
Abstract
Astrocytes are the most numerous cell type in the central nervous system. They provide structural, trophic, and metabolic support to neurons and modulate synaptic activity. Accordingly, impairment in these astrocyte functions during brain ischemia and other insults can critically influence neuron survival. Astrocyte functions that are known to influence neuronal survival include glutamate uptake, glutamate release, free radical scavenging, water transport, and the production of cytokines and nitric oxide. Long-term recovery after brain injury, through neurite outgrowth, synaptic plasticity, or neuron regeneration, is influenced by astrocyte surface molecule expression and trophic factor release. In addition, the death or survival of astrocytes themselves may affect the ultimate clinical outcome and rehabilitation through effects on neurogenesis and synaptic reorganization.
Keywords
Astrocytes are the most numerous nonneuronal cell type in the central nervous system and make up about 50% of human brain volume (Tower and Young, 1973). Astrocytes perform several functions that are essential for normal neuronal activity, including glutamate uptake, glutamate release, K+ and H+ buffering, and water transport. Accordingly, astrocyte function can critically influence neuronal survival during ischemia and other brain insults. Astrocytes also influence neurite outgrowth and other processes that contribute to brain recovery in the postinjury period. We review astrocyte—neuron interactions that influence brain injury and recovery, as well as factors that affect survival of astrocytes themselves. Whereas most of the literature in this area comes from studies of cerebral ischemia, many mechanisms operative in ischemia also apply to trauma, brain hemorrhage, hypoxia, hypoglycemia, and other insults.
Astrocytes are classically divided into three major types according to morphology and spatial organization: radial astrocytes surrounding ventricles, protoplasmic astrocytes in gray matter, and fibrous astrocytes located in white matter (Privat et al., 1995). Radial astrocytes are oriented perpendicular to the ventricular surfaces and have long, unbranched processes. Protoplasmic astrocytes have a bushy morphology with numerous highly branched short processes. Fibrous astrocytes display more stellate shapes, with smooth, long processes that are less branched. Astrocytes located at the border of gray and white matter regions can display a mixed morphology intermediate between fibrous and protoplasmic astrocytes. Glial fibrillary acidic protein (GFAP) and S-100β (a cytosolic Ca2+-binding protein) are commonly used as astrocyte markers (Ludwin et al., 1976; Eng et al., 2000). Glial fibrillary acidic protein is an astrocyte-specific intermediate filament protein whose expression is required for fibrous astrocyte normal function including maintenance of CNS white matter and blood—brain barrier integrity (Liedtke et al., 1996; Kakinuma et al., 1998). Of note, the majority of protoplasmic astrocytes do not express enough GFAP to stain positive with routine immunohistochemical methods (Walz, 2000), and consequently most astrocytes in gray matter are GFAP-negative with routine staining.
Astrocyte effects on brain injury are divided into those with immediate influence on cell survival and those with long-term or delayed effects that influence later recovery and function. These will be addressed in turn.
ASTROCYTE EFFECTS ON CELL SURVIVAL DURING BRAIN INJURY
Factors that influence astrocyte survival
Depending on severity and duration, brain injury may cause either infarction or selective neuronal death. “Infarction” is histologically characterized by pan-necrosis of glial and neuronal elements. By definition then, treatments or conditions that influence infarct size must influence the extent of astrocyte death. This fact is often overlooked in clinical or experimental studies of ischemia in which infarct size is used as the end point. For example, glutamate receptor antagonists (e.g., MK-801, dextromethorphan) have been shown in animal models to decrease infarct size (Choi, 1988). Astrocytes, however, unlike neurons, are not vulnerable to glutamate excitotoxicity. This implies that the action of these drugs on infarct size must be mediated by some additional or indirect mechanism.
Neurons cannot survive in the brain without close interaction with astrocytes. Consequently, in any region where astrocytes fail to survive there will necessarily be failure of neuronal survival and no possibility for synaptic remodeling. In regions where astrocytes do survive, there remains the possibility for synaptic remodeling and neurite outgrowth to compensate for loss of neurons. Astrocytes are generally more resistant to ischemia and other stressors than are neurons; there may be exceptions, however. Lukaszevicz et al. (2002) noted that protoplasmic astrocytes exhibited a more rapid loss of cell membrane integrity than fibrous astrocytes during ischemia, suggesting that certain astrocyte subtypes may be much more sensitive to ischemia and other insults. Several groups (Garcia et al., 1993; Martin et al., 1997; Liu et al., 1999) have reported that ischemia can induce loss of astrocyte marker proteins and evidence of astrocyte death before histologic evidence of neuronal death, although it is difficult to ascertain whether this reflects a true ischemic vulnerability of astrocyte subpopulations or simply differences in the rates at which the cells disintegrate after lethal insult. The mechanisms by which astrocytes may be more vulnerable than neurons have not been established, but studies (Giffard et al., 1990, 2000) using cortical astrocyte cultures suggest that astrocytes may be particularly vulnerable to acidosis (discussed further on).
Astrocyte vulnerability to hypoxia and acidosis.
Complete ischemia causes complete cessation of glucose and oxygen delivery, ATP depletion, and irreversible injury to all cell types, including astrocytes. Incomplete ischemia is the more common result of cerebral artery occlusion. Glucose delivery is partly maintained during incomplete ischemia, particularly at the margins of the vascular territory of an occluded vessel (the “ischemic penumbra”) (Obrenovitch, 1995). The residual blood flow to this region delivers more glucose than extractable oxygen, and glucose utilization is actually increased in the ischemic penumbra (Ginsberg et al., 1977; Nedergaard et al., 1986; Shiraishi et al., 1989), presumably reflecting a Pasteur response to hypoxia (Nedergaard and Diemer, 1988). Astrocytes are able to survive and function for extended periods under hypoxic conditions by glycolytic ATP production alone (Swanson, 1992). A consequence of anaerobic glucose utilization, however, is the production of lactic acid (Hochachka and Mommsen, 1983; Siesjö, 1984). The combination of hypoxia, anaerobic glycolysis, and lactic acid accumulation is characteristic of the ischemic penumbra (Obrenovitch, 1995). Although anaerobic glycolysis enables a period of continued astrocyte survival, the accumulation of lactic acid during hypoxia is likely to have deleterious effects on astrocyte metabolism. Astrocytes in culture are unable to maintain nonoxidative ATP production from glucose as pH falls below 6.6 (Swanson et al., 1997), and the failure to maintain ATP production is accompanied by irreversible astrocyte injury. Of note, hyperglycemia is found to promote infarction in most ischemic models, and this effect of hyperglycemia is likely due to increased lactic acidosis in the ischemic territory (Plum, 1983; Siesjö, 1984).
