleidervish et al, 2001). γ-Aminobutyric acidA receptors are functionally expressed in freshly isolated hippocampal astrocytes (Fraser et al, 1995; Zhou and Kimelberg, 2001), and the activation of these receptors under our recording conditions ([Cl−]i/[Cl−]o*** = 6/140 mmol/L, ECl = −80 mV) should lead to Vm hyperpolarization. We next sought to answer if elevated astrocytic GABAA activation is critical for maintaining the low level of Vm depolarization in the initial period of OGD application. However, when the GABAA receptor antagonist 10 μmol/L bicuculline was added, there were no significant effects on OGD-induced Vm depolarization and the subsequent hyperpolarization on return to normal aCSF solution (n = 5, P > 0.05, data not shown).
Finally, we examined the astrocyte Vm change in the presence of a mixture of the five inhibitors used above to see any possible aggregate contribution from astrocyte receptor and transporter activation (Figure 3B). In comparison with the control OGD, these inhibitors together produced minimal effects: a slight 8% (n = 5, P > 0.05) of further depolarization in the first phase, and a reduction of approximately 11% (n = 5, P > 0.05) of the OGD induced later depolarization plateau. It also blocked 15% (n = 5, P > 0.05) of the post-OGD hyperpolarization in the aCSF. Taken together, the OGD-induced characteristic astrocyte depolarization and post-OGD hyperpolarization seem to be only marginally affected by activation of any glutamate and GABA astrocyte receptors and transporters. Indeed, we cannot rule out that the small effects seen were indirect effects due to activation of neuronal receptors. This leaves increased [K+]o as being primarily responsible for the observed astrocyte Vm change, in accordance with the classical model of the mature astrocyte.
Glycolysis is Primarily Responsible for the Multiphase Astrocyte Membrane Potential Response to Oxygen and Glucose Deprivation
The initial 11 mins slow depolarization phase appears to be particularly interesting as it might reflect a time window within which the brain tissue has a better chance for a fully functional recovery from OGD insults. Therefore, we further explored the potential mechanisms that might account for it.
On the basis of the work of Allen et al (2005), we examined the effect of inhibitors of glycolytic and mitochondrial oxidative phosphorylation, either alone or together, on the OGD-induced astrocytic Vm changes. When glycolytic and mitochondrial ATP production was inhibited by iodoacetate (2 mmol/L), antimycin (25 μmol/L) plus rotenone (50 μmol/L), astrocytes showed a rapid depolarization (Figure 4A). Since this Vm depolarization progressed quickly and always led to an irreversible damage to the recorded astrocyte in terms of nonrecovery of Vm, we shortened the OGD exposure time to 8 mins. Even so the Vm could never be fully recovered as compared with the 30 mins OGD control experiments. Overall, as shown in Figure 4A, in the presence of full energy blockers, astrocyte Vm first showed a 27.5±4.5 mV depolarization, which was 5.5-fold greater than the amplitude of the OGD-alone-induced astrocyte Vm depolarization at the same time point (n = 6, P < 0.05). In addition, when OGD application approached 8 mins, an additional large and abrupt depolarization of 34.0±5.5 mV was observed (n = 6). After OGD withdrawal at 8 mins, the astrocyte Vm recovered gradually, but never to the normal preOGD level. These results indicate that the continued ATP production from either of these pathways plays a critical role for the observed multiphase astrocyte Vm changes in OGD.
