
Abstract
Microglia and brain infiltrating macrophages significantly contribute to the secondary inflammatory damage in the wake of ischemic stroke. Here, we investigated whether inhibition of KCa3.1 (IKCa1/KCNN4), a calcium-activated K+ channel that is involved in microglia and macrophage activation and expression of which increases on microglia in the infarcted area, has beneficial effects in a rat model of ischemic stroke. Using an HPLC/MS assay, we first confirmed that our small molecule KCa3.1 blocker TRAM-34 effectively penetrates into the brain and achieves micromolar plasma and brain concentrations after intraperitoneal injection. Then, we subjected male Wistar rats to 90 minutes of middle cerebral artery occlusion (MCAO) and administered either vehicle or TRAM-34 (10 or 40 mg/kg intraperitoneally twice daily) for 7 days starting 12 hours after reperfusion. Both compound doses reduced infarct area by ∼50% as determined by hematoxylin & eosin staining on day 7 and the higher dose also significantly improved neurological deficit. We further observed a significant reduction in ED1+-activated microglia and TUNEL-positive neurons as well as increases in NeuN+ neurons in the infarcted hemisphere. Our findings suggest that KCa3.1 blockade constitutes an attractive approach for the treatment of ischemic stroke because it is still effective when initiated 12 hours after the insult.
Keywords
Introduction
In addition to directly causing neuronal damage, focal ischemic stroke elicits a strong and long-lasting inflammatory response (Weinstein et al, 2010; Yenari et al, 2010). Activated by multiple stimuli, which include hypoxia, neuronal debris, ATP and glutamate, microglia retract their branched processes, round up and transform into ‘reactive’ microglia. Partial breakdown of the blood–brain barrier additionally promotes the infiltration of macrophages, neutrophils, and activated T cells from the blood. In both rodent models of cerebral ischemia and in histopathological studies on human postmortem brain sections activated microglia/macrophages are abundant in the infarcted area and the peri-infarct zone 18 to 96 hours after an ischemic insult (Beschorner et al, 2002; Campanella et al, 2002; Price et al, 2006), and are still present in chronic cystic stages months after a stroke (Beschorner et al, 2002). More recent positron emission tomography imaging in ischemic stroke patients demonstrated microglia activation in the peri-infarct zone on a slightly more delayed time scale: starting at 72 hours and lasting for at least 4 weeks (Price et al, 2006). While microglia can of course exert neuroprotective functions by releasing neurotrophic growth factors such as brain-derived neuroprotective factor or phagocytosing debris and potentially even invading neutrophils (Denes et al, 2007), activated microglia/macrophages are also the main source of inflammatory cytokines such as IL-1
Since interventions aiming at neuroprotection in the acute phase of ischemic stroke have largely failed in the clinic due to the fact that many stroke patients only reach medical attention several hours after the insult, more recent attempts at therapeutic intervention have refocused on inflammation because of its delayed onset. Based on studies demonstrating that immunosuppressive strategies such as general immunosuppression with cyclosporine, neutralization of IL-
Another attractive pharmacological target for inhibiting brain inflammation after ischemic stroke is the intermediate-conductance calcium-activated K+ channel KCa3.1 for which our laboratory designed a small molecule inhibitor, TRAM-34, that blocks this channel with an IC50 of 20 nmol/L and exhibits 200- to 1,500-fold selectivity over other K+ channels (Wulff et al, 2000). KCa3.1 is expressed on proliferating fibroblasts (Pena et al, 2000), on dedifferentiated vascular smooth muscle cells (Köhler et al, 2003), and on immune cells including microglia and macrophages (Hanley et al, 2004; Kaushal et al, 2007; Khanna et al, 2001), activated CCR7+ T cells and IgD+ B cells (Ghanshani et al, 2000; Wulff et al, 2004). In all these cells, KCa3.1 is part of signaling cascades that involve relatively global and prolonged calcium rises during cellular proliferation, cytokine secretion, and volume regulation (see Wulff and Castle, 2010 for a recent review). KCa3.1 channels are voltage independent and only require a small increase in intracellular calcium to open and then maintain a negative membrane potential through K+ efflux. KCa3.1 channels, thus, provide the driving force for store-operated inward-rectifier calcium channels like CRAC (calcium release-activated Ca2+ channel) or transient receptor potential channels like TRPC1 (Wulff and Castle, 2010).