Astrocytes in culture are more sensitive than neurons to extended periods of mild acidosis (pH 6.8) (Giffard et al., 1990). The reason for difference may be attributable to the fact that astrocytes, but not neurons, express an electrogenic sodium bicarbonate cotransporter that mediates an inward sodium current during acidosis. This sensitivity to acidosis may become a significant cause of astrocyte death during prolonged periods of incomplete ischemia (Giffard et al., 2000). When coupled with the ion shifts that occur during severe ischemia, astrocytes become much more rapidly sensitive to acidosis. As shown by Bondarenko and Chesler (2001), astrocytes exposed for as short as 15 minutes to acidosis (pH 6.6) in conjunction with hypoxia and the strong ion shifts characteristic of cerebral ischemia are irreversibly injured on replacement in normal medium. These results indicate that astrocytes can be killed rapidly by changes in the extracellular microenvironment that occur in settings of traumatic and ischemic brain injury.
Astrocyte sensitivity to reactive oxygen species.
Astrocytes, like other cells, are vulnerable to the reactive oxygen species (ROS) generated by ischemia—reperfusion (Abe and Saito, 1996; Robb and Connor, 1998; Hollensworth et al., 2000; Ying et al., 2000; Chen et al., 2001a). Reactive oxygen species produced by transient oxygen—glucose deprivation have been shown to cause astrocyte mitochondrial membrane depolarization and mitochondrial membrane permeability transition (Reichert et al., 2001). Reactive oxygen species have also been shown to kill astrocyte by activating the poly(ADP-ribose) polymerase-1 cell death pathway (Ying and Swanson, 2000; Ying et al., 2001). Of note, astrocytes can be rescued after poly(ADP-ribose) polymerase-1 activation by supply of metabolic substrates such as pyruvate and glutamine, which do not require metabolism by glycolysis (Ying et al., 2002). Zinc-induced astrocyte death (Swanson and Sharp, 1992) has also been linked to ROS production (Ryu et al., 2002). The effects of ROS on astrocytes are potentiated by acidosis, possibly because of Fe2+ liberation under acidosis conditions (Ying et al., 1999).
Astrocyte effects on neuronal survival during acute brain injury
Glutamate uptake.
Glutamate toxicity is an important mechanism of neuronal death in ischemia, hypoglycemia, and trauma (Choi, 1988). Clearance of glutamate from the extracellular space is accomplished primarily by Na+-dependent transporters localized on astrocytes (reviewed in Anderson and Swanson [2000]). The astrocyte Na+-dependent glutamate transporters were originally cloned from rat brain and termed GLAST and GLT-1. Human homologues of these transporters are termed EAAT1 and EAAT2, respectively. Several studies support an essential role of astrocyte glutamate transporters in maintaining extracellular glutamate concentrations below toxic levels. In cortical cultures, neuronal vulnerability to glutamate is 100-fold greater in astrocyte-poor cultures than in cultures with abundant astrocytes (Rosenberg and Aizenman, 1989), showing that uptake by astrocytes can be a liming factor in glutamate neurotoxicity. Similarly, autoradiography of [3H]D-aspartate uptake in brain slices studies showed accumulation preferentially in astrocyte processes (McLennan, 1976). The dominant role of astrocyte transporters is further supported by studies using genetic downregulation of transporter subtypes. Antisense “knockdown” of GLAST or GLT-1, but not the neuronal subtype EAAC1, produces excitotoxic neurodegeneration and increased susceptibility to seizures and injury (Rothstein et al., 1996; Tanaka et al., 1997; Watase et al., 1998). Similarly, genetic downregulation of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, was shown to exacerbate ischemic neuronal damage in rat brain (Rao et al., 2001), thereby establishing the importance of astrocyte glutamate uptake for neuronal survival during ischemia.
Glutamate uptake requires movement against a steep concentration gradient; extracellular glutamate is normally less than 10 micromolar, whereas intracellular glutamate concentrations range from 1 to 10 millimolar in neurons and glia (Erecinska and Silver, 1990). This gradient is overcome by coupling the inward movement of each glutamate ion to the inward movement of 3 Na+ ions and 1 H+ ion and the outward movement of 1 K+ (Fig. 1). The energy expenditure required to clear glutamate from the extracellular space has been estimated to account for a large fraction of total brain ATP turnover (Sibson et al., 1998). Accordingly, one potential advantage of glutamate uptake by astrocytes is that this shifts this large energy expenditure away from neurons. Given the high energetic cost of glutamate uptake, it is not surprising that complete ischemia leads to abrupt cessation of glutamate uptake and massive increase in extracellular glutamate concentrations (Benveniste et al., 1984; Choi, 1988; Swanson et al., 1994). During incomplete ischemia, however, continued supply of glucose can fuel continued ATP production by glycolysis, even in the absence of oxygen. This glycolytic ATP production appears sufficient to fuel glutamate uptake in astrocyte cultures and in vivo (Hakim, 1987; Swanson, 1992; Swanson et al., 1994). As noted previously, however, pH is a significant factor determining the ability of astrocytes to maintain ATP levels in the absence of oxygen delivery, and the ability of astrocytes to maintain glutamate uptake during hypoxia is markedly impaired by acidosis over the range created by ischemia (Swanson et al., 1995).
