
Abstract
Nitric oxide produced by the inducible nitric oxide synthase (iNOS) is believed to participate in the pathogenic events after cerebral ischemia. In this study, we examined the expression of iNOS in the brain after transient focal cerebral ischemia in mice. We detected differential expression of exons 2 and 3 of iNOS mRNA (16-fold upregulation at 24 to 72 h after middle cerebral artery occlusion, MCAO) compared with exons 6 to 8, 12 to 14, 21 to 22, and 26 to 27 (2- to 5-fold upregulation after 72 and 96 h), which would be compatible with alternative splicing. Expression levels of iNOS mRNA were too low for detection by the Northern blot analysis. Using specific antibodies, we did not detect any iNOS immunoreactivity in the mouse brain 1 to 5 days after MCAO, although we detected iNOS immunoreactivity in the lungs of mice with stroke-associated pneumonia, and in mouse and rat dura mater after lipopolysaccharide administration. In chimeric iNOS-deficient mice transplanted with wild-type bone marrow (BM) cells expressing the green fluorescent protein (GFP) or in wild-type mice transplanted with GFP+ iNOS-deficient BM cells, no expression of iNOS was detected in GFP+ leukocytes invading the ischemic brain or in resident brain cells. Moreover, both experimental groups did not show any differences in infarct size. Analysis of three different strains of iNOS-deficient mice and wild-type controls confirmed that infarct size was independent of iNOS deletion, but strongly confounded by the genetic background of mouse strains. In conclusion, our data suggest that iNOS is not a universal mediator of brain damage after cerebral ischemia.
Keywords
Introduction
The pathogenic events of cerebral ischemia comprise many inflammatory cascades, including the production of nitric oxide (NO) by the inducible NO synthase (iNOS). Excess production of iNOS-derived NO has been implicated not only in cerebral ischemia, but also in meningitis, trauma, and septic shock (reviewed by Iadecola et al, 1997; Moro et al, 2004; Gibson et al, 2005). After cerebral ischemia, proinflammatory mediators such as NO are believed to account for the recruitment of penumbral tissue into the lesion (Dirnagl et al, 1999). Large amounts of NO are believed to damage brain tissue by oxidative stress, DNA destruction, and inhibition of mitochondrial respiration (Keynes and Garthwaite, 2004). However, NO can also exert protective effects depending on the cell type, the concentration of NO, the target molecules encountered, and the temporal expression pattern after injury. For example, iNOS may participate in the induction of ischemic tolerance (for review see Dirnagl et al, 2003) and contribute to increased neurogenesis after ischemia (Zhu et al, 2003).
Mice with disruption of the iNOS gene (MacMicking et al, 1995) sustain smaller infarcts at 96 h, but not at 24 h, after focal cerebral ischemia compared with wild-type animals (Iadecola et al, 1997, Loihl et al, 1999), suggesting that iNOS plays a role in the delayed progression of ischemic brain damage. Inducible nitric oxide synthase was shown to be expressed within the ischemic territory, and iNOS immunoreactivity was detected in infiltrating leukocytes at early times after murine cerebral ischemia (Iadecola et al, 1996). However, other groups found no evidence for iNOS activity, iNOS mRNA, or protein expression after cerebral ischemia in rodents (Cash et al, 2001; Lerouet et al, 2005). Thus, the proposed deleterious role of iNOS-expressing neutrophils in the ischemic brain remains a matter of controversy. Their appearance could also be related to the scavenging of necrotic material (for review see Emerich et al, 2002).
These contradictory data suggest complex regulatory mechanisms of iNOS expression after cerebral ischemia. In this study, we aimed to determine (i) whether ischemia induces iNOS mRNA in the brain, (ii) whether blood-borne leukocytes are the major source of iNOS expression in the brain after cerebral ischemia, and (iii) whether iNOS knockout mice are protected from transient focal ischemia.
Materials and methods
Animals
All animal procedures were performed according to the local guidelines for animal research.
For ischemia, six groups of 9- to 11-week-old male mice were used, with four groups obtained from Jackson Laboratories (Bar Harbor, ME, USA): (i) iNOS knockout mice that were originally generated on a mixed background of C57BL/6 and SV129 strains by Laubach et al (1995) (B6;129P-Nos2) together with (ii) their appropriate wild-type controls provided by the supplier (B6129PF2/J), (iii) ‘Laubach’ iNOS knockout mice backcrossed onto C57BL/6 background (C57BL/6-Nos2) together with (iv) their C57BL/6 wild-types provided by the supplier (C57BL/6J). Breeding pairs of a second iNOS knockout mouse (MacMicking et al, 1995) were kindly provided by Dr S Murphy (University of Nottingham, UK) and bred as heterozygotes in our animal facility to obtain (v) homozygous iNOS knockouts together with (vi) their wild-type littermates.
Adult male C57BL/6-Nos2 and C57BL/6J mice (Charité Breeding Facility, Berlin, Germany) were used for harvesting of bone marrow (BM) cells.
Retroviral Transduction of Bone Marrow Cells and Generation of Chimeras
Harvesting of BM cells and transduction with a murine stem cell virus-based retroviral vector containing the cDNA for enhanced green fluorescent protein (GFP) were performed as described (Priller et al, 2001). Efficiency of gene transfer into clonogenic hematopoietic precursors was assessed in colony assays on methylcellulose supplemented with hematopoietic cytokines (Stem Cell Technologies, Vancouver, Canada). After transduction, 5 × 106 unsorted BM cells were transplanted into each lethally irradiated recipient mouse by tail vein injection as described (Priller et al, 2001). Reconstitution of hematopoiesis was assessed 4 to 5 weeks later by fluorescence-activated cell sorter analysis of GFP expression in peripheral blood leukocytes (Priller et al, 2001).
Focal Cerebral Ischemia and Infarct Volume Measurement
General anesthesia was maintained with 1.0% halothane in 70% N2O/30% O2. Body temperature was maintained at 37±0.5°C using a heating pad. The left middle cerebral artery was occluded (MCAO) by a silicone-coated monofilament (Heraeus Kulzer, Hanau, Germany) via the left internal carotid artery. After 45 or 60 mins, the monofilament was withdrawn for reperfusion. Mice were housed in heated cages for 2 h, and rectal temperature was measured frequently. Animals were then returned to home cages with free access to food and water. Animals were killed after 1 to 14 days, and the brains were removed, snap-frozen, and stored at −70°C. Brain sections were collected serially at 600-μm intervals, and infarct volumes were determined from 20-μm hematoxylin-stained cryostat sections with image analysis (SigmaScan Pro, Jandel Scientific, Erkrath, Germany). Volumes were corrected for postischemic edema by calculating the indirect infarct volume as the volume of the contralateral hemisphere minus the noninfarcted volume of the ipsilateral hemisphere. After a priori power analysis at a statistical power of 0.8 and an α = 0.05 (two-tailed), experimental group sizes were designed to detect effect sizes close or equal to the s.d. (standardized effect size d = 1; Cohen, 1988).
Magnetic Resonance Imaging Measurement of Cerebral Blood Flow
Acute cerebral blood flow (CBF) was measured with magnetic resonance imaging (MRI) during MCAO in iNOS knockout mice (n = 5; MacMicking et al, 1995) and wild-type controls (n = 6). Middle cerebral artery occlusion was induced as described above. Core temperature was maintained at physiologic levels with a heated water jacket during MRI measurement. Immediately after MCAO, animals were transferred into the MRI scanner (Bruker 7T PharmaScan® 70/16 with a Bruker 98/38 mm RF Coil) operating on a Paravision software platform (Bruker, Karlsruhe, Germany). Images for CBF measurement were acquired using a flow-sensitive alternating inversion recovery MRI protocol as described (Prass et al, 2007) with a slight modification: the order for slice-selective versus nonselective inversion was reversed for each consecutive slice to minimize additional T1 effects (imaging parameters: echo time = 16.2 ms; imaging slice thickness = 2 mm; image matrix = 64 × 64; field of view = 20.1 × 20 mm, repetition time = 8,300 ms; inversion parameters: inversion slab thickness = 6 mm; pulse length = 1 ms, 11 images with increasing inversion times, TI = 12 ms + i × 800 ms; i = 0, 1, 2, 3 … 10). Five 2-mm slices were imaged for each animal, covering the area from the olfactory bulb to the cerebellum. The most caudal slice was not used for calculation, because it does not belong to the MCA territory. Image acquisition (with a total duration of approximately 18 mins) was started 21 mins after MCAO induction. In a post hoc analysis, CBF was quantified as described (Prass et al, 2007). After testing for normal distribution with a Kolmogorow—Smirnow test, a pooled t-test was performed on mean hemispheric CBF values in iNOS-deficient versus wild-type mice (P < 0.05 was considered significant).