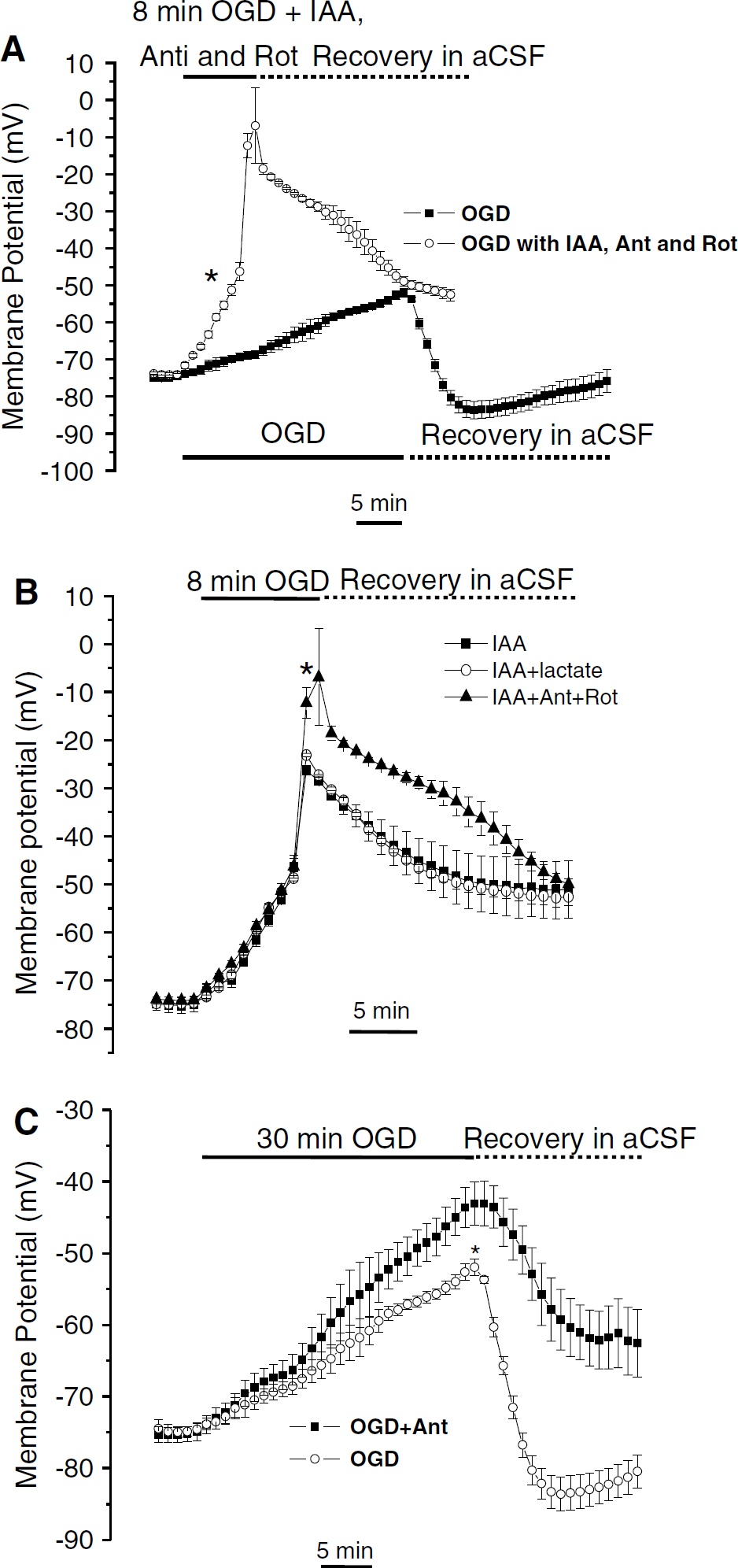
Glycolysis plays a preferential role in maintaining astrocyte Vm in the early period of OGD. Since block of energy production caused an accelerated astrocytic Vm depolarization, the OGD treatment was shortened to 8 mins. (
It has been suggested that plasma membrane Na+/ K+-ATPase is preferentially fueled by ATP from glycolysis (Rosenthal and Sick, 1992). To dissect out the relative contribution of energy from glycolytic and oxidative pathways to the astrocyte Vm changes, we applied the inhibitors separately. We found that the glycolysis inhibitor iodoacetate (2 mmol/L) produced a similar Vm changes as the three inhibitors applied together for an 8-min OGD treatment, although the maximal Vm depolarization was ~18% less (n = 6, P < 0.05, Figure 4B). Since mitochondria oxidative phosphorylation can be fueled by metabolites downstream of glycolysis, iodoacetate can also indirectly inhibit oxidative metabolism in the brain where lactate has been considered a primary substrate for fueling the neuronal mitochondrial tricarboxylic acid cycle (Schurr et al, 1988, 2006). Therefore, we further investigated the effect of iodoacetate on astrocyte Vm responses in the presence of 5 mmol/L lactate. However, the inhibition of glycolysis with iodoacetate in the presence of lactate produced the same accelerated astrocyte Vm depolarization as iodoacetate alone (Figure 4B). In contrast, when oxidative phosphorylation was selectively inhibited by antimycin for 30 mins OGD (Figure 4C), neither the duration nor the amplitude of the first phase changed in the presence of antimycin, and only the maximal Vm depolarization in the second plateau further depolarized by 30% (n = 5, P < 0.05). However, the astrocyte Vm depolarization could only be partially recovered after OGD withdrawal, and there was none of the characteristic hyperpolarization. This was probably caused by the slow reversibility of antimycin that could not be washout within a 25-min reperfusion period. Since the energy blockers should affect both astrocytes and neurons, these results show that glycolysis is the major energy source in the slice to sustain the plasma membrane ion pumps