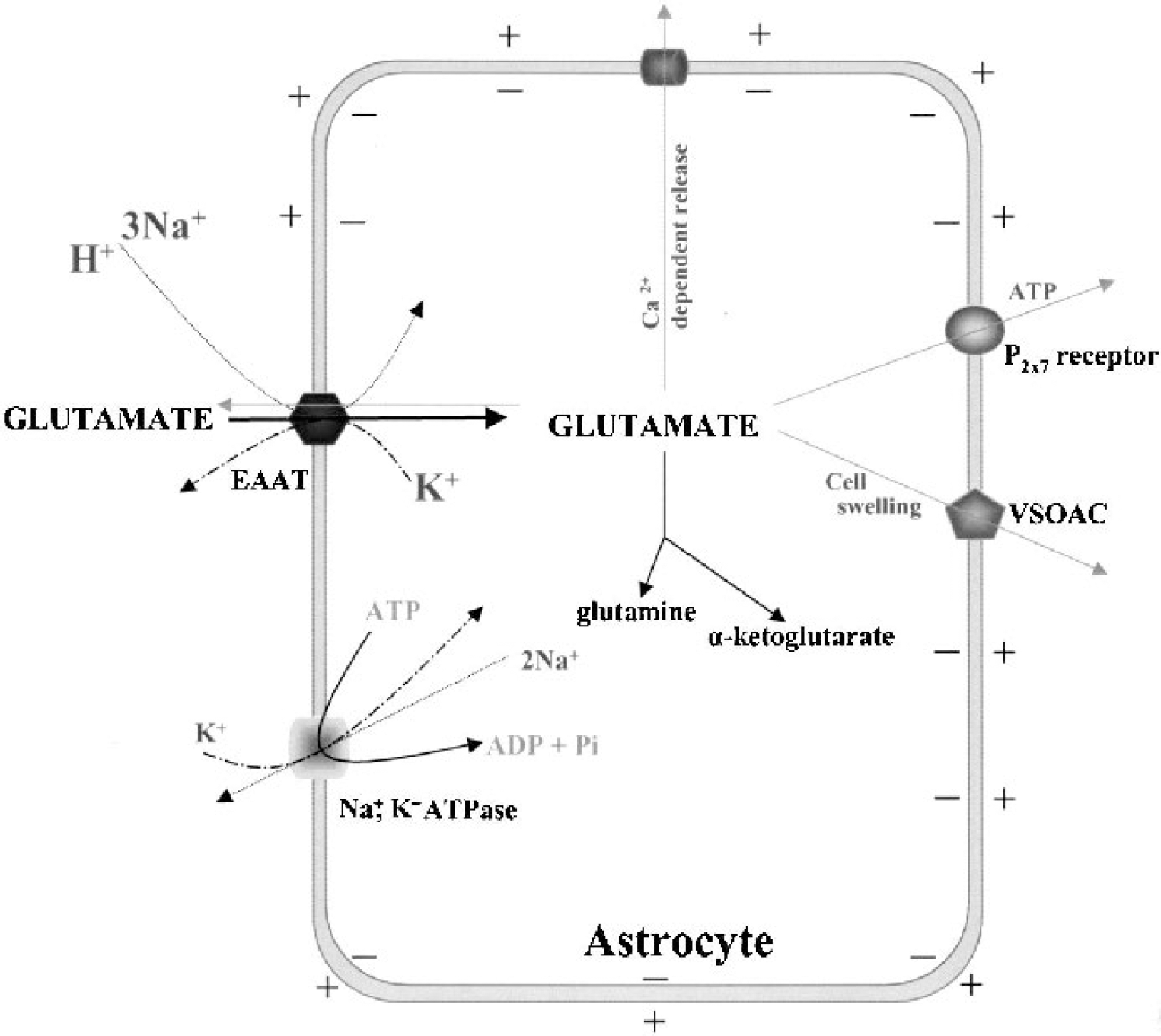
Schematic representation of glutamate uptake and release in astrocytes. Uptake of glutamate plus H+ is driven by the coupled transport of Na+ and K+ down their respective concentration gradients. The cell membrane potential also contributes to the uptake driving force because there is a net inward movement of positive charge with each glutamate transported. The membrane potential and transmembrane Na+, K+, and H+ gradients are maintained by Na+/K+ ATPase. Transporter stoichiometry may differ among the different transporter subtypes. Glutamate release can occur by “reverse” action of the transporters in conditions such as ischemia that cause ATP depletion. Glutamate can also be released by a Ca2+-dependent mechanism in response to signaling molecules such as prostaglandin E2 (PGE2) and bradykinin. Activation of P2X7 receptor channels by ATP and activation of volume-sensitive organic anion channels (VSOAC) by cell swelling allow passive glutamate release due to the large intracellular—extracellular glutamate concentration gradient. EEAT, excitatory amino acid transporters; Pi, phosphate.
Glutamate release by astrocytes.
Glutamate transporters, like other transporters, are capable of substrate movement in both the inward and outward directions. Glutamate uptake reversal was first demonstrated by Szatkowski et al. (1990), using patch-clamped Müller glia. In the setting of ATP depletion, as occurs during severe ischemia, the ATP-supported membrane gradients collapse. This results not only in cessation of uptake, but also efflux of glutamate via uptake reversal (Fig. 1). ATP depletion has been shown to cause uptake reversal in primary astrocyte cultures (Longuemare and Swanson, 1995), retina, (Zeevalk et al., 1998), spinal cord and brain slice preparations (Li et al., 1999), and in the intact brain (Seki et al., 1999). These studies suggest that the rise in extracellular glutamate in brain that occurs during energy failure may be due not only to failure of astrocyte glutamate uptake, but also to glutamate efflux from astrocytes via reversal of the astrocyte glutamate transporters. Glutamate efflux from neurons is also likely to occur during ATP depletion, and this may be a quantitatively greater than release from astrocytes in many brain regions (Rossi et al., 2000).
Glutamate efflux from astrocytes can also occur in response to specific signaling pathways (Fig. 1). Astrocytes exhibit Ca2+-dependent glutamate release in response to bradykinin (Jeftinija et al., 1996) and prostaglandin E2 (Bezzi et al., 1998) in a manner that is sensitive to botulinum B neurotoxin. An apparently distinct mechanism of glutamate release can be induced by extracellular ATP, as stimulation of the glial P2X7 receptor/channel causes significant Ca2+-independent glutamate release from mouse astrocytes (Duan et al., 2003). In addition, astrocyte swelling induced by elevated extracellular potassium or other factors can induce glutamate efflux via volume sensitive anion channels (Kimelberg et al., 1990; Rutledge et al., 1998; Longuemare et al., 1999).
Astrocyte release of metabolic intermediates.
Glutamine released by astrocytes is the major precursor for neurotransmitter glutamate synthesis (Bradford et al., 1978; Waniewski and Martin, 1986). Glutamate released at neuronal synapses is taken up by surrounding astrocytes (McLennan, 1976; Rothstein et al., 1996) and converted by glutamine synthetase to glutamine. Glutamine is in turn released from astrocytes for uptake by neurons (Broer and Brookes, 2001; Chaudhry et al., 2002), where it is metabolized back to glutamate and packaged into synaptic vesicles (Fig. 2). Pharmacological inhibition of glutamine synthetase reduces brain glutamine levels, reduces K+-evoked glutamate release (Paulsen and Fonnum, 1989), and has been reported to reduce infarct size in a focal ischemia model of stroke (Swanson et al., 1990). The cycle of glutamate carbon through neurons and astrocytes is supplemented by a similar flux of branched chain amino acids, in which transamination of leucine and α-ketoglutarate in astrocytes produces glutamate and α-ketoisocaproic acid (Yudkoff et al., 1996). α-Ketoisocaproic acid is released to the extracellular fluid and may be taken up by neurons as a substrate for the reverse reaction (Fig. 2).
