
Abstract
Glutamate excitotoxicity is a primary contributor of ischemic neuronal death and other cellular components of the neurovascular unit. Several strategies have been developed against glutamate excitotoxicity, however none of them have not shown positive results in the clinical practice so far. Nowadays, the concept of blood/brain glutamate grabbing or scavenging is well recognized as a novel and attractive protective strategy to reduce the excitotoxic effect of excess extracellular glutamate that accumulates in the brain following an ischemic stroke. The main advantage of this novel therapeutic strategy is that it occurs in the blood circulation and therefore does not affect the normal brain neurophysiology, as it has been described for other drug treatments used against glutamate excitotoxicity. In this work we report all experimental data from the beginning of our studies, focused on stroke pathology, and we describe new findings about the potential application of this therapy. Future clinical trials will allow to know the real efficacy of this novel therapeutic strategy in stroke patients.
Introduction
Improvements in the management of stroke patients in primary care and hospitals, especially in developed countries, have contributed to the reduction in mortality rates observed in recent decades.1,2 However, the incidence of stroke has not followed the same trend with a continuous increase in developed countries and an especially worrying increasing trend in underdeveloped and developing countries. A recent analysis that includes countries with different levels of development showed that the incidence in 1990 was 250.55 per 100,000 people; in 2005 this rate increased to 255.79, and in 2010 it was 257.79. 2 Demographic changes, the rise of hypertension and obesity cases, and other comorbidities could explain the increase in stroke incidence. 1 In addition, the demographic change expected in Europe for the next 50 years favored by the current economic crisis and the decrease in immigration rates suggest that this situation will become worse.
Ischemic stroke is the result of the interruption of blood supply to the brain. From a physiological point of view, immediately after stroke, the ischemic brain region can be separated in two regions: the core zone and the penumbra. 3 The core zone, which is an area of severe ischemia (blood flow below 10–25%), exhibits a loss of oxygen and glucose that results in the rapid depletion of energy stores. If the ischemia persists for a long time, this compromised area can result in the necrosis of neurons and also of supporting cellular elements (glial cells). 4 The penumbra region is defined as the hypoperfused tissue surrounding the ischemic core in which blood flow is too low to maintain electric activity but sufficient to preserve ion channels, allowing this area to remain viable for several hours. This phenomenon results because the penumbral zone is supplied with blood by collateral arteries anastomosing with branches of the occluded vascular tree. However, even cells in this region will die if reperfusion is not established during the early hours because collateral circulation is inadequate to maintain the neuronal demand for oxygen and glucose indefinitely. Therefore, the penumbra is where pharmacologic interventions are most likely to be effective.
The prognosis of ischemic stroke is well known to depend on the age of patient, comorbidity, the intensity of neurological damage, the cerebral region affected, the thrombus size, the state of collateral circulation, the elapsed time from symptom onset to recanalization, the degree of restoration of blood flow, and the possible occurrence of symptomatic brain hemorrhage. 5 Currently, the control of systemic parameters, such as body temperature, blood pressure or glycemia, has considerably improved the outcome of stroke patients both short and long term; however, in the absence of a protective therapy, an early artery reperfusion remains the primary goal of treatment in ischemic stroke,6–8 and intravenous application of tissue plasminogen activator (t-PA) within the first 4.5 h after ischemic onset is the only approved treatment thus far. This therapeutic recommendation is supported by two clinical trials,9,10 three meta-analyses,11–13 and other observational studies.14,15 Other novel methods to achieve successful recanalization of occluded arteries, such as mechanical recanalization, are being used; however, they are not completely established in clinical practice.7,16
Although the benefit of t-PA is well established, its use in developed countries is less than 5% of the patients with ischemic stroke and less than 2% in case of developing countries, mainly due to the short therapeutic window. Moreover, the selection of those stroke patients who may benefit from this therapy requires the use of complex and expensive diagnostic technologies that are not available in all hospitals.17–19
Because of the progressive increase in stroke incidence, the high morbidity and the limited therapeutic tools against stroke, seeking new alternatives that can be applied more universally and with less technological requirements is in high demand. Investing efforts to develop therapies that can be used only in a small number of centers of highly developed countries may be an objective of pharmaceutical companies but does not meet the needs of the medical community.
Glutamate excitotoxicity on the neurovascular unit
Glutamate is one of the most important neurotransmitters in the adult central nervous system (CNS). Its neurotoxic effect was initially demonstrated in the mouse retina, 20 and its critical role in the cortical spreading depression phenomenon was described in rabbit brain. 21 Subsequent studies have provided unequivocal evidence that glutamate-mediated excitotoxicity is a primary contributor to ischemic neuronal death. 22
Glutamate excitotoxicity appears when the neurotransmitter’s homeostatic balance is disrupted and levels become elevated in the extracellular fluid.
