
Abstract
Although the function of fever is still unclear, it is now beyond doubt that body temperature influences the outcome of brain damage. An elevated body temperature is often found in stroke patients and denotes a bad prognosis. However, the pathophysiologic basis and treatment options of elevated body temperature after stroke are still unknown. Cerebral ischemia rapidly induced neuronal interleukin-6 (IL-6) expression in mice. In IL-6–deficient mice, body temperature was markedly decreased after middle cerebral artery occlusion (MCAO), but infarct size was comparable to that in control mice. If body temperature was controlled by external warming after MCAO, IL-6–deficient mice had a reduced survival, worse neurologic status, and larger infarcts than control animals. In cell culture, IL-6 exerted an antiapoptotic and neuroprotective effect. These data suggest that IL-6 is a key regulator of body temperature and an endogenous neuroprotectant in cerebral ischemia. Neuroprotective properties apparently compensate for its pyretic action after MCAO and enhance the safety of this endogenous pyrogen.
In acute brain damage, the body temperature is often elevated. About half of the patients who have ischemic stroke have a core temperature more than 37.5°C in the first 3 d after onset of symptoms (Grau et al., 1995). A similar incidence of increased temperature has been reported for traumatic brain injury (Albrecht et al., 1998). Epidemiologic studies show that increased body temperature after stroke is associated with large cerebral infarcts and poor functional outcome (Reith et al., 1996; Castillo et al., 1998). The important pathogenic role of body temperature in acute brain damage is further underlined by ample experimental data. In various animal models of brain damage, hypothermia exerts neuroprotective effects, whereas hyperthermia increases the damage (Dietrich et al., 1996; Corbett and Thornhill, 2000). This led to the clinical recommendation to lower body temperature after stroke (Hacke et al., 2000). However, controlled studies of pharmacologic reduction of body temperature in stroke are lacking, and the mechanisms of regulation of body temperature after stroke are as yet unclear.
Body temperature is regulated by cytokines. In infection, the cytokine interleukin-6 (IL-6) acts as an important endogenous pyrogen. It is both a major regulator of the acute phase reaction in peripheral tissues and governs its central components including fever, anorexia, and apathy (Chai et al., 1996; Bluthé et al., 2000). Intracerebroventricular administration of IL-6 rapidly elevates the body temperature and reduces locomotor activity (Schöbitz et al., 1995). In stroke, increased concentrations of IL-6 were found in cerebrospinal fluid and in serum (Beamer et al., 1995; Tarkowski et al., 1995). Concentrations of IL-6 correlate with the infarct volume. Evidence for an induction of IL-6 in the ischemic brain comes from experimental studies. Several groups reported elevated levels of IL-6 messenger RNA (mRNA) or protein after focal cerebral ischemia in rats (Wang et al., 1995; Loddick et al., 1998; Suzuki et al., 1999). Exogenous IL-6 reduced the infarct size (Loddick et al., 1998). A neuroprotective action of IL-6 is supported by in vitro data that show that IL-6 protects primary neurons against glutamate-induced excitotoxicity (Ali et al., 2000). However, the role of IL-6 in neuronal survival has been debated. Clark et al. (2000) reported no change of infarct size in IL-6–deficient mice, and others have even observed neurotoxic effects of IL-6 (Campbell et al., 1993; Qiu et al., 1998). To further elucidate the role of endogenous IL-6 in ischemic brain damage, we reexamined cerebral ischemia in IL-6–deficient and control mice, paying special attention to body temperature.
MATERIALS AND METHODS
Animals
Interleukin-6–deficient (–/–) (Kopf et al., 1994) and C57/bl 6 control mice were housed under controlled laboratory conditions with a 12-hour dark–light cycle, a temperature of 22 ± 2°C, and a humidity of 60% to 70%. The mice had free access to standard rodent diet (C-1000; Altromin, Lage, Germany) and tap water. All mice were genotyped by polymerase chain reaction (PCR) using the following primers: 5′-TTC CAT CCA GTT GCC TTC TTG G-3′, 5′-TTC TCA TTT CCA CGA TTT CCC AG-3′, resulting in a PCR product of 170 bp in wild-type and 1.3 kbp in IL-6 –/– mice. For all experiments, IL-6 –/– and control mice were age- and sex-matched. Mice were used at an age of 3 to 9 months. They had a body weight of 30.7 ± 5.1 g, and 70% of all mice were male.
Ischemic model
For middle cerebral artery occlusion (MCAO), the filament model was used. An 8–0 nylon suture (Dermalon, Braun-Dexon, Spangenberg, Germany) was coated (Xanthopren M Mucosa; Heraeus Kulzer, Dormagen, Germany) and inserted into the common carotid artery on the left side. It was advanced into the internal carotid artery until the laser Doppler signal decreased considerably. For sham surgery, preparation of the common carotid artery was identical but the suture was not inserted. For anesthesia, halothane (1%) and N2O/O2 (70%/30%) were used. During surgery, a body temperature of 37°C was maintained, with the mice on a heating pad. In transient MCAO, mice were reanesthetized and the filament was removed after 1 h of ischemia. For laser Doppler measurements, the probe (P415–205; Perimed, Järfälla, Sweden) was placed 3 mm lateral and 6 mm posterior to the bregma. Relative perfusion units were determined (Periflux 4001; Perimed, Järfälla, Sweden). The neurologic status of mice was evaluated 24 h after surgery as has been described previously (Bederson et al., 1986).
Autoradiography and determination of infarct size
Mice were reanesthetized 24 h after onset of permanent MCAO by intravenous injection of 0.03 mg/kg · min–1 etomidat Lipuro (B. Braun, Melsungen, Germany). Local cerebral blood flow (CBF) was measured by an autoradiographic method as described by Sakurada et al. (1978). Briefly, 125 μCi/kg body weight iodo[14C]antipyrine (Biotrend, Cologne, Germany) was infused into the right femoral vein for 1 minute. In parallel, 12 to 16 scheduled arterial blood samples were taken to determine the time course of the arterial iodo[14C]antipyrine concentration. Immediately after the infusion, mice were decapitated, and the brains were quickly frozen. Coronal cryosections of the brains (20 μm in thickness) were cut every 200 μm, dried on a heating plate at 60°C, and then exposed on a Kodak MinR1 x-ray film (Kodak, Rochester, NY, U.S.A.) together with a 14C standard set for 21 d. The local CBF was calculated from the optical density with an image analyzing system (MCID; Imaging Research Inc., St. Catherines, Ontario, Canada). In the autoradiograms, the size of those areas in which CBF was below 30, 20, or 10 mL · 100 g–1 · min–1 was determined. Parallel sections were stained with a silver technique to determine the infarct size (Vogel et al., 1997). Infarct volumes were corrected for brain edema (Swanson et al., 1990).