Induction of Meningeal Inflammation and Meningitis
Male Sprague-Dawley rats (Charles River, Sulzfeld, Germany) weighing 300 to 350 g were anesthetized with thiopental sodium. Rats were placed on a heating blanket and the body temperature was maintained at 37±0.5°C. The animals were placed in a stereotaxic frame, the skin above the skull was removed, and a craniotomy was performed (0.5 × 0.3 cm) laterally and dorsally to the bregma leaving the meninges intact. The skull flap was removed and a plastic chamber (diameter 0.7 cm) was placed above the hole, fixed with dental cement, and filled with saline to avoid drying of the dura. The skin around the chamber was sutured and saline was removed using cotton flaps. Then 150 μL of 10 ng/mL interleukin-1β (R&D Systems, Wiesbaden, Germany) dissolved in 0.9% NaCl or vehicle (NaCl) was applied to the meninges. After 6 h, rats were deeply anesthetized with thiopental sodium (100 mg/mL), and brains were fixed by transcardial perfusion.
For the induction of meningitis, rats and mice were anesthetized as described above. For rats, the skin above the neck was opened, and the muscles retracted until the tissue above the cisterna magna was exposed. A soft catheter (PE-10, 0.28mm internal diameter; Becton Dickinson, Franklin Lakes, NJ, USA) was placed into the cisterna magna and the skin was closed. After 30 mins, 10 μg lipopolysaccharide (LPS) 026:B6 (Sigma, Deisenhofen, Germany) dissolved in 10 μL saline or 10 μL of saline was slowly injected as described (Ivey et al, 2005). For mice, 10 or 150 μg LPS dissolved in 10 μL saline compared with 10 μL saline was injected through the skull under general anesthesia using a Hamilton syringe. Animals were killed after 6 h by an overdose of thiopental sodium and perfusion-fixed.
Cell Stimulation
RAW cells, endothelial cells, neurons, microglial cells, and astrocytes were stimulated with 1 μg/mL LPS (Sigma, Deisenhofen, Germany). Six hours after stimulation, cells were harvested for Western blot, PCR, or were fixed for immunocytochemistry.
Cerebral Microvascular Endothelial Cells
Primary cultures of cerebral microvascular endothelial cells were prepared from 3-week-old Wistar rats as described (Freyer et al, 1999) and cultured in medium with HAM's F10, 20% fetal calf serum, 2 mmol/L L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 2.5 μg/mL amphotericin B, 0.5 μg/mL vitamin C (Sigma-Aldrich, Munich, Germany), 8 μg/mL endothelial cell growth factor (Roche, Mannheim, Germany), and 80 μg/mL heparin. All supplements were purchased from Biochrom (Berlin, Germany) unless otherwise mentioned. The endothelial cells formed confluent monolayers after 6 to 8 days and were used subsequently.
Primary Neuronal Cell Culture
Primary neuronal cultures of cerebral cortex from mouse embryos (E17) were prepared as described (Ruscher et al, 2002). Cells were plated in starter medium (neurobasal medium with B27, 100U penicillin/streptomycin/mL, 0.5 mmol/L L-glutamine, 25 μmol/L glutamate) at a density of 150,000 cells/cm2. Cultures were kept at 36.5°C and 5% CO2, and fed beginning with the fourth day in vitro with culture medium (starter medium without glutamate) by replacing half of the medium twice a week.
Glial Cell Cultures and RAW 264.7 Cells
Astroglial cell cultures were prepared according to a modified method from McCarthy and de Vellis (1980). Cortical meninges of newborn C57BL/6 mice were removed and the tissue mechanically dissected and digested (0.05% trypsin and 0.02% EDTA (ethylenediaminetetraacetic acid)) at 37°C for 15 mins. The tissue was washed twice in phosphate-buffered saline (PBS) followed by mechanical dissociation in Dulbecco's modified Eagle's medium using a pipette. The dissociated cells were seeded in 75-cm2 flasks (2 brains/flask) and grown in Dulbecco's modified Eagle's medium, 10% fetal calf serum, 1% penicillin/streptomycin, 2 mmol/L L-glutamine, and 0.1% glucose. After 8 to 10 days, cultures were shaken at 200 r.p.m. for 2 h to remove microglia. Microglial cells were collected and seeded at a density of 100,000 cells/cm2, and used for experiments after 24 h. Astrocytes were trypsinized and seeded at density of 120,000 cells/cm2 used for experiments after 48 h. Murine macrophage RAW 264.7 cells (kindly provided by Professor R Schumann, Charité, Berlin) were seeded at a density of 20,000 cells/cm2 cultured in Dulbecco's modified Eagle's medium with 10% fetal calf serum. Purity of cell cultures was high with less than 2% microglia in astroglial cultures, 10% astrocytes in neuronal cultures, and less than 1% astrocytes in microglial cultures.
Immunohistochemistry
Mice were deeply anesthetized and perfused transcardially with PBS followed by 4% paraformaldehyde in PBS. Brains and lungs were dissected, immersion fixed in 4% paraformaldehyde at 4°C overnight, cryoprotected with 30% sucrose, shock-frozen in hexane at −70°C, and stored at −80°C. Frozen tissues were cut into 20 μm sections on a cryostat and washed in PBS. Sections were preincubated in 10% normal goat serum in PBS containing 0.3% Triton X-100. Sections were washed three times in PBS and incubated at 4°C for 36 h in primary antibody solutions (10% normal goat serum, 0.3% Triton X-100). Inducible nitric oxide synthase protein was detected using rabbit anti-mouse iNOS polyclonal antibodies raised against either the N terminus (Upstate Biotechnology, Lake Placid, NY, USA) or the C terminus of mouse iNOS (Transduction Laboratories, Lexington, KY, USA) or against a synthetic peptide corresponding to murine C-terminal amino acids (affinity-purified; Chemicon, Temecula, CA, USA). All iNOS antibodies were diluted 1:100, and the rat anti-CD45 antibody was diluted 1:500 (Serotec, Düsseldorf, Germany). After two washes, sections were incubated in PBS-A (2 mg bovine serum albumin in 1 mL PBS) for 1 h and subsequently treated with the secondary antibody (biotinylated IgG, 1:2,000 in PBS-A; Vector Laboratories, Burlingame, CA, USA) at room temperature for 16 h. Two further washes were followed by avidin—biotin complex (Elite ABC, 1:1,000 in PBS-A; Vector Laboratories) at room temperature for 6 h. After another three washes and preincubation in 3,3′-diaminobenzidine solution (Sigma, Deisenhofen, Germany) containing 0.5 mg 3,3′-diaminobenzidine in 1 mL 50 mmol/L Tris buffer, pH 7.6, peroxidase activity was visualized for 3 mins after addition of H2O2 (0.015% final concentration). For CD45 immunoreactivity, final concentrations of 10 mmol/L imidazole and 0.3% ammonium nickel sulfate were added. After several washes in PBS, sections were mounted and coverslipped. Omission of primary antibodies served as negative controls.
For immunofluorescence staining, sections were treated with the primary anti-iNOS antibodies as described. After 16 h incubation with secondary antibodies (Alexa 488— goat anti-rabbit IgG 1:500 or Texas Red—goat anti-rabbit 1:200; Molecular Probes, Eugene, OR, USA), sections were mounted with Mowiol 4-88 (Hoechst, Paris, France) and examined using a fluorescence microscope.
Western Blot Analysis
Mouse brains or RAW 264.7 cells were homogenized on ice in a dounce-homogenizer (Wheaton, Millville, NJ, USA) in homogenization buffer (4 mmol/L HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.4, 250 mmol/L sucrose, 1 mmol/L EDTA, 5 mmol/L sodium azide, 2 μg/mL aprotinin, 1 μg/mL pepstatin A, 1 μg/mL leupeptin, and 0.5 mmol/L phenylmethylsulfonylfluoride). The homogenate was centrifuged at 2,500g for 10 mins, followed by centrifugation of the supernatant at 100,000g. The pellet was resuspended in 50 mmol/L Tris—HCl, pH 7.5, and protein concentration was determined with bicinchoninic acid assay (Pierce, Rockford, IL, USA). Homogenates were heated at 90°C in sodium dodecyl sulfate sample buffer for 3 mins. Forty micrograms of protein per lane was loaded on 8% sodium dodecyl sulfate-polyacrylamide gels followed by electrophoresis. After separation, proteins were electroblotted onto nitrocellulose (Schleicher & Schuell, Dassel, Germany). The membranes were blocked with 5% low-fat milk powder in PBS containing 0.5% Tween-20 for 1 h, incubated over night at 4°C with an anti-iNOS antibody (1:1,000; Chemicon), a secondary anti-mouse horseradish peroxidase-linked antibody (1:1,000; Sigma, St Louis, MO, USA), and visualized with enhanced chemoluminescence (Pierce) using ImageMaster VDS-CL (Amersham, Freiburg, Germany).