Particularly in microglia, KCa3.1 has been shown to be involved in respiratory bursting (Khanna et al, 2001), migration (Schilling et al, 2004), proliferation (Maezawa et al, 2010), and lipopolysaccharide or amyloid-
Materials and methods
Middle Cerebral Artery Occlusion With 7 Days of Reperfusion
This study was approved by the University of California, Davis, Animal Use and Care Committee and conducted in accordance with the guidelines of Animal Use and Care of the National Institutes of Health and the University of California, Davis for survival surgery in rodents. Adult male Wistar rats weighing 160 to 180 g were purchased from Charles River (Wilmington, MA, USA), acclimatized to the new vivarium for 5 to 7 days and used for the surgery when they weighed 200 to 230 g. Rats were anesthetized using box induction with 5% isoflurane and then maintained on 0.5 to 1.5% isoflurane in medical grade oxygen via a facemask. To assure consistent reduction of cerebral blood flow (CBF) throughout the procedure, we affixed a small hand-made adapter for the Laser Doppler probe (Moor Instruments, Wilmington, DE, USA) to the surface of the skull. The center of the adapter was 5 mm lateral to the central fusion line and 2.5 mm posterior to bregma. Instant adhesive and dental cement were applied to the base and around the edges of the small plastic adapter to hold the Doppler probe. The adapter with the attached probe remained in place throughout the MCAO surgery to confirm continuous occlusion and later the establishment of reperfusion. Focal cerebral ischemia was then induced by occlusion of the left middle cerebral artery (MCA) according to Zea Longa (Longa et al, 1989; O’Donnell et al, 2004). Briefly, the left common carotid artery was surgically exposed, the external carotid artery was ligated distally from the common carotid artery, and a silicone rubber-coated nylon monofilament with a tip diameter of 0.43±0.02 mm (Doccol Corp., Redlands, CA, USA) was inserted into the external carotid artery and advanced into the internal carotid artery to block the origin of the MCA (when maximum CBF reduction observed). The filament was kept in place for 90 minutes and then withdrawn and removed from the blood vessel to restore blood supply. Rats received TRAM-34 at 10 mg/kg, 40 mg/kg or vehicle (Miglyol 812 neutral oil at 1
Pharmacokinetics, Brain Concentrations, and Plasma Protein Binding of TRAM-34
TRAM-34 was synthesized in our laboratory as previously described (Wulff et al, 2000) and its chemical identity and purity checked by 1H NMR and high pressure liquid chromatography/mass spectrometry (HPLC/MS). For intravenous application, TRAM-34 was dissolved at 5 mg/mL in a mixture of 25% CremophorEL (Sigma-Aldrich, St. Louis, MO, USA) and 75% phosphate-buffered saline and then injected at 10 mg/kg into the tail vein of male Wistar rats. At various time points after the injection, ∼100 to 200
The percentage of plasma protein binding for TRAM-34 was determined by ultrafiltration. Rat plasma was spiked with 50 and 100
Assessment of Infarct Area
Rats were euthanized with an overdose of isoflurane. Blood samples for determination of electrolytes, pH, pCO2, glucose and hemoglobin (I-STAT; Abbott, Princeton, NJ, USA) were drawn from the vena cava and brains quickly removed and sectioned into eight 2-mm thick slices starting from the frontal pole. Slices were then fixed in 10% buffer formalin embedded in paraffin and sectioned at 5
Immunohistochemistry
Sections were dewaxed with xylene, rehydrated through an alcohol gradient, and heated with 10 mmol/L Na citrate (pH 6) in a microwave for 15 minutes to retrieve antigenic determinants. After treatment with 1% H2O2 to inactivate endogenous peroxidase activity and blocking with 5% goat serum in phosphate-buffered saline, the sections were incubated overnight at 4°C with the primary antibody in phosphate-buffered saline/2% goat serum. The following primary antibodies were used: KCa3.1 (1:500; AV35098, Sigma, St Louis, MO, USA), CD68 (ED1, 1:1,000; Serotec, Raleigh, NC, USA), and NeuN (1:1,000; A60, Millipore, Billerica, MA, USA). The polyclonal anti-KCa3.1 antibody, which recognizes human, rat and mouse KCa3.1 was tested for specificity with spleen and vascular sections from KCa3.1 wild-type and KCa3.1−/− mice (Si et al, 2006) obtained from the laboratory of Dr Ralf Köhler at the University of Southern Denmark in Odense. Bound primary antibodies were detected with a biotinylated donkey anti-mouse IgG secondary antibody for CD68 and NeuN, or with a biotinylated goat anti-rabbit IgG secondary antibody (both 1:500, Jackson ImmunoResearch, West Grove, PA, USA) for KCa3.1 followed by a horseradish peroxidase-conjugated avidin complex (Vectastain Elite ABC Kit, Vector Laboratories, Burlingame, CA, USA). Peroxidase activity was visualized with 3,3′-diaminobenzidine (DAB Substrate Kit for Peroxidase, Vector Laboratories). Sections were counterstained with hematoxylin (Fisher, Pittsburg, PA, USA), dehydrated and mounted with Permount (Fisher). Apoptosis was assessed with the ApopTag Peroxidase
ED1 stains CD68, a lysosomal membrane protein, which is mainly found in phagocytosing macrophages and reactive microglia (Damoiseaux et al, 1994). At 1:1,000, the antibody produced no stain on resting microglia. Infiltration of ED1+ cells was evaluated according to the method of Lehr et al (1997). Briefly, sections stained for ED1 were photographed and the resulting photos composited into whole-slide images with Photoshop. The Magnetic Lasso tool was used to outline hemisphere borders, brown pixels were selected with the magic wand tool and the number of brown pixels determined with the Histogram tool. The results are reported as brown=ED1+-positive pixels per one millimeter square area (pixels/mm2). NeuN is a DNA-binding, neuron-specific protein present in neuronal nuclei, perikarya, and some proximal neuronal processes. Strong nuclear staining suggests proper nuclear regulatory protein function representative of a healthy neuron. Sections stained for NeuN were photographed and the resulting photos composited into whole-slide images with Photoshop. NeuN and TUNEL-positive cells in the infracted hemisphere were counted with the Photoshop CS3 extended count tool.