Measurement of physiologic parameters
Before the infusion of iodo[14C]antipyrine (24 h after permanent MCAO), arterial blood pressure and pulse were monitored. Samples (150 μL per mouse) were collected for analysis of arterial blood gas, hematocrit, and glucose. To measure the core body temperature, a probe (Emitter 4000; Minimitter, Bend, OR, U.S.A.) was implanted intraperitoneally 24 hours before MCAO. Mice were kept at room temperature (21 ± 2°C). The temperature signal was recorded every minute by a receiver under the cage (Receiver/Transmitter ER-4000; Minimitter) using Vitalview 3.1 software (Minimitter). Mean body temperature of each mouse was calculated for 15-minute intervals.
Intraperitoneal implantation of the probes slightly increased mortality after MCAO. To minimize mortality in the experiments in which temperature was controlled by external warming, we implanted the probe subcutaneously. Subcutaneous body temperature was 0.46 ± 0.14°C lower than the core body temperature measured by a rectal probe (correlation coefficient 0.67, n = 49). To maintain a constant body temperature, an infrared lamp (100 W, Zoomed ZM7, Repticare ceramic infrared heat emitter; Zoomed Laboratories Inc., San Luis Obispo, CA, U.S.A.), fixed 22 cm above the bottom of the cage, was controlled by the subcutaneous body temperature of the mice. The lamp lit up if the temperature dropped to 34.9°C or lower. In these experiments, we also measured locomotor activity of the animals using the same probe.
Immunohistochemistry
Immunohistochemistry of IL-6 was carried out as described before (Suzuki et al., 1999). In brief, 6 h after permanent MCAO or sham surgery, animals were reanesthetized and perfused with phosphate-buffered saline (PBS), followed by 4% freshly depolymerized paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4 (PB). The brains were removed and postfixed for 6 h in 4% paraformaldehyde dissolved in PB before cryoprotection by sequential bathing in 10%, 15%, and 20% sucrose in PB at 4°C. The brains were then frozen in powdered dry ice. Coronal cryosections (20-μm thick) were prepared on a cryostat (Cryocut 1800; Leica Instruments GmbH, Nussloch, Germany). The sections were incubated overnight at 4°C with a polyclonal rabbit anti–mouse IL-6 antibody (1:500; Sigma, St. Louis, MO, U.S.A.). For detection, an avidin-biotinylated enzyme complex system (Vectastain ABC-Elite kit; Vector Laboratories, Burlingame, CA, U.S.A.) was used. To identify the cellular source of IL-6, double staining with monoclonal anti-neuronal nuclei (NeuN) antibody (1:250; Chemicon International, Temecula, CA, U.S.A.) was performed. Anti-neuronal nuclei were visualized with a fluorescein isothiocyanate-conjugated anti–mouse IgG antibody (Amersham, Buckinghamshire, U.K.) and IL-6 with a Texas red–conjugated anti–rabbit IgG antibody (Amersham), respectively.
Enzyme-linked immunosorbent assay
After a permanent MCAO for the times indicated the right or left brain hemispheres were homogenized in 600 μL PBS containing a cocktail of protease inhibitors (complete Mini; Roche, Mannheim, Germany) on ice. After centrifugation (12,000g, 20 minutes, 0°C), the supernatant was used for enzyme-linked immunosorbent assay. Enzyme-linked immunosorbent assay for mouse IL-6 has been described previously (Schwaninger et al., 1997).
Quantitative interleukin-6 polymerase chain reaction
After a transient MCAO of 2 h followed by the reperfusion times indicated, RNA was prepared as described before (Brambrink et al., 2000). Complementary DNA (cDNA) was synthesized from 20 μg total RNA using Superscript II reverse transcriptase and oligo-dT primer, and purified by using PCR-purification columns (Qiagen, Hilden, Germany). Quantitative PCR was performed using the LightCycler system (Roche Diagnostics) as described previously (Brambrink et al., 2000). In brief, the formation of double-stranded products is measured by real-time fluorescence detection of intercalated SYBR-green, which allows for quantitation of double-stranded PCR products in the exponential phase of the PCR. cDNAs were serially threefold diluted to obtain four concentration points for a standard curve. The concentration of IL-6 cDNA in different samples was normalized to levels of cyclophilin. The annealing temperature chosen was 60°C. Measurements of cyclophilin and IL-6 were done at 84°C to exclude potential formation of primer-dimers. The following primers were used for PCR amplification: cyc5, ACCCCACCGTGTTCTTCGAC; acyc300, CATTTGCCATGGACAAGATG (Cyclophilin, PCR product 295 bp); mIL6s, CAAGAACGATAGTCAATTCCAG; mIL6as, CATTCCAAGAAACCATCTGGC (IL-6, PCR product 975 bp). The specificity of the PCR reaction was checked by melting-point determination and agarose gel electrophoresis. Values were averaged from at least three different dilutions per sample.
Cell culture and detection of apoptosis
PC12 cells were obtained from DSMZ (Braunschweig, Germany). They were grown on a 24-well plate (106 cells per well) and were treated with human recombinant IL-6 (50 ng/mL; Bachem, Heidelberg, Germany) for 24 h as indicated. After washing cells with serum-free medium, Dulbecco's modified Eagle Medium with or without serum was added and cells were cultured for another 24 hours in the presence or absence of IL-6 (50 ng/mL). Apoptosis detection using fluorescence activated cell sorter (FACS) analysis was performed according to Nicoletti et al. (1991). In brief, cells were harvested with a rubber policeman, centrifuged at 200g for 5 minutes. After washing the pellet in PBS, the cells were resuspended in PBS and fixed in 70% ethanol on ice for 30 minutes. Then cells were centrifuged and washed in PBS. After another centrifugation, the cell pellet was stained in 1 mL of DNA staining solution (10 mL PBS, 200 μg propidium iodide, 2 mg RNAse A) for 30 minutes at room temperature in the dark. Fixation with ethanol resulted in the best differentiation of apoptotic and viable cells. Flow cytometry was performed on FACScan (Becton Dickinson Biosciences, Heidelberg, Germany). Debris and large cell aggregates were excluded. To visualize nuclear morphology, PC12 cells were stained with Hoe 33258 (50 ng/mL in PBS).