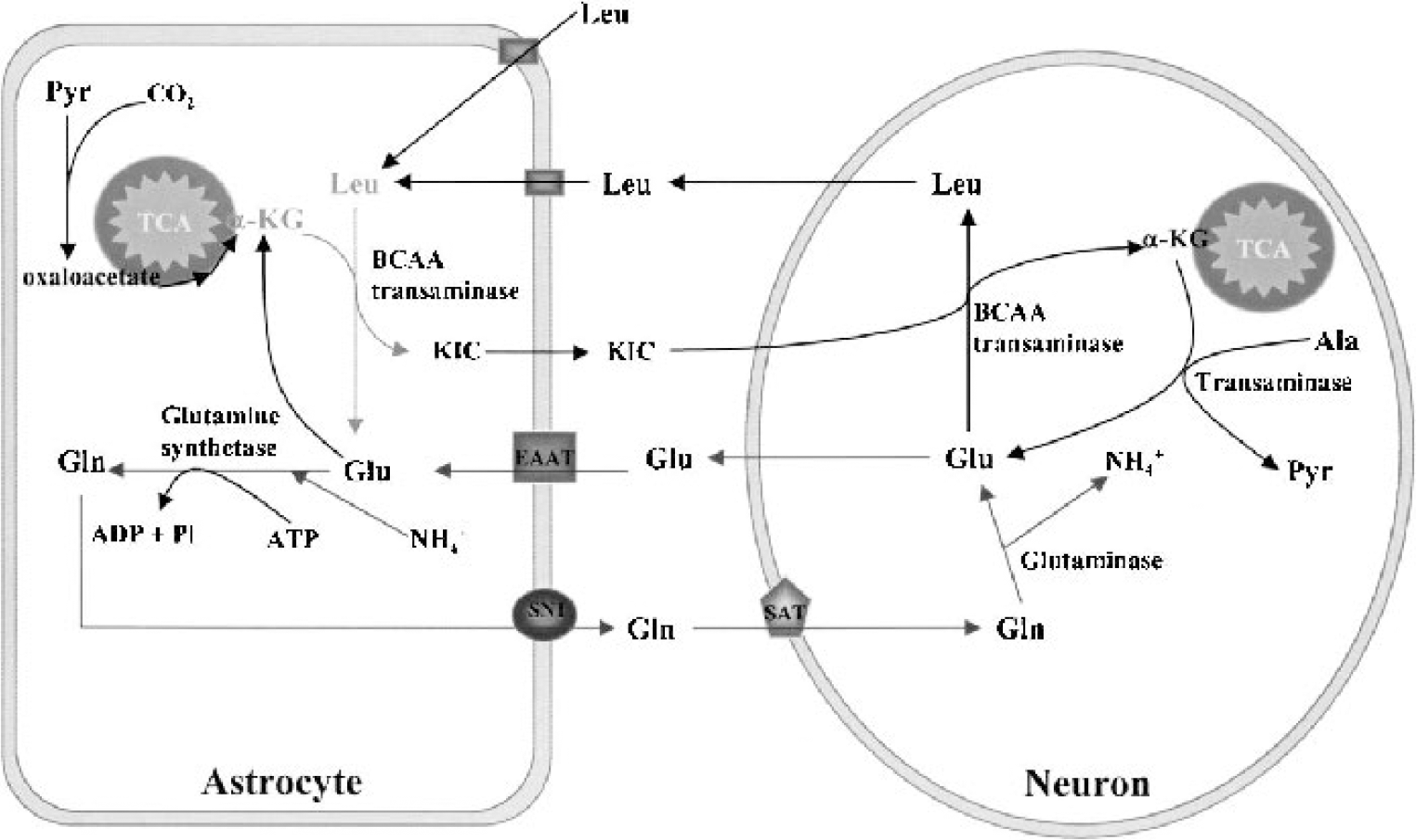
Astrocytes support neuronal glutamate metabolism. Glutamate (Glu) released during neurotransmission is taken up primarily by neighboring astrocytes through excitatory amino acid transporters (EAAT). A fraction of astrocyte Glu is converted to glutamine (Gln) by glutamine synthetase, which is abundant in astrocytes and absent from neurons. Gln is released from astrocytes via system N transporters (SN1) and taken up by neurons through sodium-coupled amino acid transporters (SAT). Gln is deaminated to Glu by mitochondrial glutaminase in neurons. Neuronal Glu is also formed from α-ketoglutarate (α-KG), which is derived in part through a second astrocyte—neuron shuttle system. Astrocytes take up leucine (Leu) and other branchedchain amino acids through a Na+-independent neutral amino acid transporter. The amino group of Leu is transferred to α-KG by branched-chain amino acid (BCAA) transaminase to yield Glu and α-ketoisocaproate (KIC). KIC can then be transferred to neurons for formation of α-KG by the reverse reaction. Pyr, pyruvate; Ala, alanine; Pi, phosphate.
Lactate, alanine, citrate, and α-ketoglutarate are released by astrocytes (Fig. 3) and can fuel neuronal metabolism (Shank and Campbell, 1984; Schurr et al., 1988; Sonnewald et al., 1991; Westergaard et al., 1993; Tsacopoulos and Magistretti, 1996). Neither the extent nor the significance of these metabolic interactions between astrocytes and neurons is sufficiently understood at present. Because these compounds are all metabolized oxidatively, it is unlikely that they could sustain neuronal function during ischemia. Evidence suggests, however, that these processes may be important for neuronal survival during severe hypoglycemia, where astrocyte glycogen stores can serve as a carbon source (Swanson and Choi, 1993; Wender et al., 2000).