Statistical Analysis
Statistical analyses of infarct area, neurological deficit scoring and immunohistochemistry were performed with one-way analysis of variance (Origin software) followed by
Results
Middle Cerebral Artery Occlusion With 7 Days of Reperfusion Induces Substantial Activation of Microglia/Macrophages Expressing KCa3.1
After first performing filament MCAO with reperfusion in both male Sprague-Dawley and male Wistar rats, we chose Wistar rats for our experiments because in agreement with several reports on the differing cerebrovascular anatomy of different rat strains (Aspey et al, 2000; Dittmar et al, 2006) we found infarcts in this strain less variable than in Sprague-Dawley rats. Next, we needed to determine the optimal length of reperfusion for our experiments as we intended to evaluate whether KCa3.1 blockade reduces inflammatory damage. In keeping with previous studies investigating the time course of immune cell infiltration after MCAO in rodents and ischemic stroke in humans (Beschorner et al, 2002; Campanella et al, 2002), 48 hours of reperfusion only induced mild-to-moderate inflammation as measured by the number of ED1+ (=CD68+)-activated microglia/macrophages (data not shown), while 7 days of reperfusion resulted in a dramatic increase in ED1+ cells in the infarcted brain areas as shown in Figure 1, which depicts paraffin-embedded sections from a 90-minute MCAO with 7 days of reperfusion. Staining of serial sections with a polyclonal anti-KCa3.1 antibody, which did not produce any stain on lymphoid and vascular tissues from KCa3.1−/− mice, revealed strong KCa3.1 expression on cells with the round or ‘ruffled’ shape characteristic of reactive microglia/macrophages in the infarcted areas. While the two stains clearly were on the same cells, they did not strictly colocalize because CD68 is a lysosomal protein (Damoiseaux et al, 1994), whereas the KCa3.1 channel is expressed on the plasma membrane and traffics through the endoplasmic reticulum (ER). As seen on the microvessel in the right panels of Figure 1, KCa3.1 protein was also detectable on vascular endothelial cells in keeping with the known expression of KCa3.1 in vascular endothelium and its role in the endothelium-derived hyperpolarizing factor (EDHF) response (Busse et al, 2002; Si et al, 2006). The abundantly present KCa3.1+ microglia in the infarcted area suggests that KCa3.1 inhibition might be of therapeutic benefit for curbing the secondary inflammatory damage in the wake of ischemic stroke.
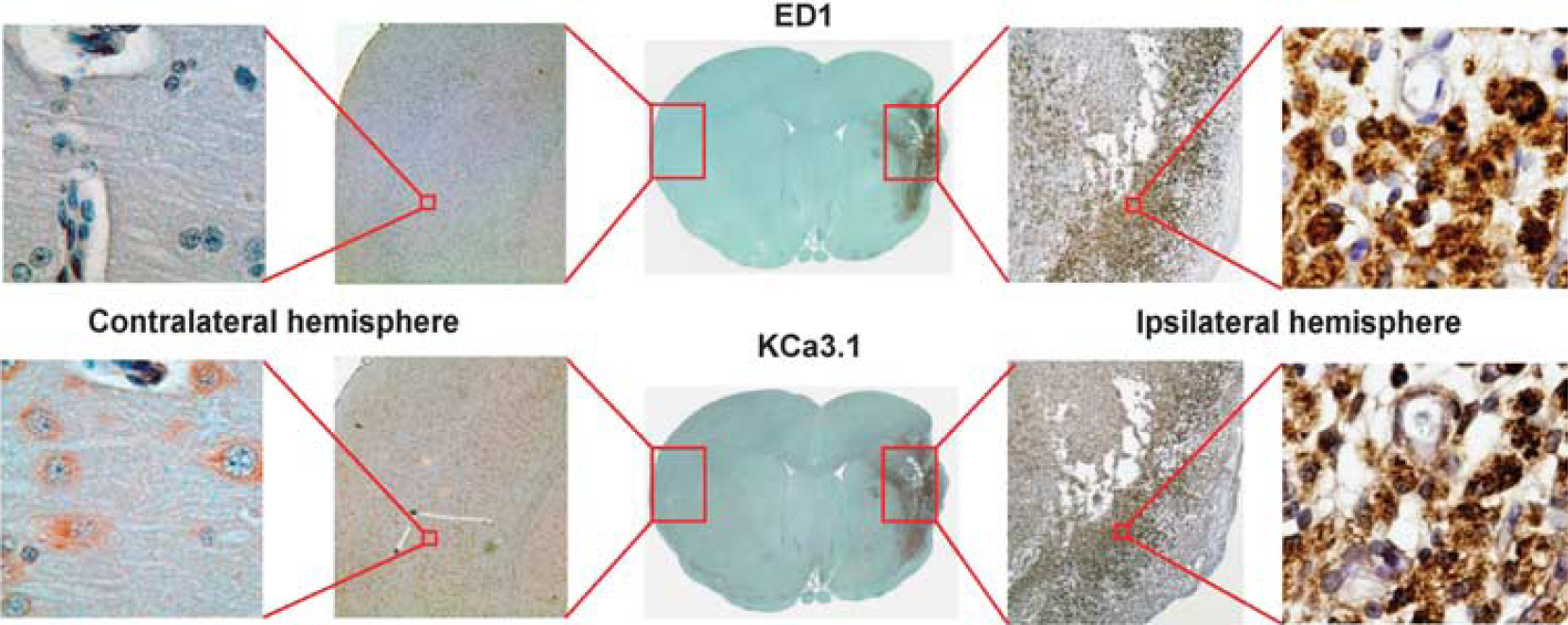
Activated microglia/macrophages in infarcted brain areas express KCa3.1. Staining of serial paraffin-embedded sections from a 90-minute middle cerebral artery occlusion (MCAO) with 7 days of reperfusion for ED1 and KCa3.1.