Cortical neurons were prepared from fetal mice (E16) and plated on a previously established bed of cortical glia (1 × 105 neurons per cm3), as previously described (Rose et al., 1993). Cells were cultured in Eagle's minimal essential medium (Earle's salts with 2 mmol/L glutamine) supplemented with 10% fetal bovine serum, 10% horse serum, and 20 mmol/L glucose. After 5 to 7 d in vitro, growth of nonneuronal cells was halted by exposure to 5 μmol/L uridine and 5 μmol/L (+)-5-fluor-2′-deoxyuridine for 24 h. After 14 d in vitro, cells were pretreated with 50 ng/mL recombinant mouse IL-6 (Becton Dickinson Biosciences) for 8 h and then exposed to N-methyl-
Immunoblot
After permanent MCAO, protein extracts of mouse brain tissue were prepared as described (Qi et al., 2001). One hemisphere was homogenized in 1.5 mL lysis buffer. For detection of cleaved poly (ADP-ribose) polymerase (PARP), protein extracts were subjected to sodium dodecyl sulfate–polyacrylamide electrophoresis (SDS-PAGE) using 10% gels and transferred to nitrocellulose (Hybond, enhanced chemiluminescence [ECL] nitrocellulose; Amersham, Freiburg, Germany). Nitrocellulose filters were incubated for 2 h in 5% nonfat dry milk dissolved in Tris-buffered saline (TBS; 20 mmol/L Tris-HCl, pH 7.6, 137 mmol/L NaCl) and were then incubated with a mouse specific antibody against cleaved PARP (#9544; Cell Signaling, Beverly, MA, U.S.A.), diluted 1:1,000 in TBS containing 0.1% Tween-20, overnight at +4°C. For IL-6, immunoblot protein extracts were resolved on a 12% SDS-PAGE. The primary antibody, goat anti-mIL6 (M-19; Santa Cruz, Heidelberg, Germany), was used in a dilution of 1:200. The secondary antibody was a horseradish peroxidase–conjugated donkey anti-goat (sc-2020, 1:2,000; Santa Cruz). Antibody–antigen complexes were detected with ECL reagents (Amersham).
Statistics
Groups were compared by two-tailed Student's t-test or univariate analysis of variance. For comparison of survival, log rank analysis was performed using SPSS (SPSS, Inc., Chicago, IL, U.S.A.) software. The level of statistical significance was set at P < 0.05 if not stated otherwise.
RESULTS
Focal cerebral ischemia rapidly induces IL-6 expression: already 2 h after MCAO, IL-6 mRNA accumulation in the brain was significantly elevated compared with sham-operated mice (Fig. 1A). Interleukin-6 mRNA accumulation increased in both the ischemic and in the contralateral hemisphere, but the increase in the ischemic hemisphere was more pronounced. Because protein translation is inhibited in ischemia (DeGracia et al., 2002), we investigated whether the increased IL-6 mRNA levels are also reflected at the protein level. Interleukin-6 concentration in the brain measured by enzyme-linked immunosorbent assay was significantly elevated after cerebral ischemia compared with sham-operated controls (Fig. 1B). Interleukin-6 was elevated also in the contralateral hemisphere. The IL-6 induction after cerebral ischemia was further supported by immunoblots, which showed a band of the correct size (25 kd) in brain extracts after 6 h of permanent cerebral ischemia but not in sham-operated mice (data not shown).
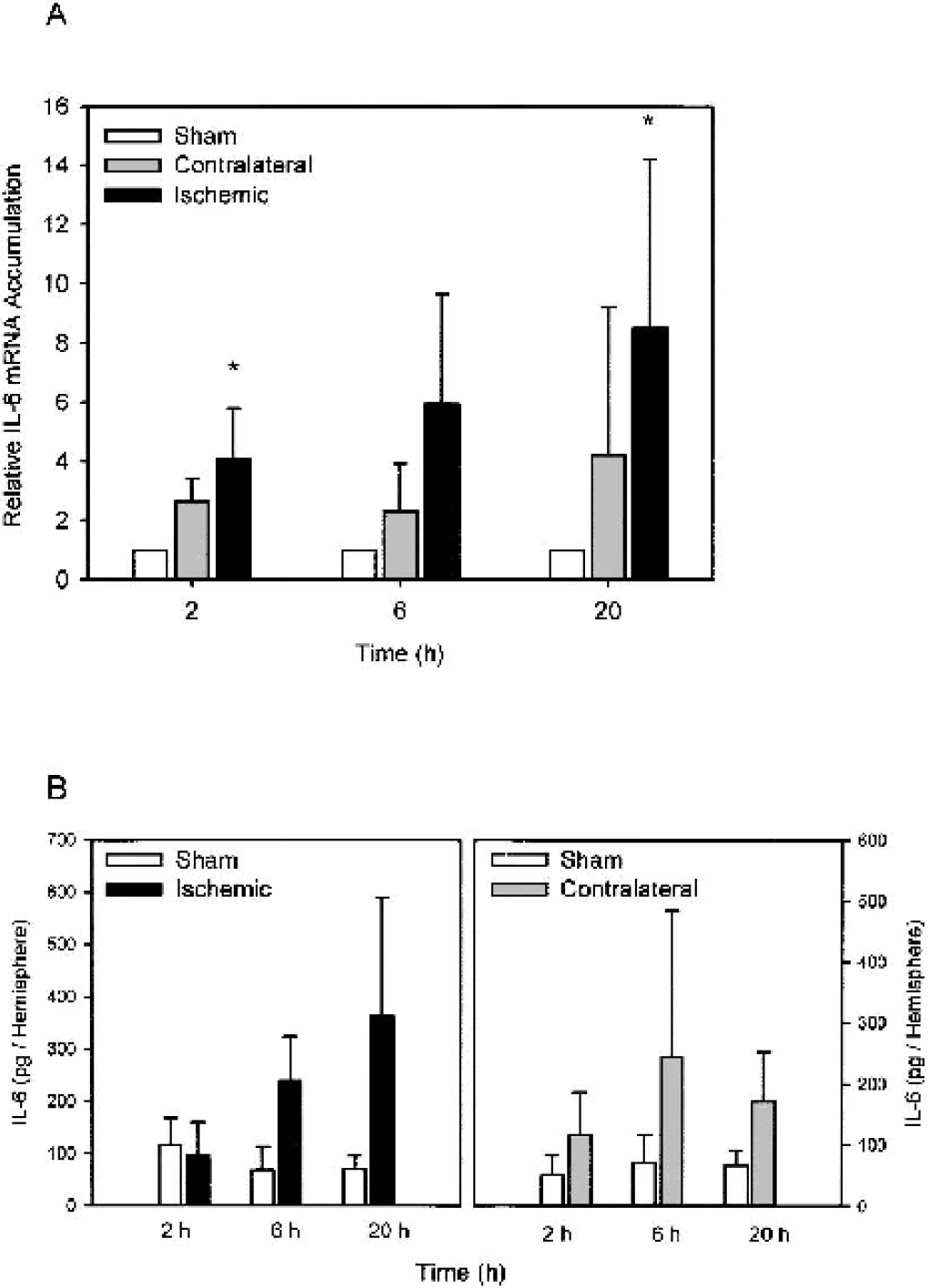
After cerebral ischemia, interleukin-6 (IL-6) is induced in both the ischemic and the contralateral hemisphere.