Preparation of Total RNA and Reverse Transcription
At 6 h, 24 h, 3 days, and 4 days after MCAO, experimental animals along with sham-operated controls (n = 5 per group) were killed using an overdose of ether. Brains were removed rapidly, and the dissected hemispheres were snap-frozen in liquid nitrogen. Total RNA was prepared from tissue specimens using Trizol (Gibco BRL, Gaithersburg, MD, USA) according to the manufacturer's protocol. Total RNA (2.5 μg) isolated from each specimen was reverse-transcribed using random hexamers and moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega, Mannheim, Germany).
Quantitative Real-Time Polymerase Chain Reaction
Expression of iNOS mRNA in each sample was normalized for RNA preparation and reverse transcriptase reaction on the basis of its glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA content. Amplification products in GAPDH and iNOS reverse transcriptase PCR were detected using the LightCycler-FastStart DNA-Master SYBR-Green-I Kit (Roche Molecular Biochemicals, Penzberg, Germany). For amplification and detection, we used the LightCycler Relative Quantification Software (Roche Molecular Biochemicals). We performed the following quality checks: (1) for establishing the real-time PCR, the amplification products of GAPDH and iNOS mRNA were cloned (TA-cloning vector) and sequenced (Cycle sequencing; Amersham Pharmacia Biotech, Little Chalfont, UK). (2) After each real-time PCR, amplification products were checked by melting curve and visualization on agarose gel. All samples were amplified in duplicate from the same RNA preparation and the mean values of the respective crossing points (Cp) were considered. For determination of PCR efficiencies (E), we analyzed a serial dilution of GAPDH and iNOS cDNA and calculated E = 10[–1/slope]. The relative expression of each gene part of iNOS compared with GAPDH mRNA expression was calculated using the respective Cp according to the equation E−Cp(iNOS)(iNOS)/ E−Cp(GAPDH)(GAPDH). Table 1 summarizes the sequence-specific primers (TIB MOLBIOL, Berlin, Germany), the thermal cycling protocols, PCR efficiencies, and amplification product lengths.
Summary of the sequences of the primer pairs, the thermal cycling protocols, the PCR efficiencies, and the amplification product lengths in real-time PCR analysis of iNOS mRNA expression
Northern Blotting
iNOS-specific riboprobe: The iNOS-specific probe was generated by SP6 transcription (SP6/T7 Transcription Kit; Roche Molecular Biochemicals) of a mouse iNOS cDNA-containing plasmid. For construction of this plasmid, we generated an iNOS cDNA fragment. RNA from LPS-stimulated RAW cells was purified and reverse-transcribed. Polymerase chain reaction was performed using the iNOS cDNA as template and iNOS-specific linker primers covering the positions 176 to 195 in exon 2 (5′-cagacatggcttgcccctgg-3′) and 979 to 959 in exon 8 of iNOS (5′-gcatctggtagccagcgtacc-3′) according to the accession number AF065921.2. Thermal cycling started with 2 mins at 95°C and proceeded with 35 cycles of 98°C for 10 s, 68°C for 30 s, and 72°C for 90 s.
The forward primer was extended by a HindIII restriction site, the reverse primer by a KpnI restriction site to allow cloning into the pSPT19 vector (Roche, Mannheim, Germany). After cloning and amplification, the plasmid was purified (Qiagen, Hilden, Germany), linearized by HindIII, and repurified. The linearized DNA was transcribed and labeled with SP6 RNA polymerase using [32P]dCTP (3,000 Ci/mmoL) according to the supplier. The DNA template was digested with RNase-free DNaseI. The labeled probe was purified using ProbeQuant G-50 Micro Columns (Amersham Pharmacia Biotech, Little Chalfont, UK).
iNOS RNA standard: As a positive control and for determining the sensitivity of Northern blotting, we synthesized an iNOS RNA fragment as standard using the pSPT19 vector containing the iNOS cDNA fragment described above. The KpnI-linearized vector DNA was transcribed with T7 RNA polymerase, and the DNA template was removed with RNase-free DNaseI after transcription and purification with RNeasy Mini-Elute Cleanup-Kit (Qiagen).
Northern blotting: Total RNA was freshly prepared using Trizol (Invitrogen, Carlsbad, CA, USA). After digestion with DNaseI, mRNA was isolated (μMACS mRNA Isolation Kit; Miltenyi Biotec, Bergisch Gladbach, Germany). Purified and glyoxal-treated mRNA (10 to 20 μg), and serially diluted, glyoxal-treated iNOS RNA standard (4 ng; 400; 40; 4 pg) were separated on 1% sodium phosphate-buffered agarose gel. Electrophoretically separated RNA was capillary-blotted onto a positively charged nylon membrane (NytranN; Schleicher & Schuell). Using QuikHyb Hybridization Solution (Stratagene, La Jolla, CA, USA), the blot was hybridized at 60°C to the labeled iNOS-specific riboprobe.
Results
Differential Expression of Exons 2 to 27 of Inducible Nitric Oxide Synthase mRNA after Middle Cerebral Artery Occlusion
At 6, 24, 72, and 96 h after transient MCAO (n = 5 per group), iNOS mRNA expression was examined both in the ipsilateral ischemic and contralateral non-ischemic hemisphere of wild-type C57BL/6 mice compared with sham-operated controls (n = 5). iNOS mRNA levels were determined relative to the mRNA levels of the housekeeping gene, GAPDH, which did not exhibit significant changes after transient MCAO.
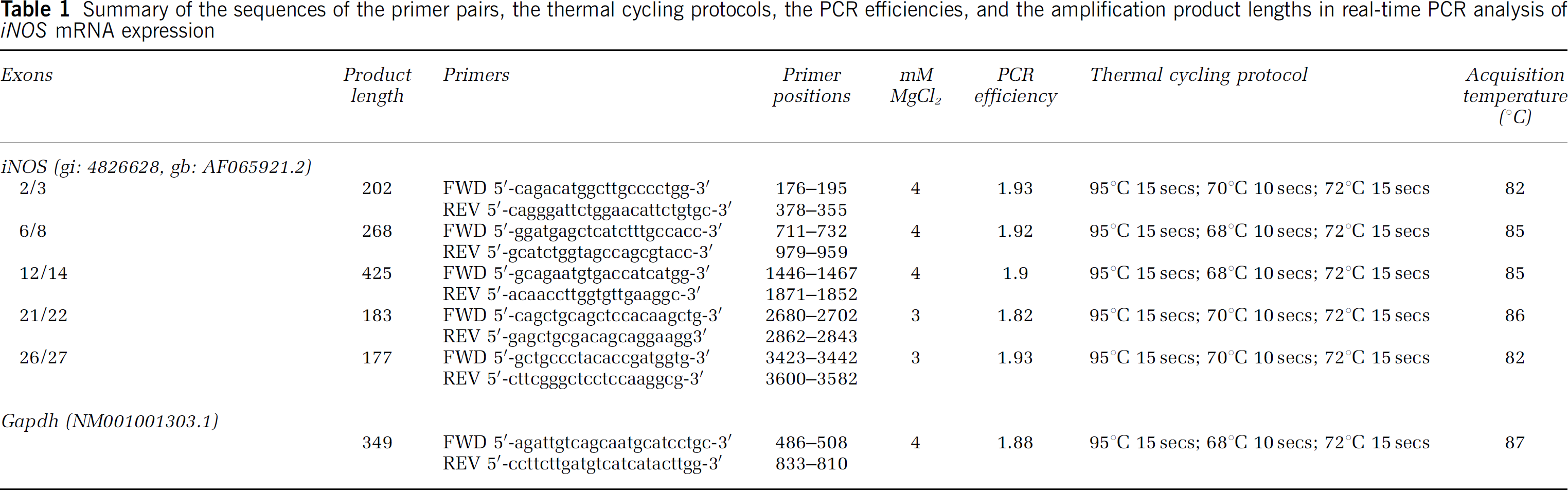
Expression of the iNOS gene was determined using five pairs of iNOS-specific PCR primers. Using primers corresponding to sequences of exons 2 and 3, a 16-fold induction of gene expression was detected after 24 h, which remained elevated up to 72 h and returned to a six-fold increase over sham control levels after 96 h (Figure 1A). The increase in mRNA expression was statistically significant after 24 and 72 h, but not after 96 h compared with sham-operated animals (Dunnett's multiple comparison test, P < 0.05). Using primers for exons 6 to 8, 12 to 14, or 21 to 22, no iNOS mRNA induction was detected in the ischemic hemisphere after 6 h, and only a discrete 1.5- to 2-fold increase was detected after 24 h, which did not reach statistical significance (Figure 1B to 1D). However, 3- to 4-fold increases in iNOS mRNA expression were observed after 72 h using primers for exons 6 to 22. A similar induction profile was found for the portion of the iNOS gene containing exons 26 to 27 with maximum iNOS mRNA levels at 72 h, although mRNA induction was not detected during the first 24 h (Figure 1E). Induction of iNOS mRNA exons 6 to 27 was statistically significant after 72 h (Dunnett's multiple comparison test, P < 0.001), and for exons 6 to 8, 12 to 14, and 26 to 27 after 96 h (Dunnett's multiple comparison test, P < 0.001). Induction of iNOS mRNA was undetectable in the contralateral non-ischemic hemisphere at any time point after MCAO using primers for exons 6 to 8, 12 to 14, 21 to 22, and 26 to 27. However, a mild 2- to 3-fold induction was detectable using primers for exons 2 and 3 (Dunnett's multiple comparison test, P > 0.05). As a positive control, we also assessed iNOS mRNA expression in the dura mater of rats challenged with an intrathecal injection of 100 ng LPS, and found a 45-fold induction of iNOS mRNA within 6 h after administration of LPS (Figure 1F). Sham animals injected with saline did not display an induction of iNOS mRNA (Figure 1F).