Pharmacokinetics and Brain Permeability of TRAM-34
To reduce brain inflammation the KCa3.1 blocker TRAM-34 should ideally reach pharmacologically active concentrations in the brain. To address this question and to determine TRAM-34's pharmacokinetics in rats, we established an HPLC/MS assay to measure TRAM-34 concentrations in plasma and tissue. After intravenous administration at 10 mg/kg, total TRAM-34 plasma concentrations fell from a peak of 40
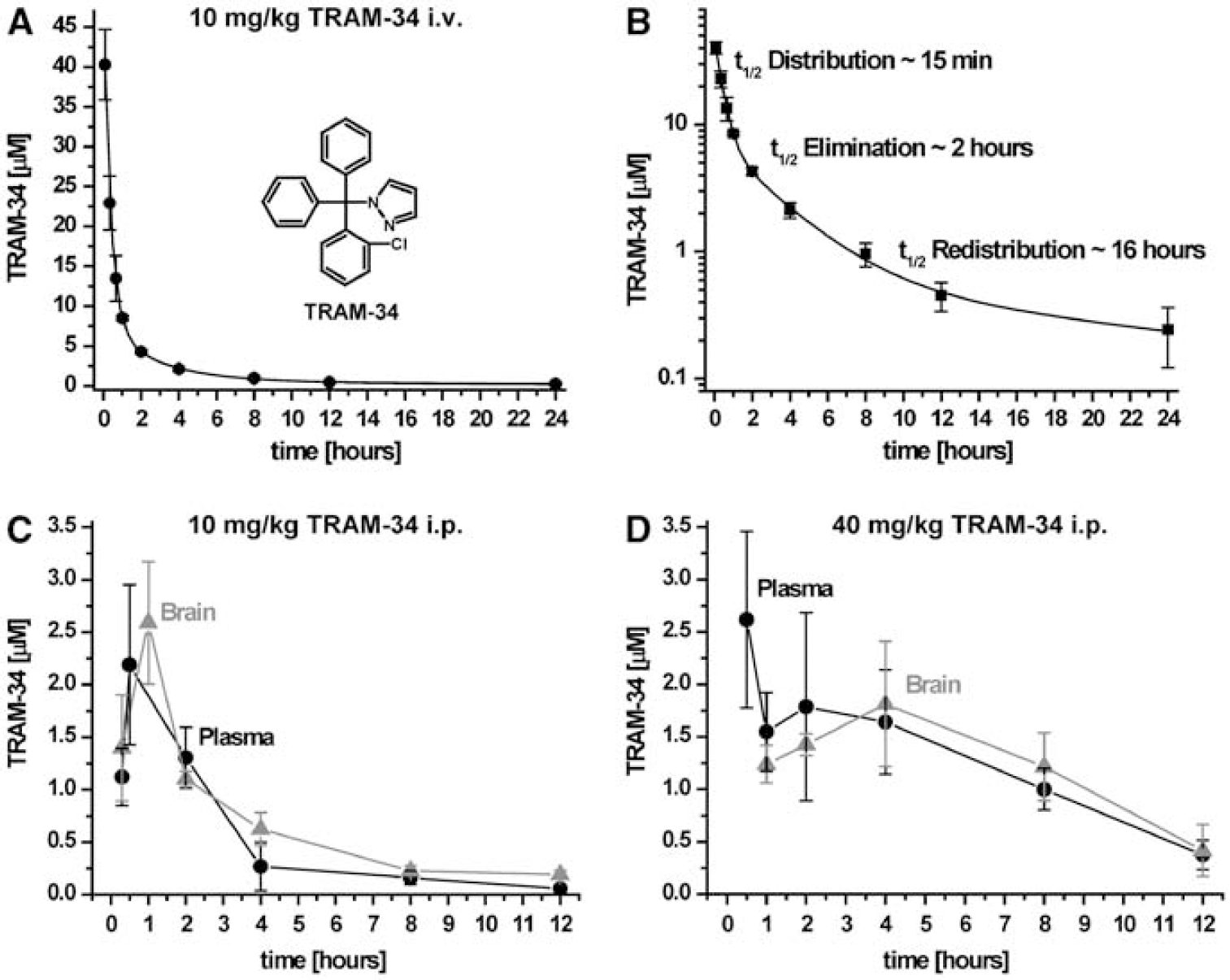
Pharmacokinetics of TRAM-34 in rats. (
Taken together, these results demonstrate that TRAM-34 has reasonably good pharmacokinetics in rats and effectively reaches the brain even when the blood–brain barrier is intact (
KCa3.1 Blockade With TRAM-34 Reduces Infarction and Microglia Activation in Middle Cerebral Artery Occlusion With 7 Days of Reperfusion When Treatment Is Started 2 Hours After Reperfusion
In preliminary experiments, we induced relatively mild infarcts by reducing CBF by only 50% (control: 51.2±8.2% flux reduction, mean±s.d.,
KCa3.1 Blockade with TRAM-34 Reduces Infarction in Middle Cerebral Artery Occlusion With 7 Days of Reperfusion When Treatment Is Started 12 hours After Reperfusion
Encouraged by the above-described results suggesting that KCa3.1 blockade can indeed reduce infarction and microglia activation, we then performed a second set of MCAO experiments where we reduced CBF more severely and evaluated infarct area by H&E staining instead of the quicker and more cost-effective triphenyltetrazolium chloride (TTC) method. H&E is more accurate for infarcts older than 48 to 72 hours because the reactive oxygen species produced by the massive numbers of activated microglia present in ‘aged’ infarcts can oxidize TTC leading to an underestimation of the infarct area. However, in order to be able to prepare undamaged 5
As in our previous experiments, male Wistar rats were subjected to 90 minutes of MCAO with 7 days of reperfusion and then treated with either TRAM-34 at 10 or 40 mg/kg or vehicle twice daily starting 12 hours after successful reperfusion. CBF reduction was 67.3±9.6% in the controls (