Immunohistochemistry showed an induction of IL-6 expression 6 h after MCAO, especially in the periischemic region (Figs. 2B and 2E to G), but also in the ischemic core (Fig. 2C), and the contralateral cortex (Fig. 2D). Double staining with the neuronal marker NeuN revealed that IL-6–positive cells were neurons (Figs. 2E to J). This is in accordance with previous findings in the rat (Suzuki et al., 1999). In the border zone of the ischemic core there were also IL-6–positive cells that did not stain for NeuN (Fig. 2H to J). Comparing conventional cresyl violet staining and IL-6 immunohistochemistry in adjacent slices, it was obvious that not all neurons in the cerebral cortex express IL-6 (data not shown).
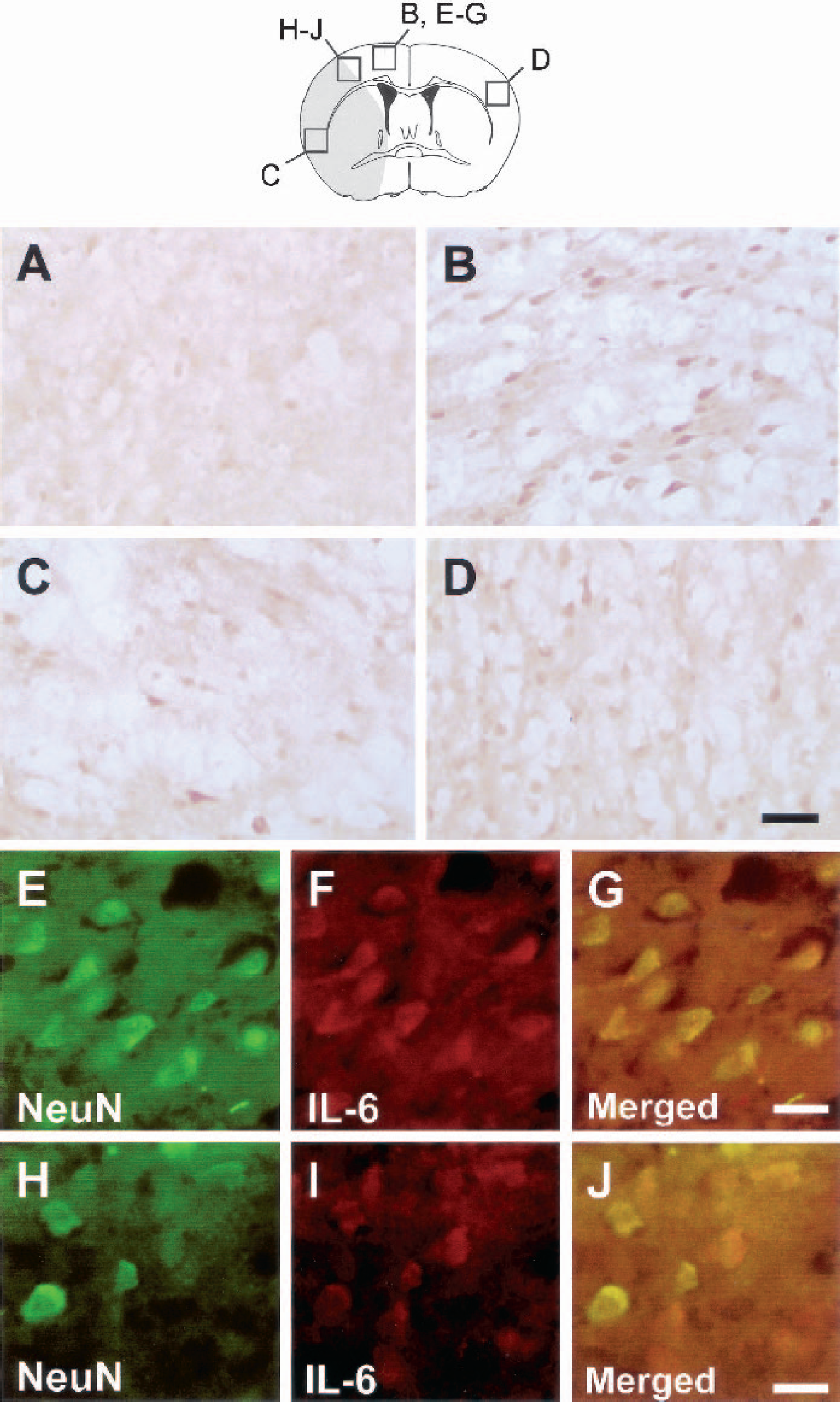
Neurons express interleukin-6 (IL-6) after focal cerebral ischemia in the mouse. Mice were killed 6 h after sham operation or permanent focal cerebral ischemia.
To investigate the function of IL-6 in cerebral ischemia we used IL-6–deficient mice. Because the genetic background has a profound influence on the pathologic outcome after cerebral ischemia, IL-6–deficient mice were backcrossed on a C57/bl 6 genetic background for more than 10 generations. C57/bl 6 mice served as a control group. Interleukin-6–deficient mice show some immunologic deficits but are otherwise viable and healthy (Kopf et al., 1994). All the physiologic parameters that are usually monitored in studies of cerebral ischemia did not differ substantially between IL-6–deficient and normal C57/bl 6 mice during cerebral ischemia (Table 1). In addition, we compared the perfusion deficit between IL-6–deficient and normal mice by iodo[C14]antipyrine autoradiography and did not find any difference either (Table 1). However, the infarct size also did not differ between IL-6–deficient and normal mice after 24 h of permanent ischemia (Table 1), a finding that is in accordance with a recent study by Clark et al. (2000).