in early OGD, which is consistent with a recent hippocampal neuron study by Allen et al (2005). It seems reasonable to infer that the continued fueling of the Na+/K+-ATPase by glycolytic ATP in early OGD can largely maintain the normal ion gradients thereby limiting astrocytic Vm depolarization, but that the increased Na+/K+-ATPase activity on reperfusion is impaired (see the next section).
Enhanced Na+/K+-ATPase Activity is Responsible for Post-oxygen and Glucose Deprivation Astrocyte Membrane Potential Hyperpolarization
We always observed a transient hyperpolarization of astrocytic Vm in the post-OGD reperfusion period. Coincidently, previous studies from rat hippocampal slices have shown that extracellular K+ undershoots its prehypoxic level during the post-OGD reperfusion (Müller and Somjen, 2000; Chao et al, 2006). The mechanism underlying the decreased [K+]o is often ascribed to increased activation of the Na+/K+-ATPase owing to intracellular accumulation of Na+. This could occur in both astrocytes and neurons. To examine whether increased Na+/K+-ATPase activity in post-OGD was responsible for the observed hyperpolarization, we used the specific Na+/K+-ATPase blocker ouabain (100 μmol/L). In the first experiment, ouabain was applied for 1 min immediately after OGD simultaneously with control aCSF. This resulted in a partial astrocyte Vm recovery, which never returned to the preOGD level (data not shown). To show directly that the hyperpolarization was sensitive to ouabain, we added ouabain for 1 min, starting at 4 mins after the perfusion solution was shifted back to control aCSF. This prevented the hyperpolarization that would otherwise have occurred after replacement of OGD solution by normal aCSF (Figure 5). However, the astrocyte Vm then depolarized in the recovery aCSF solution and did not return to the control astrocyte Vm level after removal of ouabain, presumably owing to slow reversibility of the inhibition of a high-affinity ouabain component of the Na+/K+-ATPase seen in rat brain (Berrebi-Bertrand et al, 1990). These data indicate that the hyperpolarization of astrocyte in post-OGD recovery was meditated by the activation of the Na+/K+-ATPase.