23
Whereas extracellular glutamate levels are increased during neuronal damage, in normal physiological conditions, high concentrations of neurotransmitters in the brain are stored intracellularly. The concentration of extracellular glutamate is tightly regulated to maintain physiological concentrations through sodium-dependent transporters (also known as excitatory amino acid transporters: EAATs).
23
Reuptake of glutamate from synaptic junctions after neuron excitation normally involves the participation of EAATs located on astrocytes, which bind and remove the neurotransmitter for processing and recycling.
24
Alternatively, when extracellular concentrations become elevated, sodium-dependent transport located on the antiluminal surface of brain capillary endothelial cells is able to transfer glutamate from the extracellular space.
25
When glutamate accumulates in the endothelial cells to a concentration that exceeds plasma levels it is moved via facilitated diffusion through the luminal side into the blood stream (Figure 1). In this regard, the endothelial regulation of CNS glutamate concentration can occur despite unfavorable concentration gradients from CNS to plasma.26,27
Homeostasis of glutamate in neurovascular unit. Glutamate is maintained at approximately 1 μM in the brain interstitial and cerebrospinal fluid (CSF), this concentration is more than 100 higher inside of brain cells (∼10 mM) and synaptic vesicles (∼100 mM). This is due to the presence of Na+-dependent excitatory amino-acid transporters (EAATs) not only on neuronal (EAAT1) and astroglial cells (EAAT2 and EAAT3) but also on the brain vasculature (EAAT1, EAAT2, EAAT3). In the astrocyte, glutamate enters in the tricarboxylic acid (TCA) cycle or is converted to glutamine by the enzyme glutamine synthetase. Glutamine is then released to the presynaptic neuron where it is converted to glutamate and packaged into vesicles for further release. It seems evident that uptake of extracellular glutamate into endothelial cells (EC) via EAATs is also an important step in the glutamate homeostasis. When the endothelial glutamate concentration becomes higher than the blood glutamate concentration, glutamate is transported into the blood by means XG− transporters that exist only on the luminal membrane in a position that facilitates blood excretion of glutamate from the brain. ECs may also utilize glutamate as an energy substrate. EC may catalyze the conversion of glutamate to α-ketoglutarate (α-KG) and enter the TCA cycle in the mitochondria to form pyruvate, which may then be converted to lactate in the cytosol and transported through monocarboxylate transporter 1 (MCT-1) in the luminal membrane to the blood. Adapted from Cederberg et al.
28
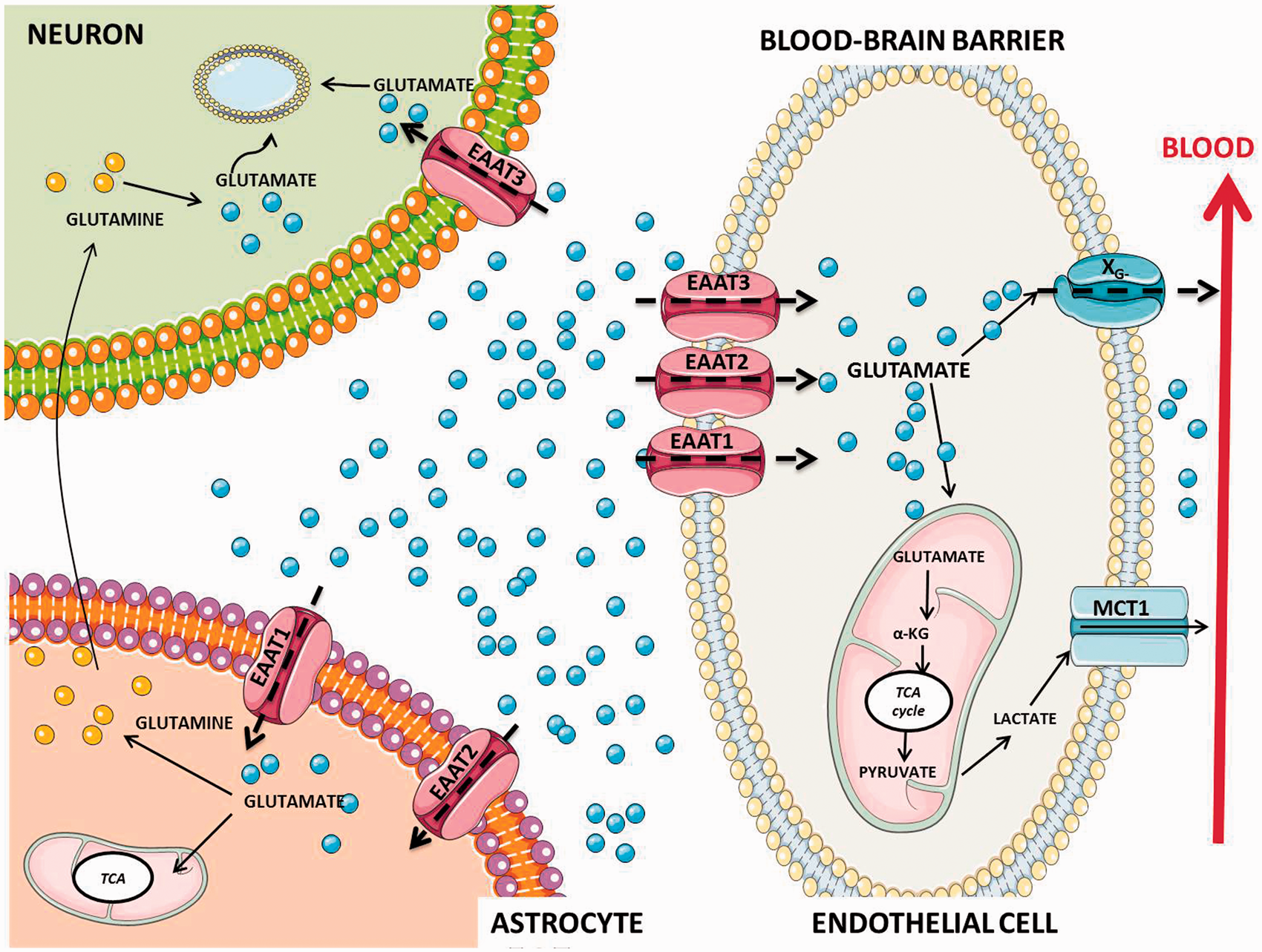