Physiological parameters

MCAO, middle cerebral artery occlusion.
Physiological parameters (±s.d.) measured by I-STAT in venous blood samples drawn after reperfusion from the tail vein or at sacrifice on day 7 after MCAO surgery from the vena cava. No significant differences were found between MCAO+vehicle (
As shown in Figure 3, treatment with TRAM-34 resulted in a significant reduction in H&E defined lesion area with the mean infarct size (Figure 3B) being reduced from 22.6±3.6% in the controls (
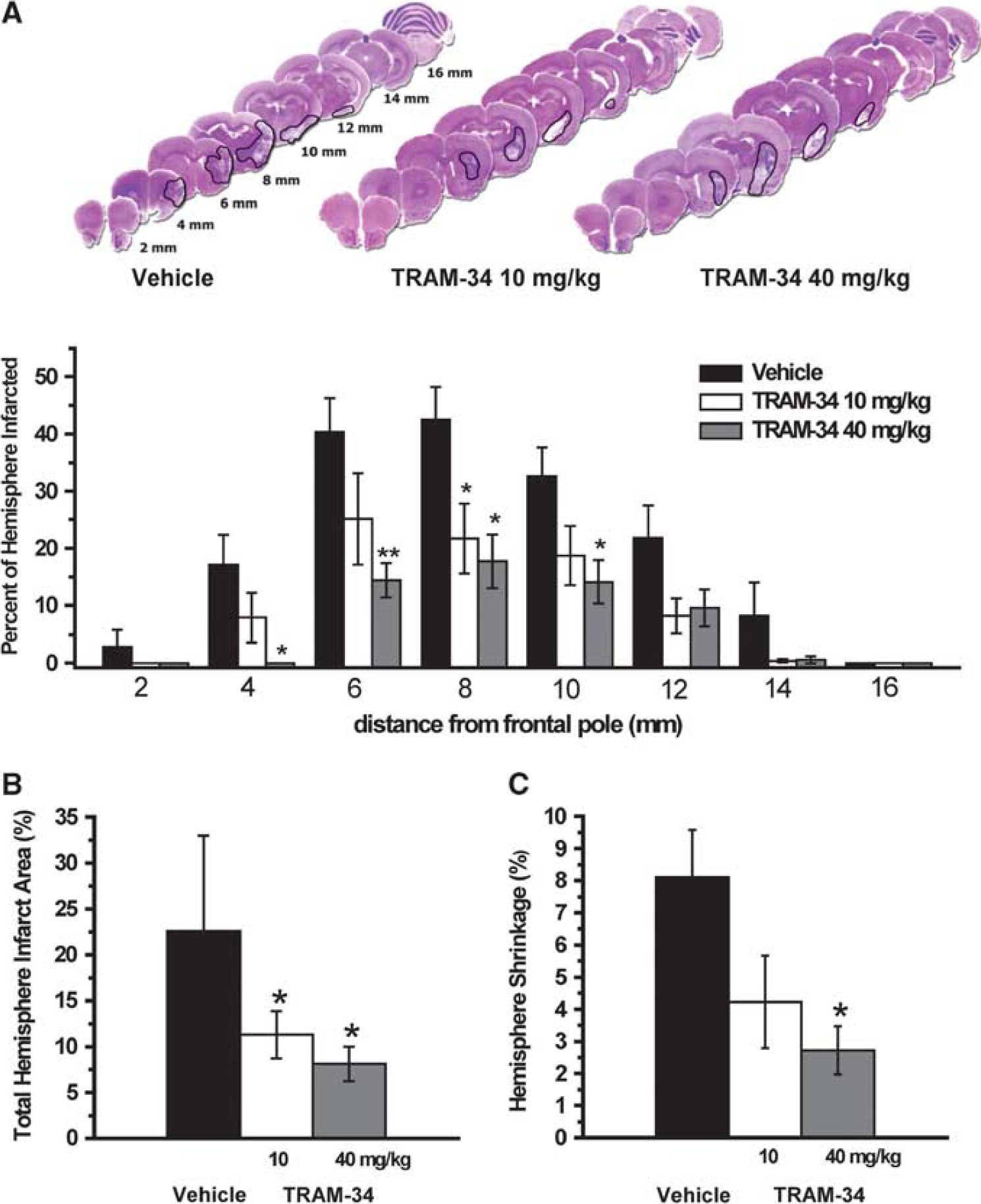
Effect of TRAM-34 on infarct area in rats subjected to 90 minutes of middle cerebral artery occlusion (MCAO) with 7 days of reperfusion. (
KCa3.1 Blockade With TRAM-34 Reduces Neurological Deficit, Microglia Activation and Neuronal Death
Using both a 4-score neurological evaluation scale shown to correlate well with infarct sizes in the frontoparietal cortex (Menzies et al, 1992) and a 14-score tactile and proprioceptive limb-placing test (De Ryck et al, 1989), rats were evaluated for neurological deficit 12 hours after MCAO and then every 24 hours for 7 days. The combination of both tests was chosen since filament MCAO in rats induces infarction not only in the major MCA territory, the lateral and parietal cortex, but also in the underlying striatum. Rats subjected to MCAO exhibited an average deficit score of 3 in the 4-score system (Figure 4A) and a score of 4 in the tactile and proprioceptive 14-score system (Figure 4B) 12 hours after MCAO. These scores slowly improved in vehicle-treated animals to an average of 2 in the 4-score and an average of 8 in the 14-score system by postsurgery day 7 probably reflecting the resolution of edema and partial compensation. (Please note that a normal rat has a score of 0 in the 4-score and a score of 14 in the 14-score system.) TRAM-34 treatment with 40 mg/kg started at 12 hours after reperfusion started to significantly improve neurological deficit in both the 4-score and the 14-score systems from day 5 or day 4 on and on day 7-treated