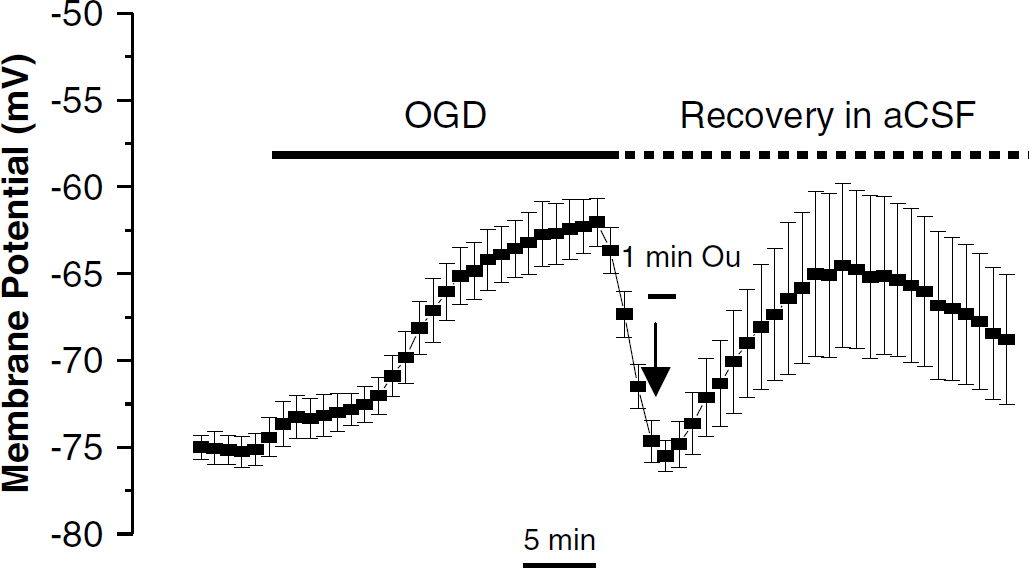
Inhibition of transient post-OGD astrocyte Vm hyperpolarization by Na+/K+-ATPase inhibitor ouabain. The plot shows the averaged astrocytes responses (n = 6) to a 30 mins of OGD treatment with addition of 100 μmol/L ouabain (Ou) applied for 1 min after the bath solution was switched to normal aCSF for 4 mins. The transient post-OGD hyperpolarization was completely inhibited with this ouabain treatment. The error bars represent means±s.e.m. for the Vm value at the given time point.
Effect of Pipette ATP on Membrane Potential Responses of the Recorded Astrocyte to Oxygen and Glucose Deprivation
Astrocytes are more resistant to ischemic insults than neurons (Petito et al, 1998). In this study, we found that astrocyte Vm depolarization fully recovered from 30 mins OGD treatment when reperfused, whereas in a parallel study on hippocampal neurons, we found that alterations in neuronal excitability became irreversible after a short 10 to 15 mins of OGD treatment (Zhang HQ et al, unpublished data). Under our experimental conditions, glucose and oxygen were removed only from the bath solution. However, the energy for the patched astrocytes was maintained by the presence of ATP/ GTP in the electrode solution as is commonly performed (Allen et al, 2005; Lipski et al, 2006). Thus, the recorded astrocytes possibly behave as sensors to OGD-induced changes in the extracellular space (Allen et al, 2005). It seemed important, therefore, to compare these results with ATP/GTP-free electrode solution conditions. Surprisingly, we found that astrocyte Vm depolarization was actually less when ATP/GTP was not present in the electrode solution (Figure 6A). The amplitudes of the three phases of Vm depolarization were 4.8±0.8, 5.9±1.8, and 1.8±1.4 mV (n = 5), respectively, which are significantly smaller than in the presence of ATP/ GTP (n = 5, P < 0.05). In the recovery aCSF without ATP/GTP in the electrode, the astrocyte actually remained hyperpolarized (–6.9±1.1 mV more negative than to pre-OGD Vm) and the Vm did not return to the initial control Vm level over the 25 mins recovery time period measured.