Physiologic parameters and infarct volume in permanent middle cerebral artery occlusion
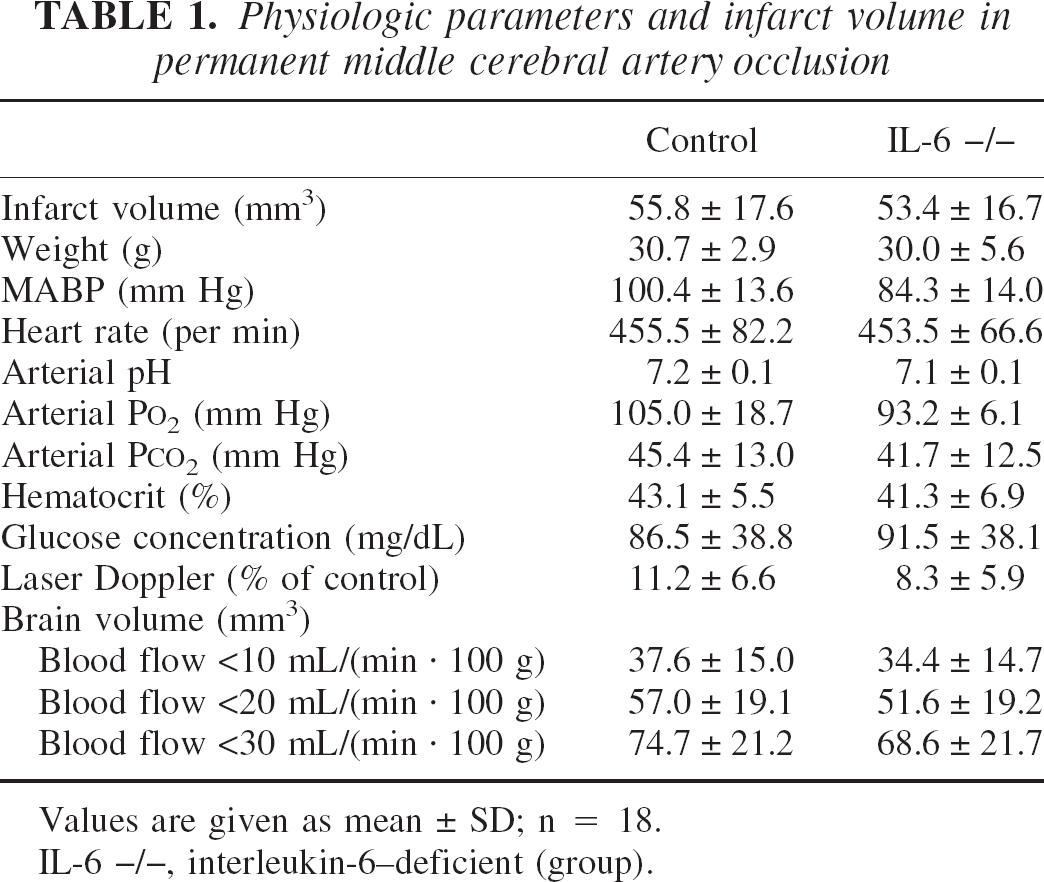
Values are given as mean ± SD; n = 18.
IL-6 –/–, interleukin-6–deficient (group).
Thanks to the recent advent of telemetric techniques, we were able to monitor the body temperature continuously. After sham surgery, there was no difference in body temperature between normal and IL-6–deficient mice (Fig. 3A). Although a small increase in the body temperature after stroke has been reported in rats (Li et al., 1999), we found a decrease in the core body temperature of control mice to 33.0 ± 1.6°C (Fig. 3B). Surprisingly, the body temperature of IL-6–deficient mice dropped to 26.7 ± 1.7°C during the 24-h period after the onset of ischemia (P < 0.05, n = 5, t-test; Fig. 3B). Nevertheless, 90% of the IL-6–deficient animals survived for 24 h after surgery.
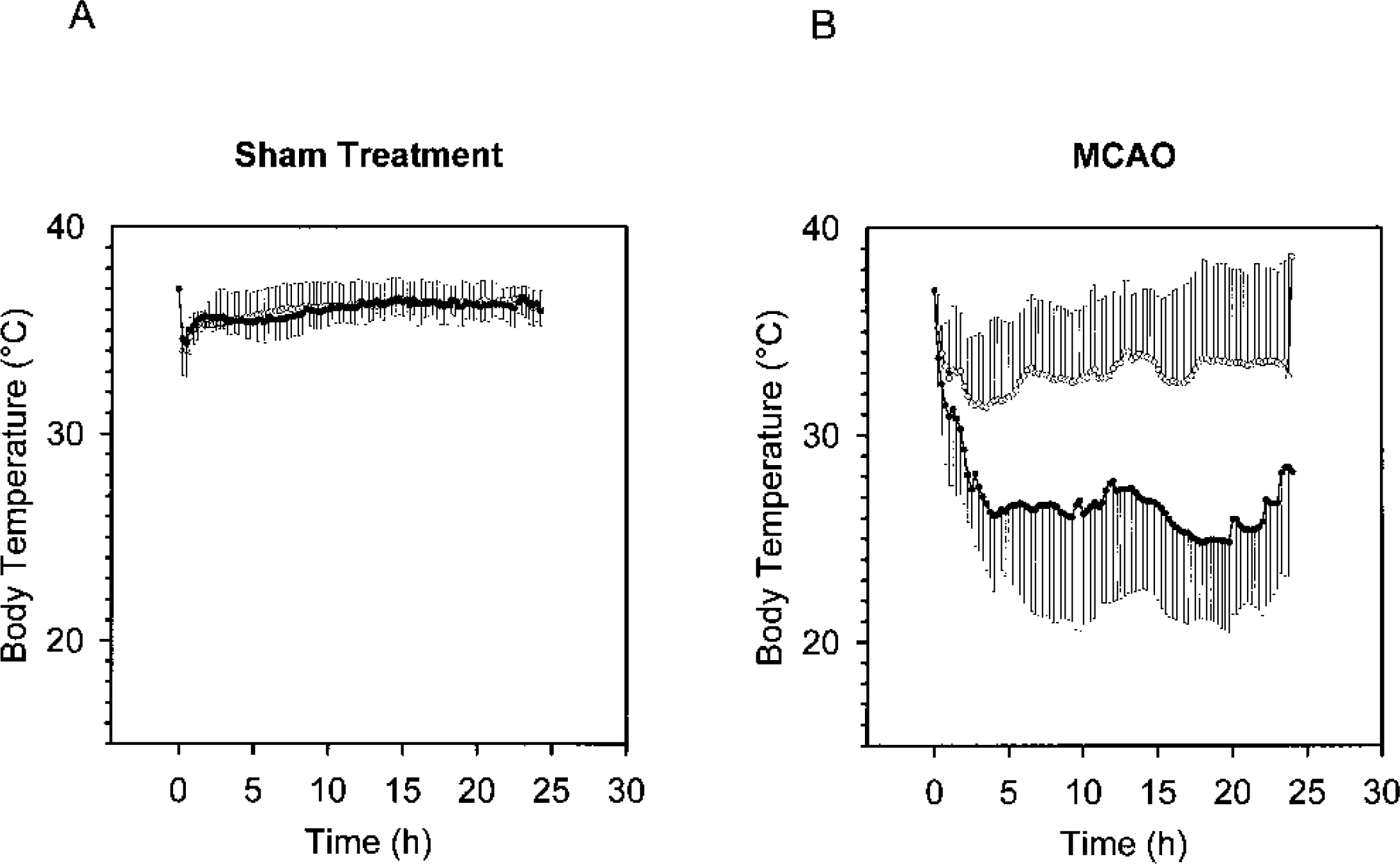
Interleukin-6 (IL-6)-deficient mice develop hypothermia after middle cerebral artery occlusion (MCAO).