Quantitative real-time polymerase chain reaction analysis of iNOS mRNA levels 6, 24, 72, and 96h after MCAO in mice compared with sham-operated animals (n = 5 per group, data are means + s.d.). iNOS-specific PCR primers corresponded to sequences of (
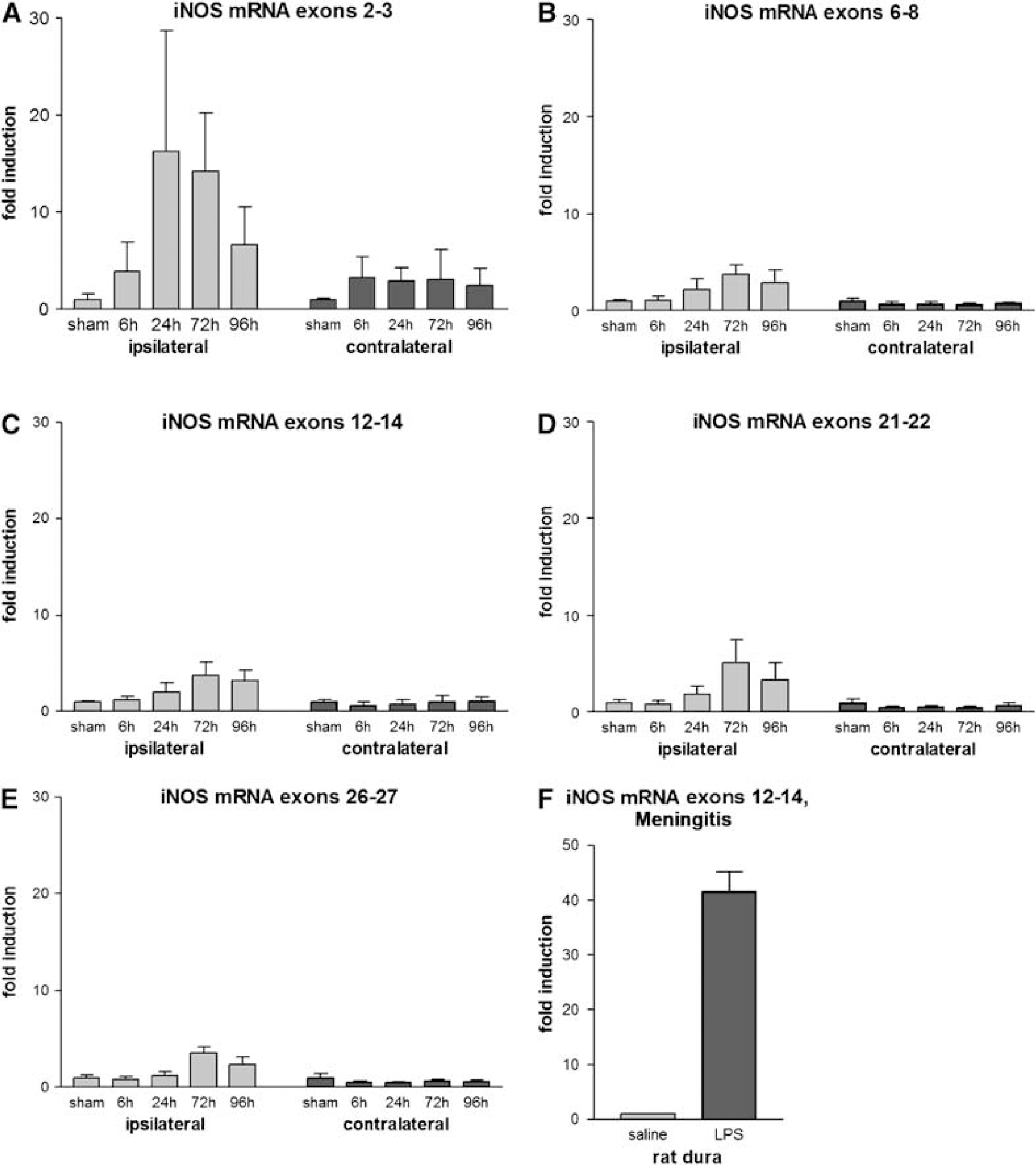
To search for potential splice variants of iNOS mRNA, Northern blot analysis with an iNOS-specific riboprobe was performed (Figure 2). Detection of 4 pg of serially diluted iNOS mRNA standard as positive control showed high sensitivity of the Northern analysis (Figures 2A to 2D). In contrast to unstimulated cells, RAW cells stimulated with LPS for 6 h clearly expressed a single band of iNOS mRNA (Figures 2E and 2F). However, no expression of iNOS mRNA was detected when purified mRNA from ischemic brain tissue 24 and 72 h after MCAO was subjected to Northern blot analysis (Figures 2G to 2M). Thus, the expression of iNOS mRNA in the ischemic brain was too low to permit detection of splice variants using Northern blot analysis.
Detection of iNOS mRNA expression in brain using Northern blot analysis with a 32P-labeled murine iNOS-specific riboprobe. Serially diluted iNOS RNA standard of smaller size (822 bp, exons 2 to 8) than the full-length iNOS mRNA (
Cell Type-Specific Expression of Inducible Nitric Oxide Synthase mRNA after Lipopolysaccharide Stimulation
To determine which cell types in the brain might contribute to iNOS mRNA expression in response to inflammation as occurring after cerebral ischemia, we challenged rodent primary cultures of cerebral microvascular endothelial cells, cortical neurons, microglia, astrocytes, and the murine macrophage cell line RAW 264.7 with 1 μg/mL LPS for 6 h (data not shown). Using exon-specific primer pairs, significant iNOS mRNA induction was detected in LPS-stimulated microglia (~400-fold), RAW cells (~150-fold), and astrocytes (~50-fold). Lipopolysaccharide-treated cultured neurons exhibited almost no induction of iNOS mRNA (< 1.5-fold), and iNOS mRNA expression in endothelial cells was highly variable. However, the specific ischemia-induced differential expression of certain parts of the iNOS gene was not detected in any of the cell cultures stimulated with LPS.