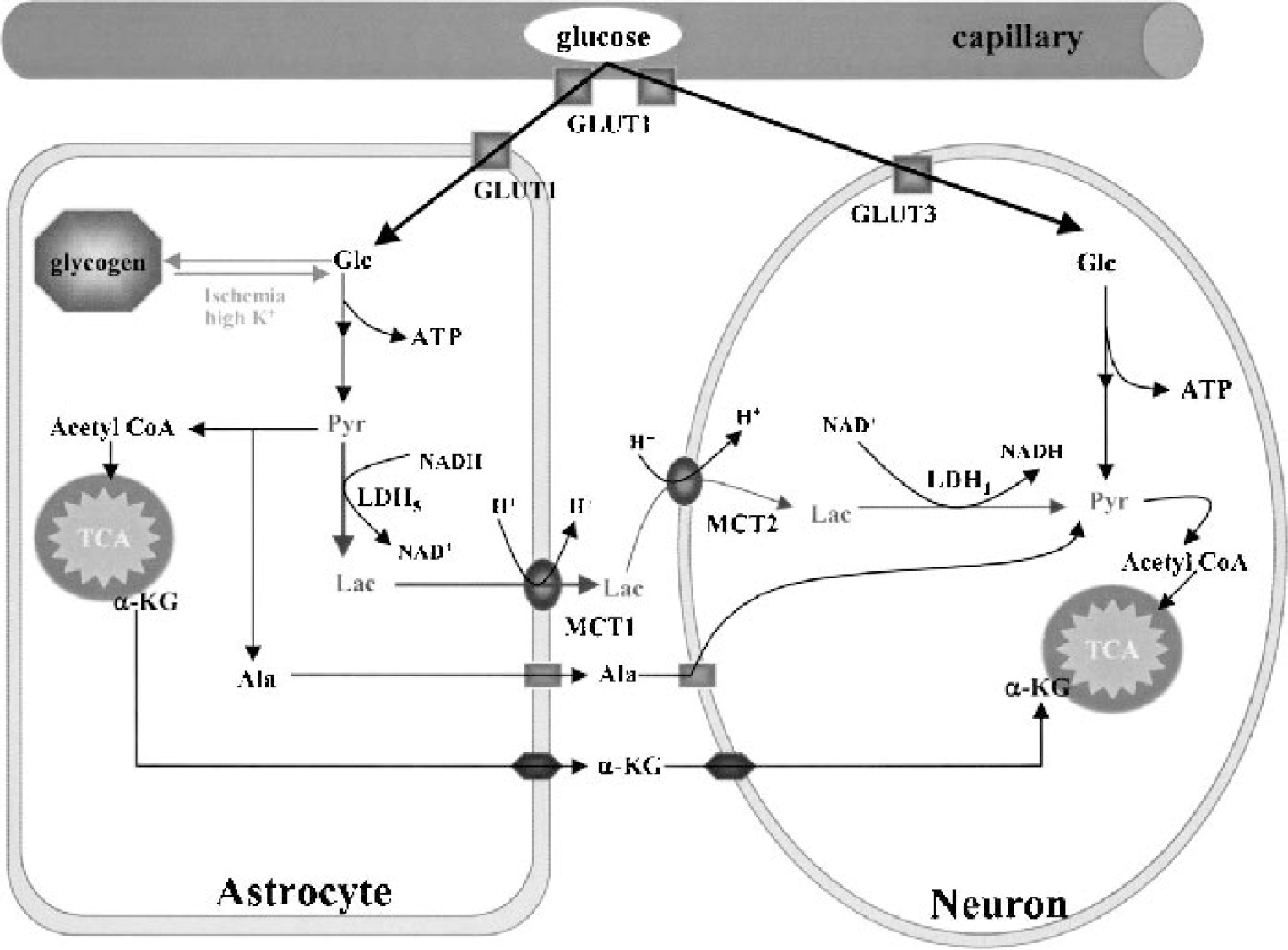
Astrocytes supply substrates for neuronal energy metabolism. Glucose transfer across endothelial cells occurs by facilitated transport through GLUT1. Glucose can then be directly taken up by neurons through GLUT3 or by astrocytes through GLUT1. Glycogen is the major energy reserve of the brain and is localized predominantly in astrocytes. Lactate (Lac) derived either from astrocyte glucose or glycogen may be shuttled to neurons via monocarboxylate transporters (MCTs). Astrocytes can also release metabolites such as alanine (Ala) or α-ketoglutarate (α-KG) for neuronal uptake. Glc, glucose; Acetyl CoA, acetyl coenzyme A; Pyr, pyruvate; LDH, lactate dehydrogenase.
Scavenging of oxygen free radicals by astrocytes.
Reactive oxygen species are a major cause of ischemic brain injury (Chan, 1996). Glutathione is the principal antioxidant in brain (Dringen, 2000), and brains depleted of glutathione are more sensitive to ischemic injury (Mizui et al., 1992). Astrocytes contain greater concentrations of glutathione and enzymes involved in glutathione metabolism than do neurons (Slivka et al., 1987; Yudkoff et al., 1990; Makar et al., 1994; Wilson, 1997). Similarly, glucose flux through the pentose phosphate pathway in astrocytes is twice that of neurons and increases three times as much as in neurons during H2O2 exposure (Ben-Yoseph et al., 1996). These factors indicate that astrocytes are more capable of scavenging ROS than neurons and suggest that oxidant-scavenging mechanisms in astrocytes may function to support neuronal survival. In support of this idea, neurons cultured in the presence of astrocytes are more resistant than neurons cultured alone to injury induced by nitric oxide, hydrogen peroxide, or superoxide (Desagher et al., 1996; Lucius and Sievers, 1996; Tanaka et al., 1999; Xu et al., 1999). Moreover, astrocytes depleted of glutathione show reduced ability to protect neurons from oxidant injury (Drukarch et al., 1997; McNaught and Jenner, 1999; Chen et al., 2001b). The effects of astrocyte glutathione may be mediated in part by astrocyte support of glutathione in neurons. Recent evidence suggests astrocytes contribute to neuronal glutathione content by an indirect route (Fig. 4). Glutathione is released by astrocytes and cleaved to the dipeptide CysGly (Dringen et al., 1999), which in turn is cleaved to free cysteine for uptake into neurons as a substrate for neuronal glutathione synthesis (Dringen et al., 2001).