To investigate whether the low core body temperature in IL-6–deficient mice has an impact on the outcome of cerebral ischemia, body temperature after surgery was adjusted in both groups by external warming. Using a warming lamp and telemetric measurement of the body temperature, body temperature could be strictly regulated (35.7 ± 0.4°C in IL-6–deficient mice and 35.2 ± 0.1°C in controls, ns). This temperature is comparable with the average temperature of control mice after sham surgery (36.2 ± 0.2°C, Fig. 3A). However, with a fixed body temperature, the mortality of IL-6–deficient mice was significantly increased (Fig. 4A). The mortality in experimental cerebral ischemia likely reflects the infarct size (Kondo et al., 1997; Huang et al., 2000), suggesting that IL-6–deficient mice had larger infarcts than controls did when the body temperature was maintained at a constant level. However, with the short survival (Fig. 4A) we were not able to compare the infarct size in the two groups directly. Therefore, we induced a transient cerebral ischemia of 1 h with a subsequent reperfusion phase of 20 h, which resulted both in smaller infarcts and in a nearly normal survival. As in permanent MCAO, the infarct size of control and IL-6–deficient mice did not differ (data not shown). However, if body temperature was controlled by telemetric feedback, the infarct volume was significantly larger in IL-6 knockout mice (51.3 ± 5.2 mm3, n = 9) than in controls (31.1 ± 5.0 mm3, n = 8, P < 0.02, t-test; Fig. 4B). In comparison, volumes of the right hemisphere in IL-6 –/– mice and control mice were identical (199.4 ± 12.4 mm3 in IL-6–/–, 186.2 ± 24.0 mm3 in control mice). Furthermore, the neurologic impairment was worse in IL-6–deficient mice (neuroscore of 2.7 ± 0.5 vs. 1.2 ± 0.6 in controls, P < 0.05, t-test). This shows that endogenous IL-6 reduces infarct size if the confounding variable of body temperature is controlled. Ischemic damage is due to necrosis and apoptosis. To address the question about which of the two death forms is