rats displayed a score of 0.5 in the 4-score and of 12 in the 14-score system (Figures 4A and 4B). The lower TRAM-34 dose of 10 mg/kg only significantly improved neurological deficit in the more grade 14-score system and despite showing a positive trend toward improvement failed to significantly reduce deficit in the 4-score system. Interestingly, the deficit that high-dose TRAM-34-treated animals consistently failed to recover was forelimb placement without vision in keeping with the fact that their infarcts were mostly restricted to the striatum. Vehicle-treated animals in contrast exhibited both forelimb and hindlimb impairment on day 7, indicating infarction in both the frontoparietal cortex and the striatum.
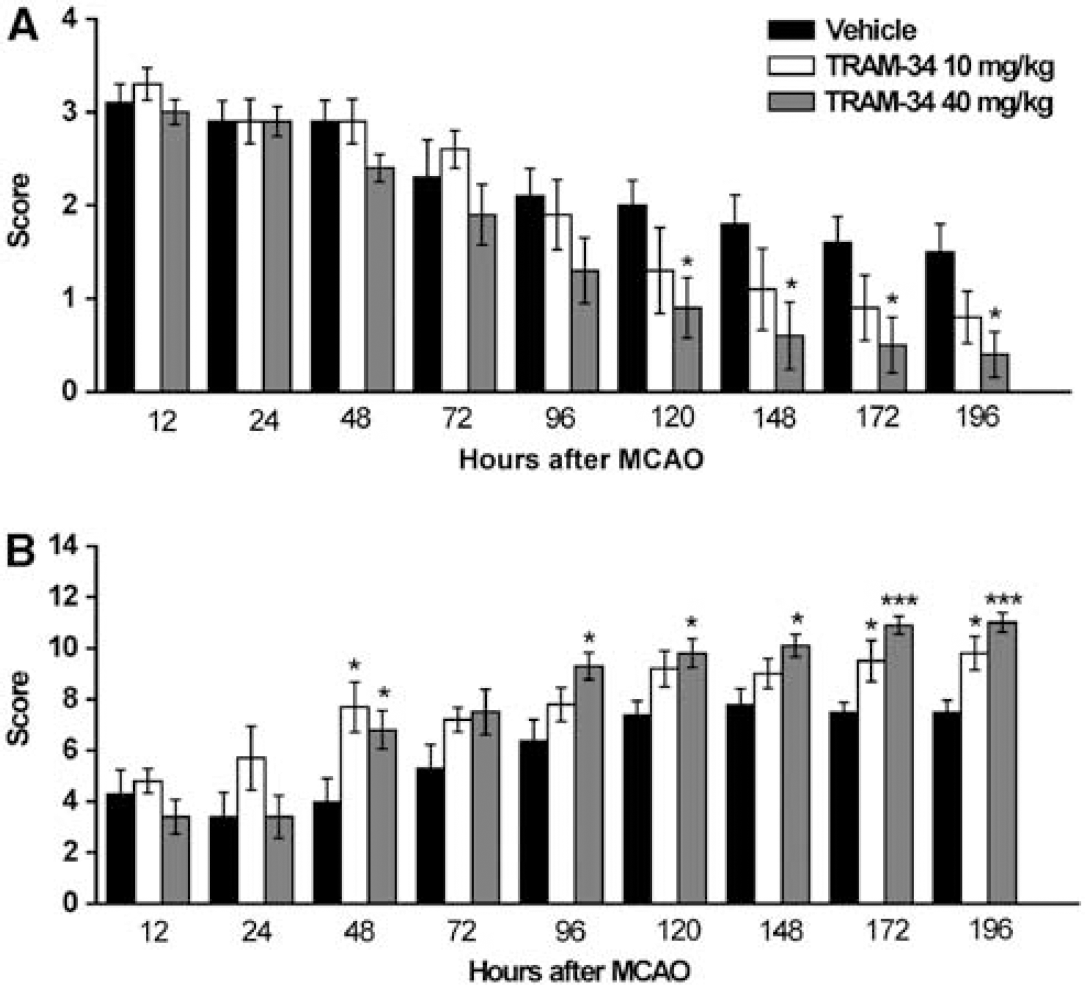
Effect of TRAM-34 on neurological deficit. (
To determine if the delayed TRAM-34 application (12 hours after MCAO) also reduced microglia/macrophage activation similar to what we had previously seen when treatment was started 2 hours after reperfusion (Supplementary Figure 1), we stained sections from the center of the infarct in the 8- and 10-mm slices from all animals in the vehicle, low-dose and high-dose TRAM-34 group for ED1+ microglia and determined the ED1+ area according to the pixel-based method by Lehr et al (1997). While the delayed administration of 10 mg/kg TRAM-34 did not result in a significant reduction in microglia activation, the higher TRAM-34 dose of 40 mg/kg reduced the ED1+ area from 3,770.6±594.2 to 1,632.6±363.75 pixels/mm2 (
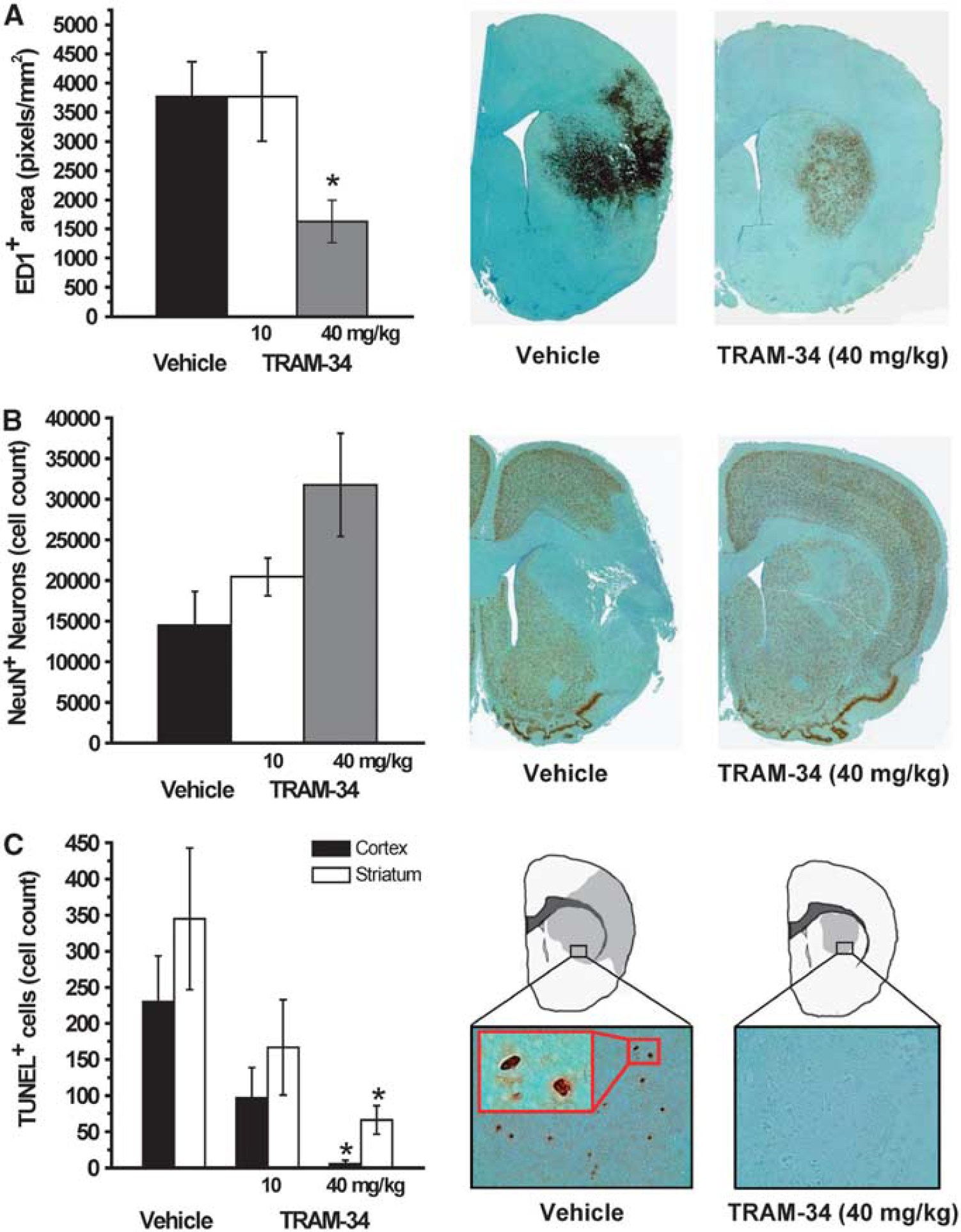
Effect of TRAM-34 on microglia activation and neuronal survival 7 days after middle cerebral artery occlusion (MCAO). (
Discussion
The calcium-activated K+ channel KCa3.1 has an important role in several microglia functions such as respiratory burst (Khanna et al, 2001), migration (Schilling et al, 2004), and microglia-mediated neuronal killing