Absence of Neuroprotection in Bone Marrow Chimeras with Tissue-Specific iNOS Gene Expression and Lack of iNOS Protein Expression after Transient Focal Cerebral Ischemia
To analyze whether infiltrating leukocytes or resident brain cells (microglia, astrocytes) are the major sources of iNOS expression and subsequent tissue damage after transient focal cerebral ischemia, we generated four groups of BM chimeric mice: wild-type mice transplanted with BM cells from wild-type (wt → wt) or from iNOS-deficient mice (iNOS−/−→wt), and iNOS knockout mice transplanted with BM cells from iNOS-deficient (iNOS−/− → iNOS−/−) or from wild-type (wt→iNOS−/−) mice. The BM chimeras with tissue-specific iNOS gene expression were subjected to MCAO for 45 mins, followed by reperfusion for 4 days. Physiologic parameters did not differ between the groups during MCAO (data not shown). Surprisingly, none of the four groups of chimeras displayed significant differences in infarct volumes (means±s.d.): wt → wt = 34.1±12.0 (n = 6); iNOS−/− → iNOS−/− = 34.5±14.8 (n = 6); wt → iNOS−/−= 34.6±14.9 (n = 11); iNOS−/− → wt = 30.3±13.0 mm3 (n = 9). The wt → wt group served as control for the calculation of the difference in means (DM) and the 95% confidence interval (CI) of the difference in means, resulting in the following data (in mm3): iNOS−/− → iNOS−/−: DM = 0.45, CI = −17.01 to 17.91; wt → iNOS−/−; DM = 0.57, CI = −13.84 to 14.98; iNOS−/− → wt: DM = −3.76, CI = −18.08 to 10.57.
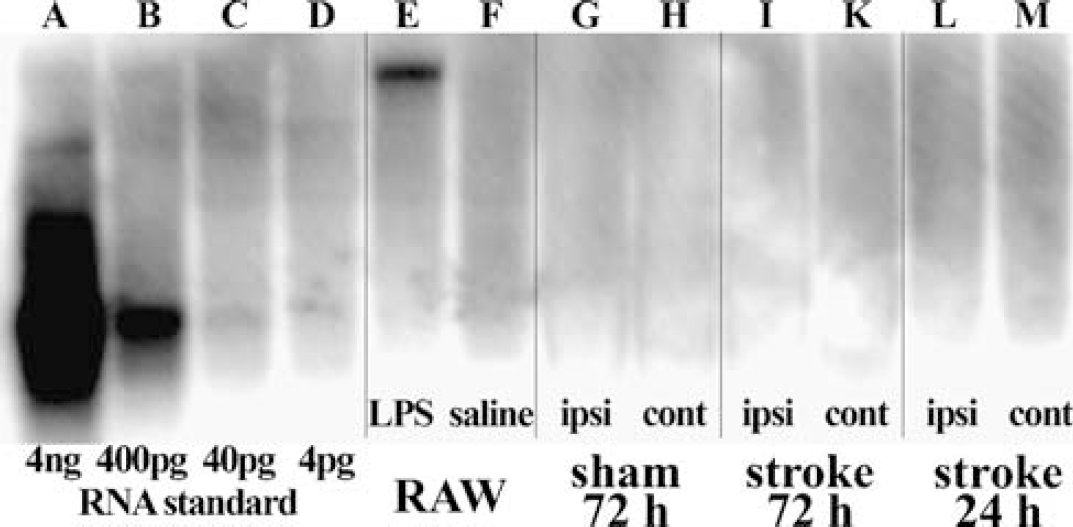
We also generated GFP BM chimeric mice by transplanting GFP-transduced BM cells from wild-type mice into myeloablated wild-type recipients. In these chimeras, on average, 80% of peripheral blood leukocytes expressed GFP at 4 to 5 weeks after transplantation (data not shown). When expression of iNOS was examined in GFP BM chimeras after 45 mins of MCAO and reperfusion for 1, 3, 4, 5, and 14 days (n = 4 per group), we detected robust infiltration of GFP+ BM-derived cells into the ischemic hemispheres starting at 1 day and persisting for 14 days after MCAO (Figure 3A, top panel; Figure 3B). However, no iNOS immunoreactivity was detectable using three different antibodies against iNOS in these GFP-expressing cells or in resident brain cells in ipsilateral (Figure 3A, bottom panel; Figure 3B) and contralateral hemispheres after MCAO. These findings are in line with the very modest induction of iNOS mRNA after MCAO using quantitative real-time PCR, and the absence of iNOS mRNA expression after MCAO using Northern blot analysis (see Figures 1 and 2). To rule out that the antibodies were too insensitive to detect iNOS, we performed several positive controls, including Western blot analysis of mouse brain homogenates. The polyclonal antibody raised against the C terminus of murine iNOS (Chemicon) recognized a single band of the appropriate size of ~130 kDa in mouse brain homogenate (Figure 4A). Furthermore, unstimulated RAW cells did not express basal levels of iNOS (Figures 4B and 4B’), but iNOS protein was detected in these cells after treatment with 1 μg/mL LPS for 6 h using immunocytochemistry and Western blot analysis (Figures 4C and 4C’). Inducible nitric oxide synthase immunoreactivity was also observed in leukocytes infiltrating the meninges of rats 6 h after local administration of the proinflammatory mediator interleukin1β (Figure 4D). Induction of meningitis using subdural injection of 10 μg LPS also caused a strong induction of iNOS protein expression in dura mater, leptomeninges, and superficial