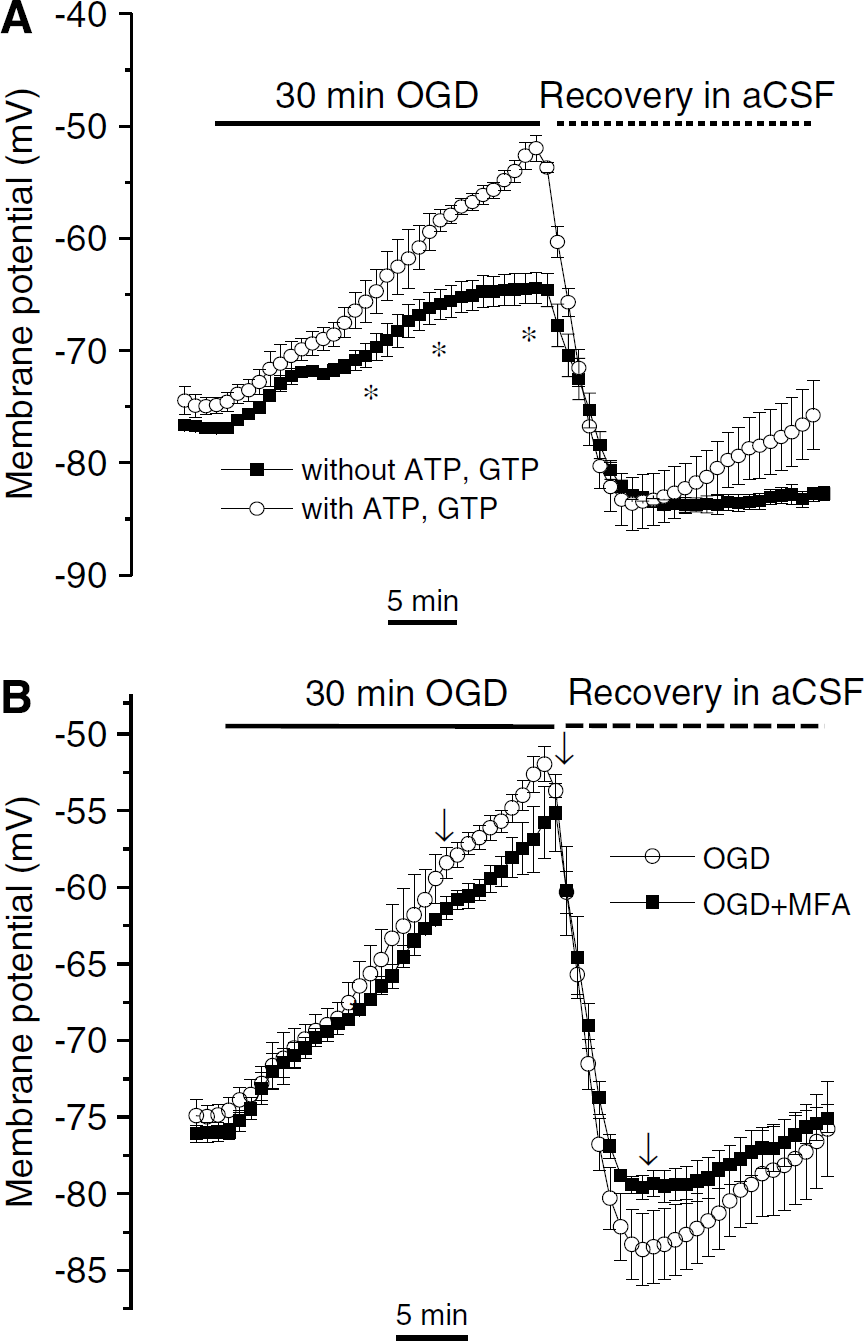
Lack of dependence of OGD-induced astrocyte Vm changes on intracellular ATP/GTP and gap junction communication. (
Lack of Effect of Gap Junction Inhibition on Membrane Potential Response of the Recorded Astrocytes to Oxygen and Glucose Deprivation
Mature astrocytes in vivo are extensively coupled through gap junctions (Theis et al, 2005; Schools et al, 2006). This interastrocyte coupling has been considered to be important for potassium spatial buffering (Kofuji and Newman, 2004) and metabolic support to neurons (Farahani et al, 2005). In acute brain slices, astrocytic gap junctions are probably continuously open during ischemia conditions, which might contribute to the gradual expansion of the ischemic lesion area (Cotrina et al, 1998). In the acute phase of OGD insults, it is unknown whether free exchange of ion, metabolites, and other ischemic-induced products through gap junction contributes to the multiphasic astrocytic Vm responses. We sought to answer this question by studying astrocyte Vm responses in the presence of the gap junction blocker, MFA. We have shown that 100 μmol/L MFA substantially inhibits gap junction coupling in rat hippocampal slices (Schools et al, 2006). In the presence of 100/ μmol/L MFA in OGD solution, astrocytes responded with the same multiphasic Vm depolarization and post-OGD hyperpolarization (Figure 6B), none of the small Vm changes in the presence of MFA reached statistical significance (P > 0.05). Accordingly, we conclude that the continued opening of gap junction channels does not play a significant role for the observed multiphsic astrocyte Vm responses in the acute OGD model.