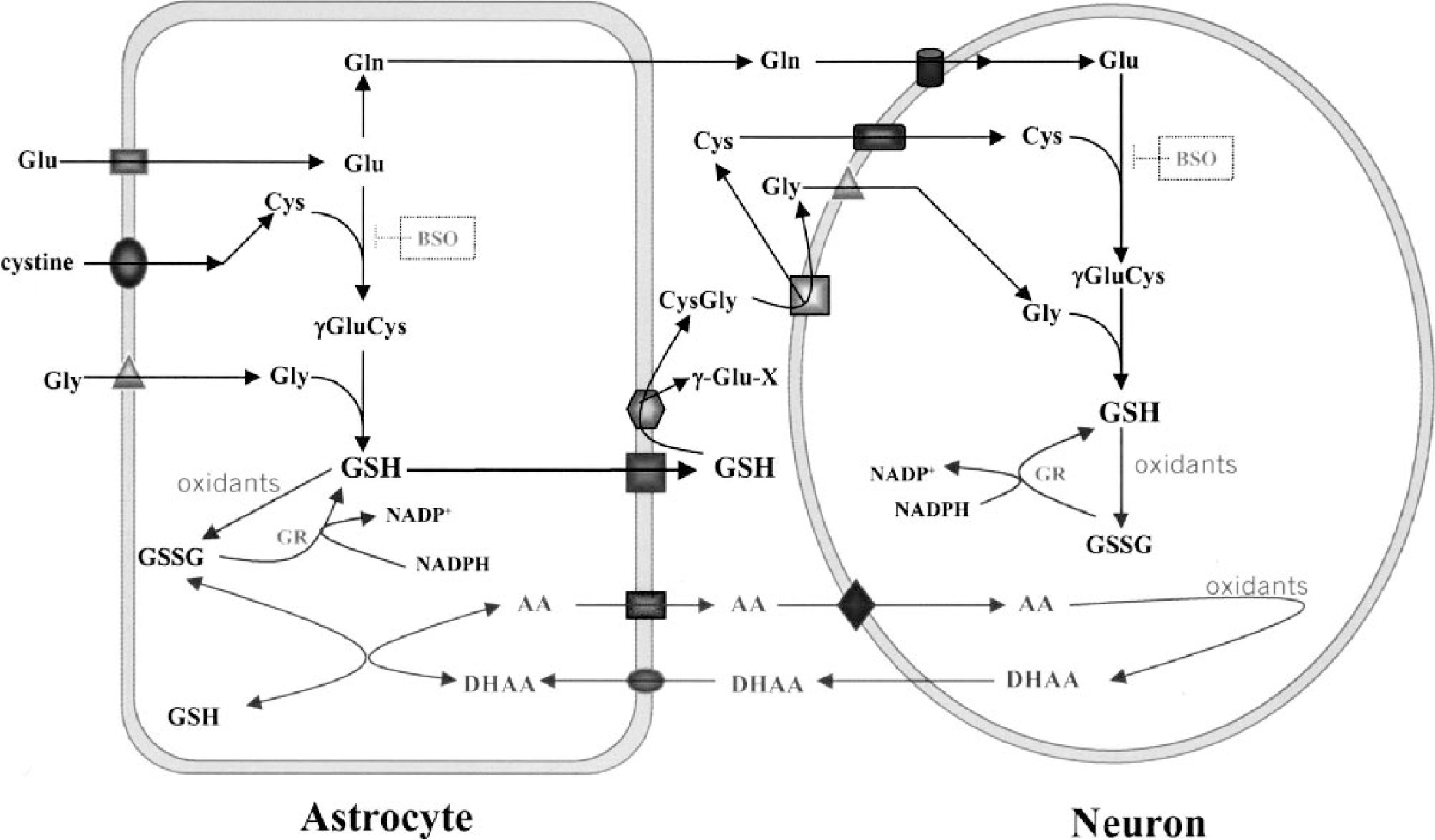
Redox coupling between astrocytes and neurons. The tripeptide γ-glutamyl-cysteinyl-glycine (GSH) is synthesized in two steps. γ-Glutamylcysteine (γGluCys) is catalyzed by γ-glutamylcysteine synthetase (GCS), which can be irreversibly inhibited by buthionine sulfoximine (BSO). Glycine is added to the dipeptide by GSH synthetase. Astrocytes maintain neuronal GSH by providing all three precursor amino acids. GSH released by astrocytes is hydrolyzed by the ectoenzyme γ-glutamyl transpeptidase (- GT) to form CysGly dipeptide. CysGly is further hydrolyzed by dipeptidase to yield glycine and cysteine, which are independently taken up for neuronal utilization. Cysteine availability is normally the rate-limiting step in neuronal GSH synthesis, whereas cystine is used by astrocytes through Na+-independent transporter X−c. GSH and ascorbate (AA) are oxidized to GSSG and dehydroascorbate (DHAA) during reduction of free radicals. GSSG is reduced back to GSH by the action of glutathione reductase (GR) in a reaction coupled to NADPH. The GSH/GSSG redox state is also coupled to the AA/DHAA redox state by both enzymatic and nonenzymatic processes. Astrocytes take up DHAA from extracellular space and convert it to AA using either GSH or NADPH as reducing agents. AA is released by astrocytes and taken up by neurons via Na+-dependent ascorbate transport (SVCT2). Gln, glutamine; Glu, glutamate; Cys, cysteine; Gly, glycine.
Like glutathione, ascorbate is an important antioxidant that is present in brain at millimolar concentrations (Rice, 2000). Ascorbate can directly react with oxidants and can also serve as a cofactor for reducing (recycling) oxidized glutathione and α-tocopherol. Evidence suggests an ascorbate cycle between neurons and astrocytes (Fig. 4). Neurons release oxidized ascorbate (dehydroascorbate) for uptake by astrocytes, which then convert it to ascorbate and in turn release ascorbate for neuronal uptake (Siushansian and Wilson, 1995; Siushansian et al., 1997; Wilson, 1997; Daskalopoulos et al., 2002). Dehydroascorbate passes across the blood—brain barrier readily and is converted to ascorbate, presumably in astrocytes. Treatment with dehydroascorbate has been shown to attenuate ischemic brain injury, suggesting this cycle is a significant aspect of astrocyte—neuron interactions during ischemia (Huang et al., 2001).
Astrocyte buffering of extracellular K+ and H+.
Elevations in extracellular K+ concentrations during ischemia may contribute to cell swelling, glutamate release, and anoxic depolarizations (spreading depression). Astrocytes exhibit both passive and active mechanisms for limiting elevations in extracellular K+ (Walz and Hertz, 1983). Passive movement of K+ from the extracellular space into astrocyte cytoplasm is augmented by electrical coupling between glial cells; local increases in extracellular K+ cause local increases in intracelluar astrocyte K+, which in turn result in a K+ current through the coupled cells that redistributes K+ throughout the glial syncytium (“spatial buffering”) (Kuffler et al., 1966; Karwoski et al., 1989). More marked elevations in extracellular K+ trigger active uptake of K+, via the action of astrocyte Na+/K+ ATPase (Walz and Hertz, 1982). Activity-induced elevations in extracellular K+ are significantly amplified under conditions where the astrocyte population is depleted (Sykova et al., 1992); the net effect of astrocyte K+ buffering during ischemia, however, is unknown and difficult to predict. Local attenuation of extracellular K+ increases may limit or slow neuronal depolarization and glutamate release. Conversely, astrocyte spatial buffering of K+ may contribute to the phenomenon of ischemic spreading depression (Nedergaard and Hansen, 1993), which has been shown to extend the area of infarction during focal cerebral ischemia (Mies et al., 1993; Busch et al., 1996).