We assume that the effects of TRAM-34 in our MCAO experiments were primarily mediated through inhibition of KCa3.1 on microglia because the majority of cells with activated microglia/macrophage morphology in ischemic brain have been described to arise from resident microglia and not from hematogenous macrophages infiltrating the brain (Schilling et al, 2003). In lipopolysaccharide-activated cultured rat microglia, TRAM-34 has previously been reported to greatly reduce neurotoxic activity through inhibition of p38 mitogen-activated protein kinase activation, reduction of iNOS induction, and suppression of nitric oxide production (Kaushal et al, 2007). TRAM-34 has further been found to inhibit amyloid-β oligomer induced microglia-mediated neuronal killing in organotypic brain slices as well as damage to postsynaptic elements (Maezawa et al, 2010). Based on these results, we hypothesize that the lipophilic TRAM-34, which effectively crosses the blood–brain barrier as shown in our study, exerts similar effects
We are aware of the fact that our study solely relies on pharmacological evidence for proposing KCa3.1 as a potential therapeutic target for reducing inflammatory damage in stroke. Future studies using genetic manipulation of KCa3.1 would be expected to further clarify the role of KCa3.1 in the pathophysiology of ischemic stroke and will also need to address the question of whether KCa3.1 blockade indeed preferentially affects detrimental microglia functions and not phagocytosis of apoptotic and necrotic neurons as observed by Kaushal et al (2007) after optic nerve transection in rats.
In general, KCa3.1 seems to be relatively safe as a therapeutic target. Two independently generated KCa3.1−/− mice (Begenisich et al, 2004; Si et al, 2006) were both viable, of normal appearance, produced normal litter sizes, did not show any gross abnormalities in any of their major organs and exhibited rather mild phenotypes: impaired volume regulation in erythrocytes and lymphocytes (Begenisich et al, 2004) and a reduced EDHF response together with an 8- to 14-mm Hg increase in blood pressure (Si et al, 2006; Brähler et al, 2009). Pharmacological blockade of KCa3.1 also seems to be safe and well tolerated. As mentioned in the Introduction, TRAM-34 