Discussion
By applying OGD solutions to acute hippocampal slices and examining the changes in astrocyte Vm in this simulated ischemia model with patch-clamp recording, this study provides novel information concerning the acute astrocyte Vm responses to OGD and their underlying mechanisms.
Astrocytic Membrane Potential Behaves as a K+ Sensor to Oxygen and Glucose Deprivation-Induced Ambient K+ Changes
The predominant expression of leak K+ channel by mature astrocytes used in this study allowed astrocytes to behave as K+ electrodes sensing [K+]o change in physiologic conditions (Orkand, 1991). However, in the ischemic brain, not only K+ but also various neurotransmitters are also released into the extracellular space. The nonlinear astrocyte Vm depolarization, therefore, could be caused by differential release of K+ or transmitters at different phases of OGD. We now show that only ~10% of the Vm depolarization could be attributed to the activation of ionotropic receptors and electrogenic transporters (Figure 3). This leave OGD-induced [K+]o increase from both astrocytes and neurons primarily responsible for the observed multiphasic astrocyte depolarization.
Previous sharp electrode intracellular recordings showed that hippocampal astrocytes in situ responded to hypoxia with Vm depolarization, which reflected the increase in [K+]o (Müller and Somjen, 2000; Leblond and Krnjevic, 1989). Early in vivo cerebral ischemia studies showed that [K+]o could reach as high as 50 to 80 mmol/L (Somjen, 1979), and this [K+]o increase was not linear with time but could be subdivided into three phases (Hansen, 1985). In a recent study using acute cortical slices (Chao et al, 2006), OGD-induced increased [K+]o followed a similar trend as in the in vivo studies. These clearly indicate that the in vitro OGD model achieves a comparable ischemic condition as in vivo ischemia in terms of changes in extracellular K+. The similarity between reported OGD-induced phasic [K+]o changes and observed astrocyte Vm depolarization supports the view that astrocytic Vm mostly reflects OGD-induced [K+]o changes.
Glycolytic ATP Supply Determines Oxygen and Glucose Deprivation-Induced Multiphasic Astrocyte Membrane Potential Depolarization
The mammalian brain can tolerate a lack of blood supply for no more than 10 mins, which is followed by an irreversible functional neuronal damage (Nedergaard and Dirnagl, 2005). In a hippocampal pyramidal neuron study, a similar time period of 8 mins was determined for reaching OGD-induced anoxic depolarization, an indication of fuel exhaustion causing ion gradient breakdown (Allen et al, 2005). Interestingly, astrocytes first responded to OGD with a small depolarized Vm plateau lasting for ~11 mins (Figure 2). In OGD-treated hippocampal slices, glycolytic ATP was found to be the major energy fuel that could maintain neuronal excitability for ~8 mins (Allen et al, 2005). Consistently, we showed that glycolytic ATPs are primarily responsible for maintaining the small initial astrocyte Vm depolarization, as inhibition of glycolysis caused an immediate large depolarization (Figure 4B). With continued oxygen supply, neuronal activity in rat hippocampal slices could be well maintained by lactate in the absence of glucose and inhibition of glycolysis (Schurr et al, 1988; Izumi et al, 1994), indicating that lactate is the major fuel for neuronal mitochondrial tricarboxylic acid cycle to maintain neuronal functions (Schurr, 2006). With oxygen deprivation, however, we showed that 5 mmol/L lactate failed to reverse the marked astrocyte depolarization due to iodoacetate in OGD solution, as expected from lactate requiring oxygen for its metabolism. This conclusion was strengthened further by showing that inhibition of the respiratory chain by antimycin or rotenone did not produce any effect on the early astrocyte Vm depolarization and a modest increased depolarization at later times of the OGD. Since loss of ion gradients is believed to be a central initiating change in ischemia pathophysiology (Lopachin et al, 2001; Martin et al, 1994), the initial slow astrocytic Vm depolarization plateau reflects a time period when [K+]o is only increasing modestly and therefore indicates a time window within which the brain tissue has a better chance for a fully functional recovery from OGD. Although glycogen is primarily stored in astrocytes (Brown et al, 2004; Wender et al, 2000), and astrocytes show markedly enhanced glycolysis on neuronal activation (Kasischke et al, 2004), the question of whether glycolytic ATP in astrocytes is primarily responsible for the limited initial astrocyte Vm depolarization needs further experimental elucidation under ischemic condition. Meanwhile, enhanced neuronal mitochondrial oxidative phosphorylation in response to elevated neuronal activation was also reported in brain slices (Brennan et al, 2006), but this would not be expected to occur under OGD conditions.