The primary cause of acidosis during ischemia is the production of lactic acid in the course of the glycolytic production of ATP and its utilization (Rehncrona et al., 1981; Hochachka and Mommsen, 1983). Glucose utilization and lactic acid production are increased during incomplete ischemia (Ginsberg et al., 1977; Rehncrona et al., 1981; Nedergaard et al., 1986; Shiraishi et al., 1989). Although astrocytes can actively sequester H+ by a variety of mechanisms and thereby reduce acidification of the extracellular space (Deitmer and Rose, 1996), it is not known whether these active mechanisms can function during ischemia or whether they are capable of buffering the massive acidosis resulting from ischemia. A fundamental problem is that active transport processes under the hypoxic conditions of incomplete ischemia can be fueled only by anaerobic glycolysis, such that any effect of active H+ uptake on extracellular pH may be negated by a resultant net increase in H+ production. Kraig and Chesler (1990), however, reported that, during ischemia, the internal pH of some astrocytes may fall as much as an order of magnitude below the extracellular pH, and postulated that this could occur passively as a consequence of astrocyte membrane characteristics (but see also Siesjo et al., 1990; Ekholm et al., 1991). Although astrocyte H+ sequestration by either active or passive means could have beneficial effects on neuronal survival, the resultant astrocyte intracellular acidification would likely have detrimental effects on astrocyte function and survival.
Astrocyte gap junction coupling.
Astrocytes are extensively coupled to one another via gap junctions that permit intercellular exchange of ions and small molecules such as Ca2+ and inositol triphosphate (Dermietzel et al., 1991; Giaume and McCarthy, 1996). Astrocyte coupling efficiency is dynamic and modulated by a variety of neurotransmitters, cytokines, and other bioactive substances (Nagy and Li, 2000; Rouach et al., 2000). Although increased Ca2+, decreasing pH, and oxygen free radicals inhibit the coupling (Bolanos and Medina, 1996; Martinez and Saez, 2000), at least a subset of gap junctions remain open in viable astrocytes under ischemic conditions (Cotrina et al., 1998).
There are conflicting data regarding the effects of astrocyte gap junction coupling on ischemic injury. Inhibition of astrocyte gap junction communication has been reported to increase neuronal vulnerability to oxidative injury in culture (Blanc et al., 1998). Similarly, Siushansian et al. (2001) reported that mice with reduced expression of connexin 43 showed increased ischemic infarct size. The authors suggested that compromised gap junction coupling reduces astrocytes' ability to efficiently remove toxic concentrations of glutamate and K+. In contrast, studies by Lin et al. (1998) indicate that gap junction communication may amplify ischemic damage by providing a conduit for the propagation of proapoptotic death signals between dying and viable astrocytes. This provides an attractive explanation for the observation that infarct margins are sharply delineated in brain, though clearly the inconsistent findings in this area indicate that much more needs to be learned. An additional consideration is that some gap junction “hemichannels” open to the extracellular space rather than to coupled cells. Recent studies (Contreras et al., 2002) suggest that these hemichannels may contribute to cell death and be regulated differently than coupled gap junctions.
Astrocyte influences on the blood—brain barrier and brain edema.
Brain edema can contribute to injury after cerebral ischemia by impairing local or global cerebral blood flow and by physically disrupting long fiber tracts. Brain edema can be classified as vasogenic, due to trans-capillary movement of water and solutes across the blood—brain barrier, or as cytotoxic, due to osmotic swelling of cellular elements (Klatzo, 1967; Fishman, 1975). Astrocytes play a role in both of these processes. Astrocyte processes (end-feet) cover much of the abluminal capillary surface and thereby form part of the physical blood—brain barrier (Abbott et al., 1992). Astrocytes also contribute to function of the blood—brain barrier by inducing formation of tight intercellular junctions between capillary endothelial cells and by regulating the expression and function of several endothelial transporters (Dehouck et al., 1990; Sun and O'Donnell, 1996; Hayashi et al., 1997). After ischemia, vasogenic edema and blood—brain barrier disruption may occur by several mechanisms, including active solute transport, vesicular transport, opening of paracellular channels, and physical disruption of astrocyte—endothelial junctions (Westergaard et al., 1976; Petito, 1979; Abbott, 2000; del Zoppo and Hallenbeck, 2000). Digestion of blood—brain barrier matrix proteins by astrocyte matrix metalloprotease 2 and other matrix metalloproteases contributes to the physical disruption of the blood—brain barrier (Mun-Bryce and Rosenberg, 1998; Rosenberg et al., 1998, 2001).
Ultrastructural studies show that pericapillary astrocyte end-feet are the first cellular elements to swell during brain ischemia (Dodson et al., 1977). Astrocyte swelling can result from uptake (cotransport) of extracellular K+, Cl−, and Na+ with subsequent osmotic movement of water (Su et al., 2002), impaired extrusion of ions by failure of the membrane Na+/K+ ATPase, or from increased production of cytosolic solutes by breakdown of macromolecules. Recent studies (Venero et al., 2001) suggest that aquaporins (AQPs) are the primary route by which water moves in and out of astrocytes in response to these osmotic changes and that AQPs play an important role in astrocyte swelling and cerebral edema formation. Aquaporins are a family of membrane proteins that mediate rapid transmembrane transport of water (Venero et al., 2001; Agre et al., 2002). The direction of water movement through AQPs is osmotically driven. Ten AQPs have been cloned, each with distinct cellular location. Some AQPs such as AQP1 and AQP4 are only permeable to water, whereas others such as AQP9 are also permeable to small solutes such as lactate (Tsukaguchi et al., 1999). Aquaporin-4 is abundantly expressed in astrocyte pericapillary end-foot processes, suggesting a role in brain water balance (Nagelhus et al., 1998; Rash et al., 1998). Mice with genetic knockout of AQP4 show less astrocyte swelling, reduced brain edema, and better neurologic outcome after focal ischemia (Manley et al., 2000), supporting a causative role for astrocyte AQP function in these processes. There is, in addition, an upregulation of AQP4 and AQP9 on reactive astrocytes after ischemia (Vizuete et al., 1999; Badaut et al., 2001), although the functional significance of these changes remains to be established.
Astrocyte influences on secondary injury and recovery
Within a few hours of virtually any type of brain injury, surviving astrocytes in the affected region begin to exhibit hypertrophy and proliferation, termed reactive astrogliosis (Ridet et al., 1997). This response is fortified by the migration of microglia and macrophages to the damaged area. Reactive astrocytes increase the expression of their structural proteins, GFAP and vimentin (Eng et al., 2000). Many other proteins are also upregulated. Copper—zinc superoxide dismutase, glutathione peroxidase, and metallothionein are increased in reactive astrocytes after ischemia (Liu et al., 1993; Takizawa et al., 1994; Neal et al., 1996; Campagne et al., 2000), indicating an enhanced capacity to reduce ROS. Similarly, astrocytes express the inducible form of heme oxygenase in response to ischemia and other brain insults (Geddes et al., 1996; Takeda et al., 1996). Heme oxygenase is the first step of heme metabolism and may be important in preventing heme iron participation in metalcatalyzed free radical production, particularly after conditions such as trauma or hemorrhagic stroke that liberate hemoglobin into the brain parenchyma.