influenced by endogenous IL-6, we investigated the cleavage of poly(ADP ribose) polymerase (PARP) by caspase-3, which is considered to be a hallmark of apoptosis (Pieper et al., 1999). As has been reported before (Sugawara et al., 2002), the 89-kd, cleaved product of PARP accumulated in the ischemic brain hemisphere 20 h after onset of ischemia (Fig. 4D). This rise of the PARP cleavage product on ischemia was markedly higher in IL-6–deficient mice, suggesting that the IL-6 deficiency promotes apoptosis in the ischemic brain.
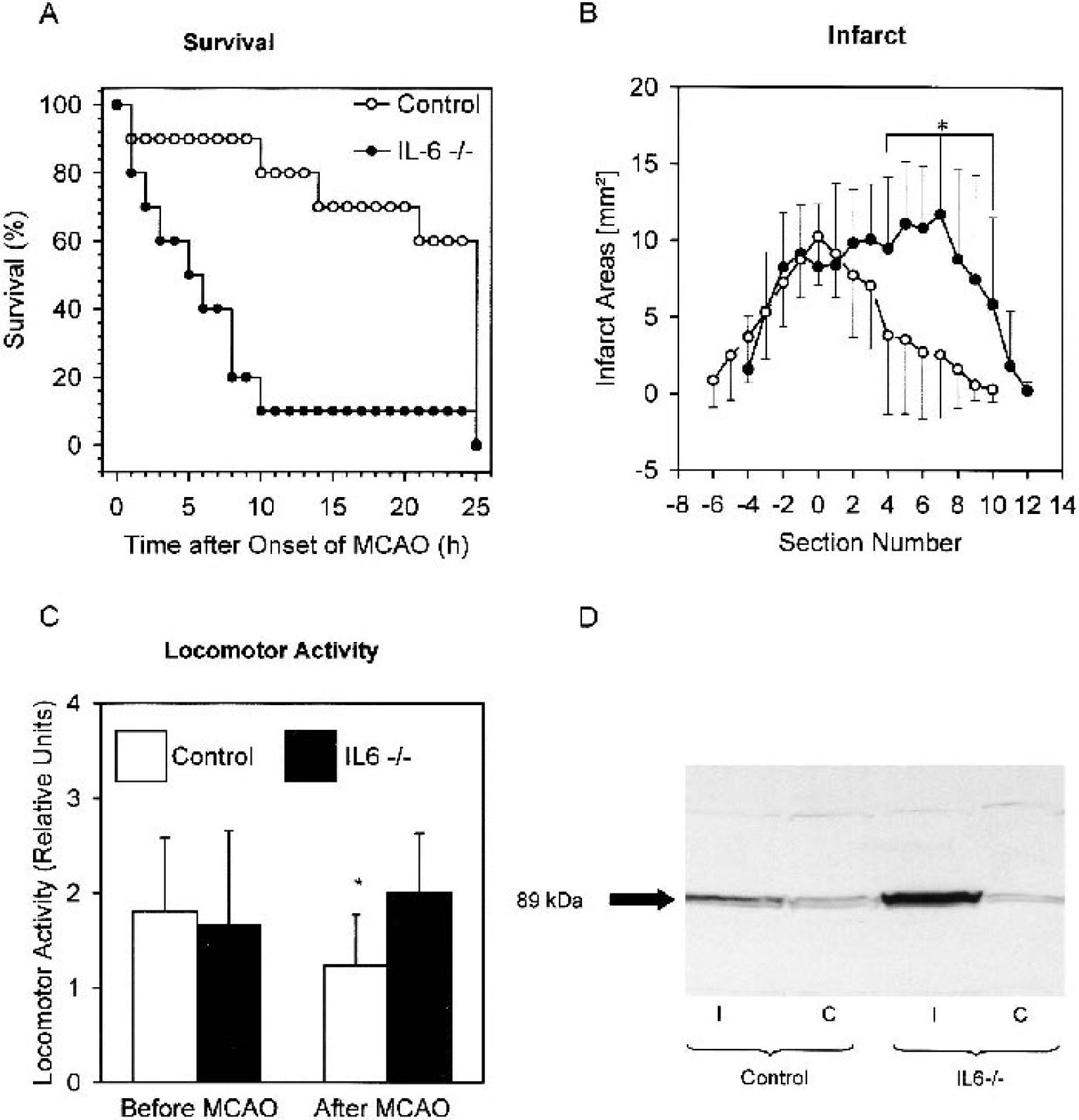
With controlled body temperature, interleukin-6 (IL-6) deficiency leads to a worse outcome after focal cerebral ischemia.
The spontaneous locomotor activity was reduced in control mice after MCAO (Fig. 4C) but did not correlate with the neurologic impairment or infarct size (data not shown), as has also been observed by others (Hattori et al., 2000). As in other disease models, reduced locomotor activity after cerebral ischemia seems to reflect a form of sickness behavior. Locomotor activity after MCAO was significantly higher in IL-6–deficient mice than in controls, although IL-6–deficient mice had larger infarcts (Fig. 4B). This suggests that IL-6 expression also induces sickness behavior after cerebral ischemia. Similar findings have been obtained in a sepsis model (Bluthé et al., 2000).
Direct neuroprotective properties of IL-6 were further investigated in a model of apoptosis of the neuron-like cell line PC12 (Greene et al., 1987). Serum withdrawal induced apoptosis in naive PC12 cells as shown by fluorescence-activated cell sorter (FACS) analysis (Fig. 5A) and nuclear condensation (Fig. 5B). Interleukin-6 (50 ng/mL) prevented apoptosis in this in vitro model (Figs. 5A and B). Interleukin-6 also exerted neuroprotective effects in postmitotic primary neurons (Fig. 5C). Interleukin-6 (50 ng/mL) protected cortical neurons against spontaneous and NMDA-induced cell death.
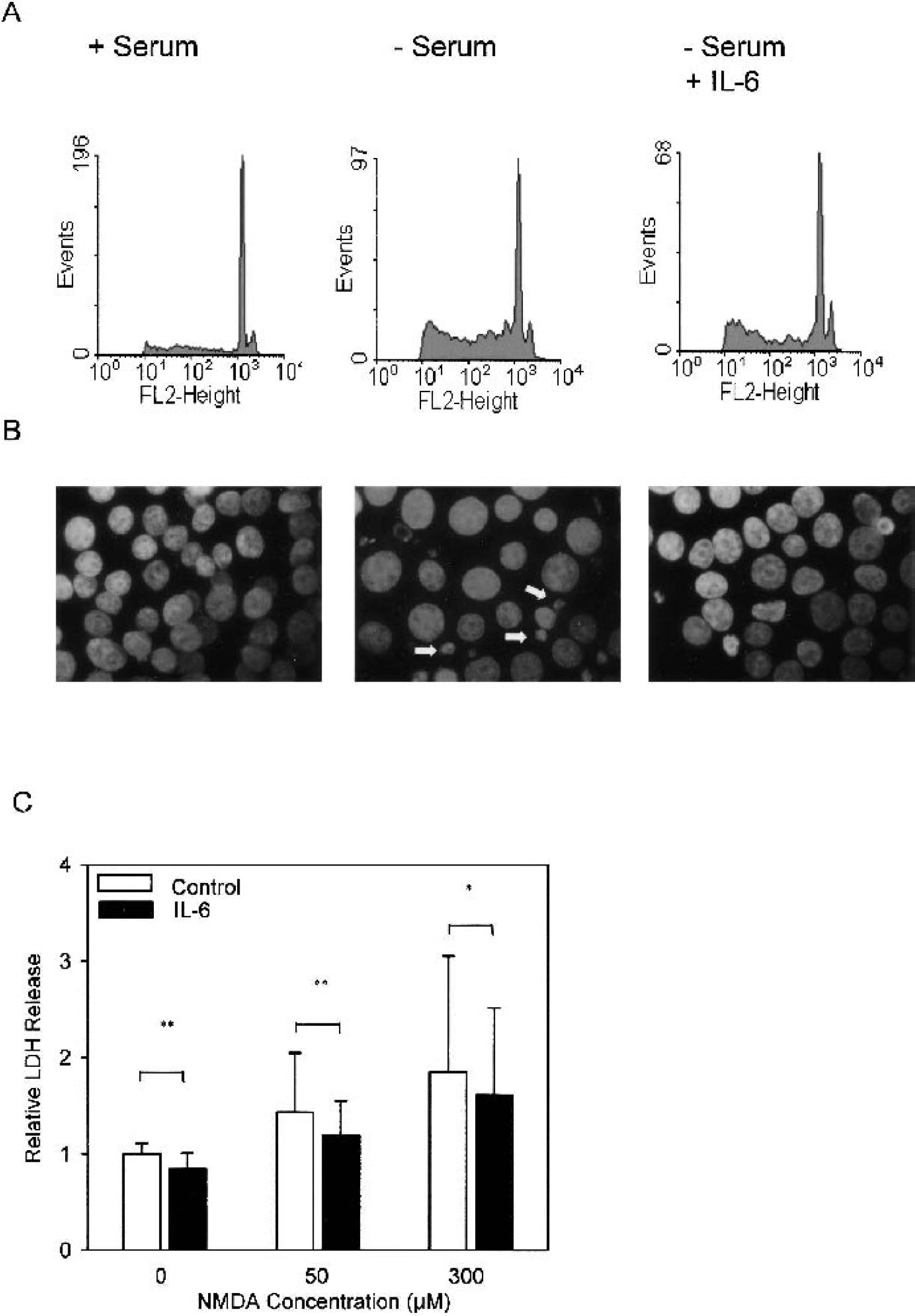
IL-6 has a neuroprotective effect in vitro.