cortical layers (Figure 4E). No expression of iNOS was observed in dura mater, leptomeninges, and cortex of rats injected with saline (Figures 4D and 4E’). Interestingly, 150 μg LPS (Figure 4F), but not 10 μg LPS, was found to induce iNOS protein expression in the mouse brain. No iNOS immunoreactivity was observed in mice after application of saline (Figure 4F’). For an additional positive control of iNOS expression in mouse tissue (Figures 4G to 4K), lung sections were used from an animal with stroke-associated pneumonia at 72 h after MCAO as described by Prass et al (2003). Pneumonia was associated with a massive infiltration of CD45-immunoreactive inflammatory cells (Figure 4G), which showed iNOS immunoreactivity (Figures 4H and 4K).
Absence of iNOS immunoreactivity (
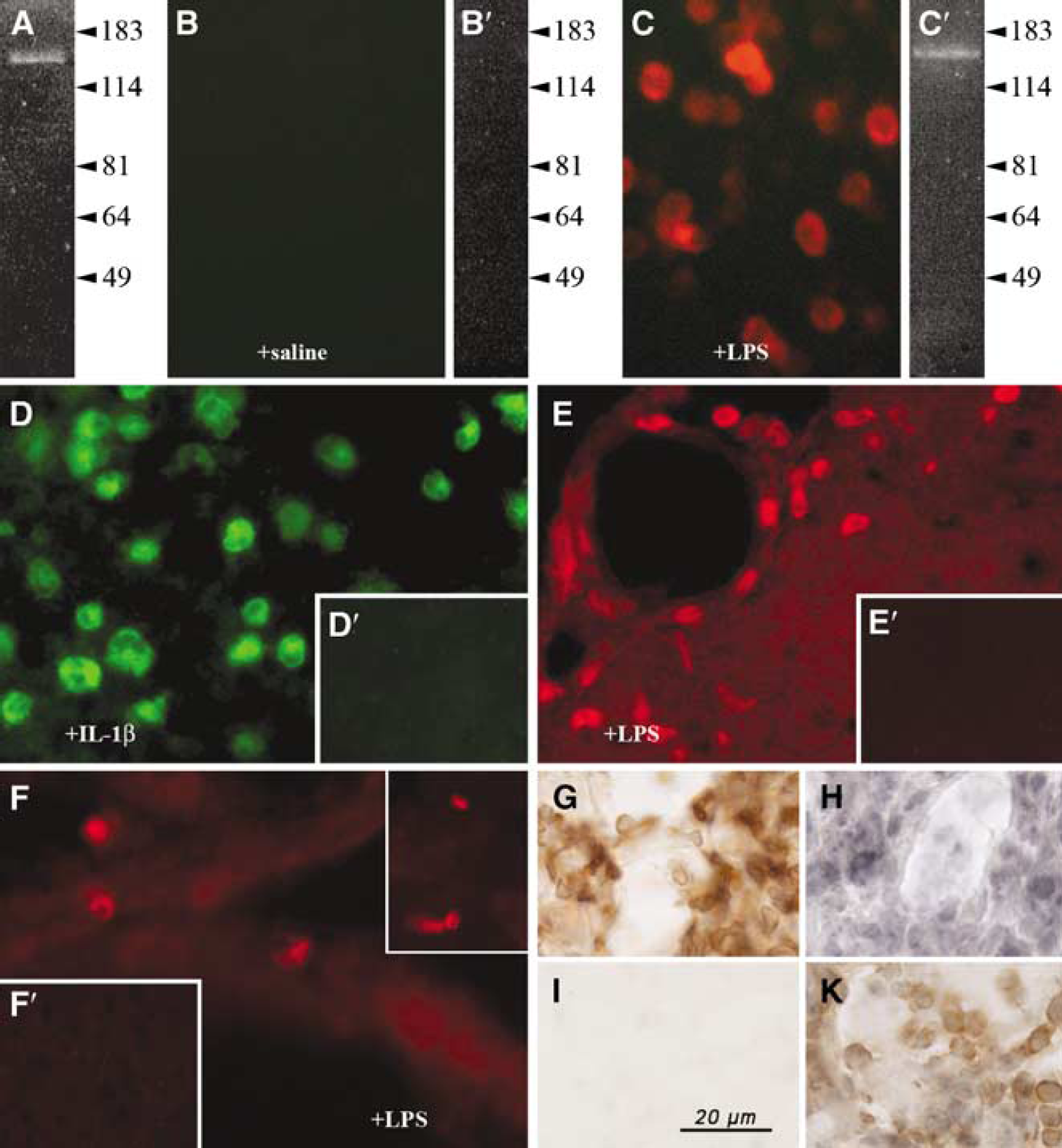
Inducible nitric oxide synthase protein expression in vivo and in vitro. The antibody raised against the C terminus of murine iNOS (Chemicon) recognized a single band of the appropriate size of ~130 kDa in Western blots of mouse brain homogenate (
Inducible Nitric Oxide Synthase Deficiency Does Not Reduce Infarct Size after Transient Focal Cerebral Ischemia in Mice
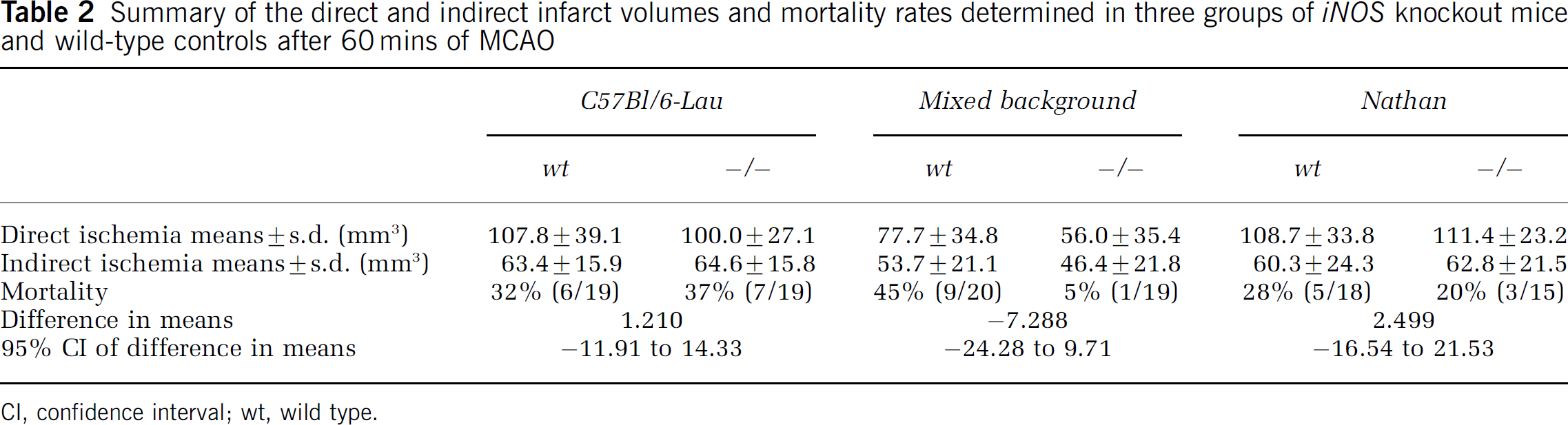
As we did not observe differences in the stroke sizes of BM chimeric wild-type versus iNOS knockout mice, we repeated the experiments in nontrans-planted wild-type and iNOS-deficient mice using a more severe model of MCAO to determine confounding effects of the BM transplantation procedure. After 60 mins of left MCAO and 96 h of reperfusion (Figure 5, left panel), male C57BL/6-NOS2 mice generated by Laubach et al (1995) displayed infarct volumes (n = 12; 64.6±15.8 mm3) that were not significantly different from those of their appropriate male C57BL/6J controls (n = 13; 63.4±15.9 mm3). Direct and indirect infarct volumes, mortality as well as differences in means with CIs of these mice and of the groups described below are summarized in Table 2. The size of the ischemic lesion produced by MCAO was comparable with that observed in other studies of focal ischemia in mice (Dubal et al, 2001). We chose 96 h of reperfusion because other groups reported a reduction in infarct size in iNOS null mice generated by MacMicking et al (1995) compared with wild-type mice at this time point, but not at 24 h after permanent cerebral ischemia (Iadecola et al, 1997; Loihl et al, 1999). As in our BM chimeric mice, no iNOS immunoreactivity was detectable in C57BL/6-NOS2 or C57BL/6J at 96h after MCAO (data not shown).
Summary of the direct and indirect infarct volumes and mortality rates determined in three groups of iNOS knockout mice and wild-type controls after 60 mins of MCAO
CI, confidence interval; wt, wild type.
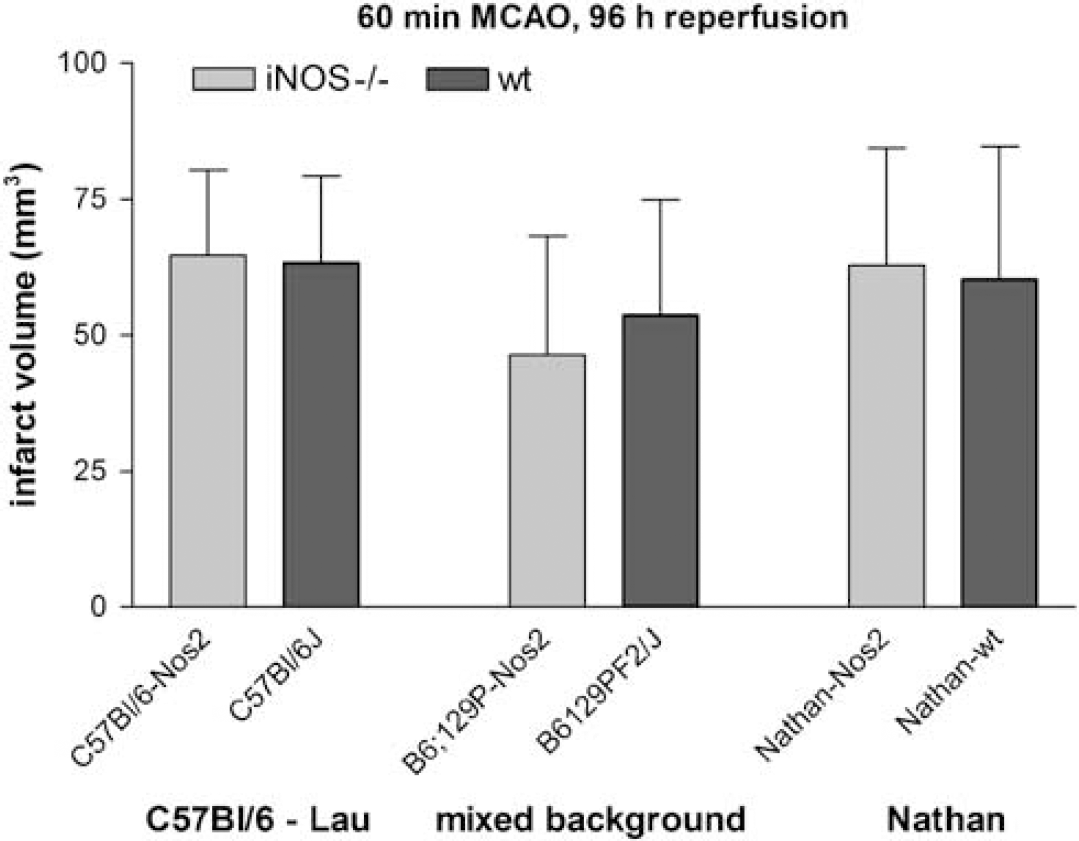
Indirect infarct volumes after MCAO in three different types of male iNOS knockout mice and their appropriate wild-type controls. Left panel: mice generated by Laubach et al (1995) maintained on a pure C57BL/6 background (number of animals: ***C57BI/6-Nos2 = 12, C57BI/6J = 13); middle panel: mice generated by Laubach et al (1995) on a mixed genetic background of C57BL/6 and SV129 strains (B6;129P-Nos2 = 18, B6129PF2/J = 11), right panel: mice generated by MacMicking et al (1995) (Nathan-Nos2 = 12, Nathan-wt = 13). Note that none of the different types of male iNOS knockout mice showed protection from cerebral ischemia compared with wild-type controls. Infarct sizes were comparatively smaller in B6;129P-Nos2 compared with C57BI/6-Nos2.