exhibits an excellent selectivity over other ion channels (Wulff et al, 2000) and was ‘clean’ in a Hit Profiling screen on 32 neuronal receptors and transporters (Toyama et al, 2008). Daily administration of TRAM-34 did further not induce any toxicity in a 28-day toxicity study in mice (Toyama et al, 2008) or in a 28-day or a 6-month toxicity study in rats (see Supplementary Toxicity data for this paper). There have also been no reports about toxicity for the structurally related KCa3.1 blocker ICA-17043, which was developed by Icagen Inc. (Durham, NC, USA) and which has been in clinical trials for both sickle cell anemia and asthma (Wulff and Castle, 2010). ICA-17043 was found to be both effective and safe in phase-2 clinical trials (Ataga et al, 2008), but the phase-3 trials were stopped in 2007 apparently due to a lack of efficacy in reducing sickling crises. We would like to point out here that long-term pharmacological KCa3.1 blockade with TRAM-34 in mice (Toyama et al, 2008) or dose-escalating studies with ICA-17043 in 28 otherwise healthy patients with sickle cell disease did not increase blood pressure or lead to electrocardiogram changes (Ataga et al, 2008; Ataga and Stocker, 2009). TRAM-34 has further been found not to delay influenza virus clearance in rats in contrast to dexamethasone (Toyama et al, 2008), demonstrating that KCa3.1 blockers are relatively mild immunosuppressants and are not likely to increase the risk of infections.
In conclusion, we here propose KCa3.1 blockade as a novel therapeutic principle to reduce detrimental brain inflammation after ischemic stroke and possibly other neurological diseases with an inflammatory component.
Footnotes
HW is an inventor on a University of California patent claiming TRAM-34 for immunosuppression. However, since no pharmaceutical companies expressed any interest in the subsequently filed disclosure claiming TRAM-34 for ischemic stroke the University of California decided not to file an addendum to the TRAM-34 patent with this indication.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.