DISCUSSION
Within a few hours after cerebral ischemia, the cytokine IL-6 is upregulated in the mouse brain. Previous studies in rats support this finding (Wang et al., 1995; Loddick et al., 1998; Suzuki et al., 1999). Induction of IL-6 is a common feature of various acute brain diseases and is not restricted to cerebral ischemia (Jüttler et al., 2002). Our experiments now show that IL-6 is an important regulator of the spontaneous body temperature during cerebral ischemia. Mice lacking IL-6 developed a profound hypothermia that could only be detected when the postischemic body temperature was closely monitored. Although body temperature is known to be a major determinant of infarct size, for methodologic reasons in most studies of experimental stroke, body temperature was controlled only during surgery but not afterwards. Only a few studies report the control of the postischemic body temperature (Loddick and Rothwell, 1996; Nogawa et al., 1997). The insertion of a rectal temperature probe causes stress in rodents and in itself increases body temperature (van der Heyden et al., 1997). However, the failure to monitor body temperature after surgery may dramatically influence the interpretation of results. In the case of IL-6, overlooking hypothermia in IL-6–deficient mice after surgery led to the erroneous conclusion that IL-6 did not have a direct influence on acute ischemic injury (Clark et al., 2000).
Why should mice lacking IL-6 develop hypothermia after MCAO although they were normothermic beforehand? A possible explanation is that both antipyretic and pyretic factors are produced during cerebral ischemia, IL-6 being one of the latter. An antipyretic cytokine that is produced in the ischemic brain is tumor necrosis factor-α (Liu et al., 1994). In a mouse model of sepsis, it has been clearly shown that IL-6 and tumor necrosis factor-α may have opposing effects on body temperature (Leon et al., 1998). A deficiency in pyretic IL-6 may thus give way to the hypothermic effect of other cytokines. Control mice were also mildly hypothermic after MCAO, in contrast to rats (Li et al., 1999). Hypothermia after MCAO in wild-type mice has also been observed by others (Corbett et al., 2002). After MCAO, mice exhibit a transient anorexia (Herrmann and Schwaninger, unpublished observation, 2002). Although not widely realized, it has been documented that mice can enter a hypothermic state known as torpor when there is a reduced food intake. During deep torpor, mice will maintain their body temperature at 1 to 2°C above ambient, down to a minimum body temperature of 16 to 19°C (Hudson and Scott, 1979; Gavrilova et al., 1999). A light form of torpor may explain the transient hypothermia in control mice and may contribute to the deep hypothermia in IL-6–deficient mice.
From previous work in several species, it is known that postischemic hypothermia may delay or reduce ischemic damage (Zhang et al., 1993; Markarian et al., 1996). The nearly identical infarct size in the presence of a markedly lower temperature in the IL-6–deficient group compared with control animals suggests yet another effect of IL-6. Indeed, when hypothermia was controlled, it became apparent that endogenous IL-6 reduces the infarct size. This protective effect seems directly related to a neuroprotective effect because IL-6 inhibited apoptosis in in vitro models of neuronal cell death. Neuroprotective properties of IL-6 have been described before (Maeda et al., 1994; Yamada and Hatanaka, 1994; Loddick et al., 1998). However, our study provides the first definite evidence that endogenous IL-6 is neuroprotective. In cerebral ischemia, IL-6 is expressed in neurons (Suzuki et al., 1999). Neuronal expression of IL-6 is stimulated by membrane depolarization, providing a possible mechanism for the induction of IL-6 in cerebral ischemia (Sallmann et al., 2000; Suzuki et al., 2000). At the genomic level, induction of IL-6 by membrane depolarization is mediated by the GRE2 promoter element and not by the binding site for nuclear factor-κB, which actually promotes neuronal cell death in cerebral ischemia (Schneider et al., 1999; Sallmann et al., 2000). Thus, IL-6 has proven to be a neuroprotectant induced by neuronal activity, very much like the growth factor brain-derived neurotrophic factor (Ghosh et al., 1994).
In summary, IL-6 is a pyrogen and a neuroprotectant. Whatever the evolutionary advantage of fever may be, it is associated with a risk for neurons. Therefore, from an evolutionary point of view, it seems logical to endow a pyretic factor with neuroprotective properties. For treatment of acute neurodegeneration such as stroke, it would be desirable to dissociate the two effects of IL-6. Temperature regulation by IL-6 involves the circumventricular organs (Saper and Breder, 1994), an area of the brain that is not shielded by the blood–brain barrier. High-molecular-weight IL-6 antagonists, which have been described before (Savino et al., 1994; Muller-Newen et al., 1998), may therefore selectively inhibit the elevation of body temperature in the circumventricular organs without interfering with the direct neuroprotective action of IL-6. Another possibility might be to modulate the IL-6 effect on temperature regulation downstream of the IL-6 receptor. Interleukin-6–triggered fever is sensitive to cyclooxygenase inhibitors (Dinarello, 1999). Thus, cyclooxygenase inhibitors may lower the body temperature after stroke without interfering with the direct neuroprotective effects of IL-6. This may, in part, explain the reduced infarct size after genetic or pharmacologic perturbation of cyclooxygenase activity (Nogawa et al., 1997, 1998). Involvement of IL-6 or other endogenous mediators in the regulation of ischemic body temperature offers a rationale for lowering body temperature after stroke, and thereby for indirectly tackling brain damage. Recent clinical evidence suggests that the body temperature after stroke can be lowered by acetaminophen, which is a central cyclooxygenase inhibitor (Botting, 2000; Dippel et al., 2001.