Reperfusion after Oxygen and Glucose Deprivation Induces an Enhanced Na+/K+-ATPase Activity
Coincident with the observation of a [K+]o undershoot in the post-OGD reperfusion phase (Müller and Somjen, 2000; Chao et al, 2006), astrocytic Vm hyperpolarized transiently on OGD withdrawal (Figure 2). Ischemia-like insults enhance cultured astrocyte Na+/K+-ATPase activity (Stanimirovic et al, 1997) suggesting a possibility that this could be a responsible mechanism for the post-OGD astrocytic Vm hyperpolarization. By showing a complete inhibition of this hyperpolarization with addition of the specific Na+/K+-ATPase inhibitor, ouabain, we provided the first direct support of this mechanism in situ. Our results also support the idea that Na+/K+-ATPase determines the rate of extracellular K+ recovery, as was concluded from study of high-frequency electric stimulation-induced [K+]o increase in rat hippocampus (D'Ambrosio et al, 2002). However, we are presently unable to determine further the relative contribution from either neurons or astrocytes to this enhanced Na+/K+-ATPase activity, as the hyperpolarized Vm detected by patched astrocytes reflected the overall ambient [K+]o decrease from both cellular elements.
Dependence of Intracellular ATP/GTP of Astrocyte Membrane Potential Responses to Oxygen and Glucose Deprivation
In the absence of ATP/GTP in the electrode solution, we surprisingly found 50% depolarization in OGD and a persistent hyperpolarization upon reperfusion (Figure 6A). Since the intracellular chloride in our study was low, 6 mmol/L, activation of intracellular ATP-dependent volume-sensitive anion channels was unlikely to account for it (Inoue and Okada, 2007), as ATP-dependent volume-sensitive anion channels inhibition should result in a further Vm depolarization. Although a higher [Cl−]i of ~40 mmol/L has been reported in primary cultures (Kimelberg, 1990), we used a [Cl−]i according to an in vivo study wherein the intracellular chloride concentration in ‘glia’ was measured at ~6 mmol/L (Ballanyi et al, 1987). The altered astrocyte OGD response with omission of ATP/GTP from the pipette could be due to the involvement of a recently discovered leak two-pore domain K+ channel family, which we have found in parallel studies to be present in mature astrocyte K+ conductance (Zhou M et al, unpublished data). Modulation of twik (weak inwardly rectifying K+ channel)-related acid-sensitive K+ channel-1 (TASK-1), one of the two-pore domain K+ channel isoforms, by intracellular GTP-dependent active Gαq subunits has been shown recently by Chen et al (2006). Nevertheless, definitive answers to this intriguing observation require further studies.
Astrocytes are Electrophysiologically more Resilient to Oxygen and Glucose Deprivation Insults
The fact that OGD-induced astrocyte Vm depolarization was always fully recoverable from a 30-min OGD treatment is in sharp contrast to the neuronal response, wherein maximum tolerance of hippocampal neurons to OGD was 10 to 15 mins (Zhang HQ et al, unpublished data). A possible explanation for this difference lies in the high-density expression of various voltage-gated ion channels and ionotropic glutamate receptors in neurons, but not in astrocytes. Although these channels and receptors are required for neuronal signaling in the physiologic brain, they are also vulnerable targets of ischemia-induced excessive accumulation of extracellular K+ glutamate-causing neuronal damage.
The resistance of astrocytes to acute OGD insults would allow astrocytes to continue homeostatic functions in the early ischemia brain for maintaining neuronal functions for an average time duration estimated in this study of around 8 to 11 mins. This may be why neurons suffer irreversible damage after an OGD period of 10 to 15 mins.