We repeated the ischemia experiments in a second iNOS knockout strain (MacMicking et al, 1995), to exclude the possibility that the lack of protection from transient focal cerebral ischemia in C57BL/6-NOS2 mice may result from a difference in the knockout strategy. In the first strain (Laubach et al, 1995), exons 12 and 13 of the iNOS gene were replaced with the neomycin resistance gene, whereas the promoter region and exons 1 to 4 were deleted in the second strain (MacMicking et al, 1995). We used male homozygous iNOS knockouts together with their male wild-type littermates. Again, after 60 mins of left MCAO and reperfusion for 96 h (Figure 5, right panel), iNOS null mice developed infarcts (n = 12; lesion size = 62.8±21.5 mm3), which did not differ in size from those of their wild-type littermates (n = 13; 60.3±24.3 mm3). The mortality rates in both groups also did not differ (Table 2).
To estimate the contribution of strain background on the outcome after transient focal cerebral ischemia, we determined infarct volumes in those iNOS knockout mice that were originally generated on a mixed genetic background of C57BL/6 and SV129 strains by Laubach et al (1995). After 60 mins of left MCAO and 96 h of reperfusion (Figure 5, middle panel), male B6;129P-Nos2 mice (n = 18; 46.4±21.8 mm3) showed no significant reduction of infarct volumes compared with B6129PF2/J wild-type controls (n = 11; 53.7±21.1 mm3). However, it is important to note that nine B6129PF2/J mice died within 96 h after MCAO (45% mortality), whereas only one B6;129P-Nos2 mouse died (5% mortality, see Table 2). This difference in mortality was statistically significant (χ2 test, P < 0.05). Again, no iNOS immunoreactivity was detectable in B6;129P-Nos2 or B6129PF2/J mice at 96 h after MCAO (data not shown). Finally, it should be noted that the infarct volumes in B6;129P-Nos2 knockout mice were significantly smaller (unpaired t-test, P = 0.02) than those in iNOS-deficient mice maintained on a pure C57BL/6 background. However, no significant differences were detected between the respective wild-type control groups.
To exclude significant differences in brain vasculature between iNOS-deficient mice and wild-type mice, we used flow-sensitive alternating inversion recovery MRI to compare the CBF after MCAO in the most commonly used iNOS knockout strain (MacMicking et al, 1995) and wild-type controls (Figure 6). Middle cerebral artery occlusion led to a drastic reduction in ipsilateral CBF both in iNOS-deficient mice (n = 5; Figure 6A, bottom row) and in wild-type controls (n = 6; Figure 6A, top row). Intraischemic CBF values were comparable to those from a previous MCAO study in mice (Prass et al, 2007). Averaging hemispheric CBF across all animals yielded ipsilateral and contralateral CBF levels, which were not different between both groups (Figure 6B).
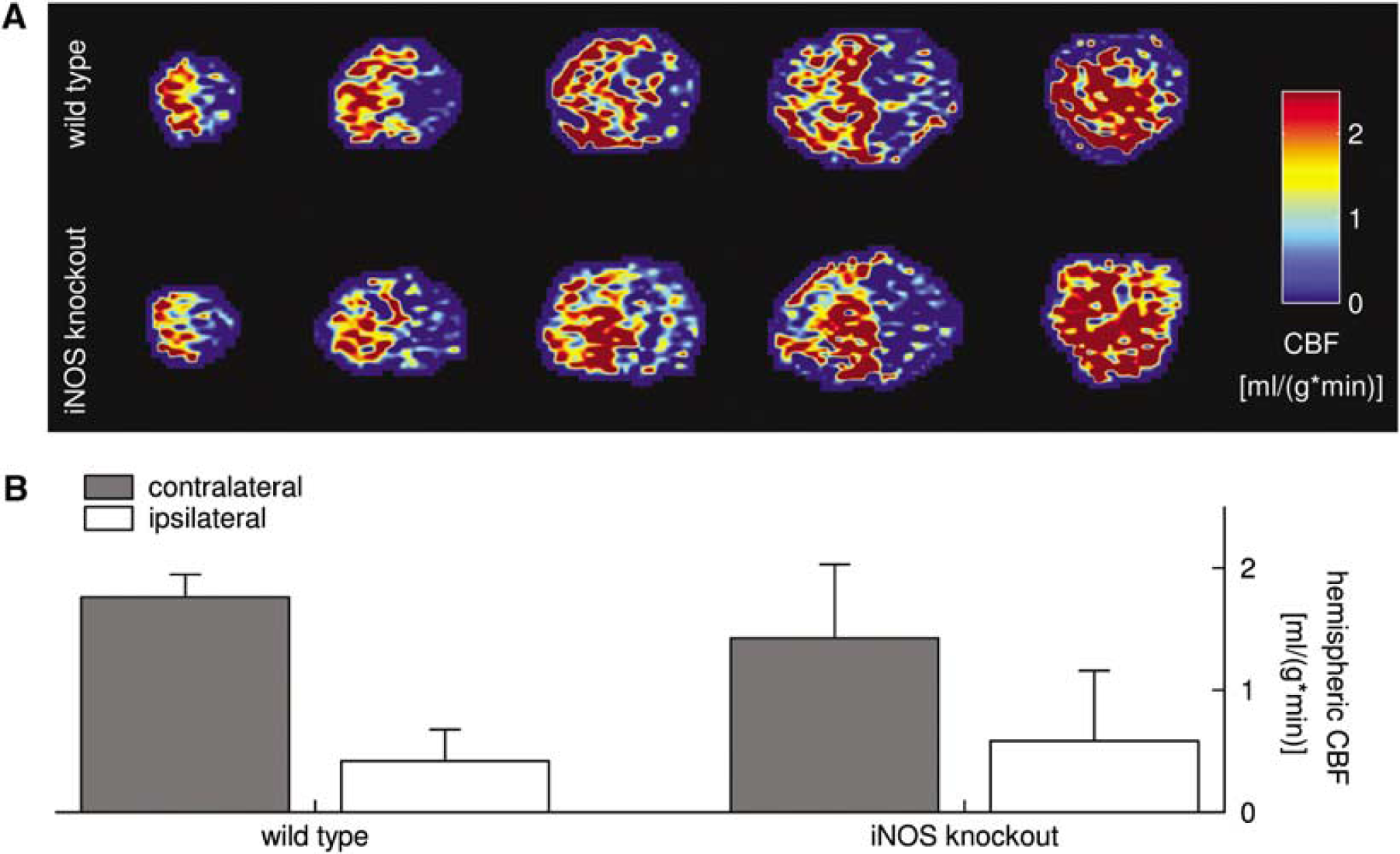
Cerebral blood flow measurement during MCAO with flow-sensitive alternating inversion recovery MRI. Exemplary CBF images of a wild-type mouse (
Discussion
Using a model of transient focal cerebral ischemia in mice, we found modest differential induction of exons 2 to 27 of iNOS mRNA in the ischemic brain, which was not accompanied by an expression of the iNOS protein. Inducible nitric oxide synthase immunoreactivity was undetectable in leukocytes invading the ischemic tissue or in resident glial cells up to 14 days after cerebral ischemia. Moreover, iNOS-deficient mice were not protected from transient ischemia compared with their wild-type controls.
Differential Expression of Exons 2 to 27 of Inducible Nitric Oxide Synthase mRNA after Transient Focal Cerebral Ischemia
We determined the expression of iNOS mRNA using five pairs of iNOS-specific PCR primers. Interestingly, distinct portions of the iNOS gene displayed a differential pattern of induction after MCAO. Exons 2 and 3 of iNOS mRNA containing the translational start signal appeared to be induced earlier (16-fold induction at 24 h after MCAO) than exons 6 to 8, 12 to 14, 21 to 22, and 26 to 27 of iNOS mRNA, which were only elevated 3- to 4-fold at day 3 after MCAO. The data support and extend a previous study pertaining to the expression of iNOS mRNA in infarct areas in a model of photochemically induced permanent ischemia in mice (Schroeter et al, 2003). The authors reported a delayed and slight increase in iNOS mRNA with an only 3- to 4-fold increase after 3 days using primers corresponding to sequences of exon 18. Iadecola et al (1997) described a strong transcriptional activation of the iNOS gene starting between 24 and 48 h and peaking at 96 h in a mouse model of permanent ischemia using a primer pair spanning exons 3 to 7.