Astrocyte inflammatory response.
The reaction of astrocytes to ischemia and other brain insults is similar in some respects to the inflammatory response of peripheral tissues. This response may be adaptive in settings such as infection, but may contribute to delayed neuronal death in settings such as stroke. One salient aspect of this response is the expression of inducible nitric oxide synthase (iNOS) (Endoh et al., 1993). Astrocyte iNOS expression is detectable within several hours and is maximal by 2 to 3 days after ischemia (Iadecola et al., 1995; Nakashima et al., 1995). Nitric oxide is an ROS that can contribute to neuronal cell death by potentiating glutamate excitotoxicity (Hewett et al., 1994) and by several other mechanisms (Dawson and Dawson, 1998). Mice genetically deficient in iNOS exhibited smaller infarct size than control, wild-type mice (Iadecola et al., 1997), although this effect may also be due to reduced iNOS expression in microglia and invading macrophages.
Astrocytes stimulated by ischemic injury also produce many cytokines, including tumor necrosis factors (TNF-α, TNF-β), interleukins (IL1, IL6, IL10), and interferons (IFN-α, IFN-β) (Feuerstein et al., 1998; Dong and Benveniste, 2001). The net effect of individual cytokines can be difficult to establish because the effects of many cytokines are strongly influenced by one another and because most cytokines have pleiotropic and cell-type specific effects. For example, IL6 and TNF-α have been shown to promote demyelination, thrombosis, leukocyte infiltration, and blood—brain barrier disruption (Feuerstein et al., 1998; Dong and Benveniste, 2001). In contrast, IL6 has been shown to protect against ischemic and excitotoxic injury (Maeda et al., 1994; Ali et al., 2000), and hippocampal neurons treated with TNF-α are less vulnerable to substrate deprivation and excitotoxicity (Cheng et al., 1994). The specific contribution of astrocyte cytokine release to these processes in vivo remains to be established.
Gliosis and barriers to axonal regeneration.
Another evolutionarily conserved function of reactive astrogliosis after brain injury may be to isolate the lesion area. Unlike most other tissues, the brain contains very few fibroblasts, so this “walling off” function is performed instead by astrocytes. At the immediate vicinity of injury, reactive astrocytes interweave their processes to form a barrier termed anisomorphic gliosis. This glial scar can be an impediment to regenerating axons (Smith et al., 1986; McKeon et al., 1991; Bush et al., 1999). Increased GFAP expression is a hallmark of reactive astrocytes and this cytoskeletal protein contributes to a barrier effect of the glial scar for axonal extension. Astrocytes cultured from GFAP-knockout mice or treated with GFAP antisense DNA show enhanced expression of laminin and become more permissive to neurite growth (Lefrancois et al., 1997; Bush et al., 1999; Menet et al., 2000, 2001; Costa et al., 2002). Reactive astrocytes additionally synthesize collagens and sulfate proteoglycans, which also impede neurite growth (Smith-Thomas et al., 1994; Ridet et al., 1997; Fidler et al., 1999; Liesi and Kauppila, 2002). Reactive astrocytes may not, however, have uniformly negative influences on recovery from ischemia. Glial cells surrounding axons have a key role in determining CNS regenerative capacity (Sivron and Schwartz, 1995; Ridet et al., 1997). Reactive astrocytes produce matrix metalloproteases and their inhibitors, which are involved in remodeling of extracellular matrix components and influencing the scar architecture after a lesion. Reactive astrocytes also promote repair of the blood—brain barrier through production of extracellular matrix components (Kakinuma et al., 1998), and targeted ablation of reactive astrocytes after stab injury was shown to prevent blood—brain barrier repair (Bush et al., 1999). It should also be noted that there is evidence for considerable heterogeneity among reactive astrocytes (Ridet et al., 1997).
Astrocyte trophic effects on neurite outgrowth and neurogenesis.
Astrocytes release a variety of trophic factors under normal conditions, and these are likely to influence neuronal survival and plasticity after brain injury. These trophic factors include nerve growth factor, basic fibroblast growth factor, transforming growth factor-β, platelet-derived growth factor, brain-derived neurotrophic factor, ciliary neurotrophic factor, and others (Ridet et al., 1997). Reactive astrocytes increase the expression of several of these, notably nerve growth factor, basic fibroblast growth factor, brain-derived neurotrophic factor, and neuregulins, which can stimulate neurite outgrowth (Schwartz and Nishiyama, 1994; Strauss et al., 1994; Mocchetti and Wrathall, 1995; Tokita et al., 2001). Reactive astrocytes also overexpress neuropilin-1 and vascular endothelial growth factor, which act in concert to promote angiogenesis after cerebral ischemia (Zhang and Chopp, 2002). Although these findings suggest that astrocytes may play an important role in functional recovery after stroke, there has not been direct confirmation of this.
The demonstration of accelerated neurogenesis after cerebral ischemia has led to the proposal that newly formed neurons may contribute to functional recovery after stroke (Liu et al., 1998; Jiang et al., 2001; Jin et al., 2001). Factors regulating neurogenesis remain poorly understood, but the essential role of astrocytes in neuronal differentiation during development suggests astrocytes may be similarly important in regulating neurogenesis. In support of this idea, it has recently been shown that astrocytes induce neurogenesis from adult neural stem cells in culture (Song et al., 2002).
CONCLUSIONS
The degree to which astrocytes are functionally intertwined with other cell types makes it difficult, and perhaps artificial, to quantify the influence of specific astrocyte functions on brain injury and repair. Nevertheless, some processes are so dominated by astrocytes that they stand out as critical, astrocyte-specific determinants of outcome after brain injury. Glutamate homeostasis is probably the chief among these, given the special role that astrocytes play in glutamate uptake and metabolism and the capacity for glutamate to kill neurons when extracellular concentrations are elevated. It has proven more difficult to assess the effect of other astrocyte influences on brain injury, such as astrocyte iNOS expression and trophic factor release, because microglia and other cell types substantially contribute to the net effects of these processes. From a therapeutics standpoint, the key question is whether manipulation of these astrocyte processes can affect outcome after brain injury. An expanding and intriguing research direction is the study of astrocyte influences on neurite outgrowth and neurogenesis after brain injury. There is now strong evidence that astrocytes play a critical role in these processes, and future studies should soon clarify whether astrocytes can be manipulated to enhance brain recovery after injury.