The differential induction profiles of certain portions of the iNOS mRNA would be compatible with the presence of alternatively spliced variants. Published data on the induction of iNOS mRNA, iNOS protein, or enzyme activity in different experimental models of stroke models vary substantially (Iadecola et al, 1997; Loihl et al, 1999; Cash et al, 2001; Lerouet et al, 2005). Bearing in mind our findings of the differential expression of individual exons of the iNOS gene after ischemic injury, splice variants of iNOS may account for these discrepancies. For example, the use of only one pair of iNOS-specific primers might result in moderate, low, or even no expression of iNOS mRNA depending on the sequences of the primers used. Alternatively spliced iNOS isoforms could be translated into proteins with different regulatory properties resulting in varying cell viability (Tiscornia et al, 2004). It is also possible that splice variants may modulate NO synthesis by regulating dimer formation (Lee et al, 1995; Eissa et al, 1998) or may switch off iNOS function as a result of the lack of an open reading frame, and alternative forms may exhibit entirely new functions unrelated to NO synthesis.
Other groups have reported the existence of alternative splicing of human iNOS mRNA (Eissa et al, 1996, 1998; Tiscornia et al, 2004). Unfortunately, expression levels of iNOS mRNA after transient focal cerebral ischemia in mice were too low for confirmation of alternative splicing by Northern blot analysis, even when using purified mRNA from the ischemic tissue. Comparison of the amounts of iNOS mRNA between LPS-stimulated RAW cells and postischemic brain suggests that 10 to 100 times higher concentrations of iNOS mRNA would be needed in the ischemic brain for detection by Northern blot analysis (H Prüss et al, unpublished observations).
Absence of Inducible Nitric Oxide Synthase Protein Expression after Transient Focal Cerebral Ischemia in Mice
Hypothesizing that the iNOS activity of invading leukocytes could play a role in the progression of ischemic damage, we selectively inactivated iNOS either in the brain or in peripheral blood cells. To this end, we generated chimeric mice by the transplantion of GFP-expressing BM cells into myeloablated recipient mice to reliably differentiate the contribution of resident cells and blood-borne leukocytes in the development of ischemic brain damage. Because activated resident microglia and blood-borne monocytes/macrophages cannot be distinguished immunohistochemically, only genetic labeling of hematopoietic cells ultimately allows to determine the origin of these cells. We generated four groups of BM chimeric mice by the transplantation of BM cells from either iNOS knockout mice or wild-type mice into myeloablated wild-type or iNOS knockout recipients. Surprisingly, the infarct sizes did not differ between the four groups of animals, leading us to question the importance of infiltrating cells for iNOS expression and delayed tissue damage in this model of cerebral ischemia. Our data are in line with earlier investigations, which showed that iNOS activity remained unchanged when leukocyte infiltration into the ischemic brain was reduced by more than 60% in a rat model of experimental neutropenia and severe cerebral ischemia (Beray-Berthat et al, 2003).
Furthermore, using three different polyclonal rabbit antibodies against different epitopes of the iNOS protein, we were unable to detect any iNOS immunoreactivity in blood-borne leukocytes or in resident brain cells after MCAO. To validate our staining protocol, we performed extensive Western blotting and immunocytochemical characterization of all three antibodies. Whereas two antibodies (Upstate and Transduction Laboratories) distinguished several bands in Western blot analyses of brain homogenates (H Prüss et al, unpublished observations; Tiscornia et al, 2004), only the Chemicon antibody detected a single band of appropriate size. It is unclear whether these additional bands correspond to spliced iNOS forms, degradation products, or proteins entirely unrelated to iNOS. Some groups have reported iNOS protein expression after murine stroke in cells invading the infarct area and in some astrocytes at later times after ischemia (Loihl et al, 1999; Iadecola et al, 1997). In contrast, others only found very low levels of iNOS immunoreactivity in the ischemic mouse brain (Schroeter et al, 2003). It is possible that these divergent findings are attributable to the different ischemia models used, that is, rats versus mice, and permanent versus transient cerebral ischemia. In a model of permanent MCAO in rats and mice, iNOS protein expression was mainly detected in neutrophils infiltrating the ischemic brain (Iadecola et al, 1995, 1997), whereas blood vessels were the predominant source of iNOS expression in rats subjected to transient MCAO, and only some neutrophils in the ischemic core were found to be iNOS-immunoreactive (Iadecola et al, 1996).
Inducible Nitric Oxide Synthase Does Not Mediate Brain Damage after Transient Focal Cerebral Ischemia
Since the absence of iNOS-immunoreactive cells after transient MCAO begged the question of the functional relevance of iNOS expression after transient focal cerebral ischemia in mice, we compared infarct volumes after transient MCAO in three different strains of iNOS knockout mice and their wild-type controls. The first strain was originally generated by Laubach et al (1995), in which exons 12 and 13 of the iNOS gene are replaced with the neomycin resistance gene. The mice were generated on a mixed genetic background of C57BL/6 and SV129 strains, and backcrossed onto C57BL/6 for more than 10 generations. In the second strain of iNOS-deficient mice, the promoter region and exons 1 to 4 were deleted in B6/129-derived animals (MacMicking et al, 1995). To estimate the contribution of mouse strain to the outcome after cerebral ischemia, we determined infarct volumes in a third strain of iNOS knockout mice. To this end, we used the mice that were originally generated on a mixed genetic background of C57BL/6 and SV129 strains by Laubach et al (1995). In contrast to previous studies (Iadecola et al, 1997; Loihl et al, 1999; Zhu et al, 2003), neuroprotection from cerebral ischemia was detected in none of the iNOS-deficient strains. It should be noted that marked inflammation is well established in our model of transient murine MCAO (Storini et al, 2005; Huang et al, 2001) and evidenced by the dramatic recruitment of GFP+ blood-borne cells into the ischemic brain in this study, suggesting that iNOS does not appear to be the key mediator of inflammation-induced damage after transient focal cerebral ischemia in mice.
Genetic Background as a Confounding Factor
Interestingly, the iNOS knockout mice on the mixed genetic background (B6;129P-Nos2) had significantly smaller infarcts compared with iNOS-deficient mice maintained on a pure C57BL/6 background. Moreover, mortality was only significantly reduced in B6;129P-Nos2 mice compared with wild-type controls. These findings suggest that the genetic background is a major confounding factor in the interpretation of infarct volumes and mortality in iNOS-deficient animals. As mouse strains have a different susceptibility to infection after cerebral ischemia (Prass et al, 2003), we cannot exclude that the different mortality in our groups resulted from varying susceptibility to postischemic pneumonia. However, it should be noted that B6;129P-Nos2 mice were not protected from LPS-induced septic death in a previous study (Laubach et al, 1995). Moreover, it is well known that mouse strains have a different susceptibility to cerebral ischemia, partly due to interstrain variability in the gross anatomy of the vasculature. Generally, C57BL/6 mice exhibit enhanced susceptibility to global cerebral ischemic injury compared with SV129 mice (Fujii et al, 1997), which might further have affected infarct volumes and mortality in our experiments.
Other Possible Confounding Factors in Previous Studies
Calcium concentration: The activity of iNOS is thought to be independent of intracellular calcium concentrations due to tightly bound calmodulin. Nitric oxide synthase activity is therefore widely considered iNOS-specific when ‘calcium-independence’ is achieved by adding 0.5 to 0.6 mmol/L ethylene glycole-bis(β-aminoethyl ether)-N,N,N‘,N‘-tetraacetic acid (EGTA) (Galea et al, 1998; Iadecola et al, 1995; Yoshida et al, 1995). However, much higher concentrations of calcium-binding agents are required to measure iNOS after cerebral ischemia (Lerouet et al, 2005). Thus, iNOS activity may have been overestimated in some studies because calcium-dependent NOS may also have been active.
Aminoguanidine: Many authors refer to aminoguanidine as an iNOS-specific inhibitor, and thus conclude that its beneficial effects result from inhibition of NO production by iNOS. However, aminoguanidine displays only partial selectivity for iNOS, and it has other neuroprotective functions that are not related to the inhibition of NOS (Ou and Wolff, 1993; Cash et al, 2001; Alderton et al, 2001).
Summary
We found differential expression of exons 2 to 27 of iNOS mRNA after transient focal cerebral ischemia in mice, which may—at least in part—explain some of the inconsistencies in the literature regarding iNOS mRNA expression after cerebral ischemia. Using GFP BM chimeric mice, we found no evidence for iNOS protein expression in invading leukocytes or in resident brain cells after transient MCAO in mice. Infarct volumes did not vary between three different iNOS knockout strains and their corresponding wild-type controls. Thus, our findings question the role of iNOS as a key mediator of inflammation-induced damage after murine cerebral ischemia.
Footnotes
Acknowledgements
We thank Sabine Cho and Katharina Stohlmann for their excellent technical assistance.
Conflict of interest
All authors declare to have no competing financial interest.