Once the glutamate is removed from the synaptic cleft for EAATs this neurotransmitter is metabolized in astrocytes via glutamine synthetase to glutamine or via glutamate dehydrogenase to α-ketoglutarate that participates in the citric acid cycle (TCA). Glutamine formed is released to the extracellular space and then removed again from the extracellular space into neurons. After that, neurons convert glutamine back to glutamate by means of the enzyme glutaminase (a metabolic process known as glutamate–glutamine cycle), and glutamate is subsequently accumulated and used in a new synapses. 28 In endothelial cells, glutamate can diffuse to the endothelial barrier into the blood stream and can be also metabolized into α-ketoglutarate, through the TCA (Figure 1).
The failure of nutrient supply after ischemia causes a neuronal depolarization leading to a massive release of glutamate to the extracellular space. In addition, due to glutamate uptake is a high energy-dependent process, this restriction of energy causes a drastic disruption of the glutamate transporters enhancing the excitotoxic effect to trigger the death of neurons. Based on the efficient capacities of astrocytes and endothelial cells to remove and metabolize glutamate and because they are less susceptible than neurons to ischemic conditions, it could be postulated that both cellular components of the neurovascular unit should be able to protect the neurons against the glutamate increase. However the experimental and clinical evidences show that any of the endogenous homeostatic mechanism are not able to stop the “tsumani” glutamate effect after ischemic stroke.
Excitotoxic cascades are not only a neuronal phenomenon but also comprise all cellular components of the neurovascular unit.
29
Thus, it has been well described that oligodendroglial lineage cells, which are one of the main cell types in cerebral white matter and important in maintaining normal neuronal function, express N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kinate glutamate receptors which make them sensitive to glutamate increase and its alteration by glutamate contributes to the neuronal damage and dysfunction.
29
Pericytes, other important cellular component of the neurovascular unit that expresses metabotropic glutamate receptors and respond to glutamate in vitro experiments, can be potentially affected by glutamate excitoxocity.
30
Finally, endothelial cells, besides glutamate transporters, also express glutamate receptors. Via these receptors it has described that in millimolar concentration range (excitotoxic levels), glutamate induces a mitochondrial reactive oxygen species (ROS) production that leads to endothelial apoptosis and blood–brain barrier (BBB) damage, therefore increasing the cerebral injury after stroke
31
(Figure 2).
Effect of glutamate excitotoxicity of glutamate on neurovascular unit. Neurovascular unit is composed of neurons, glia (astrocytes, microglia, and oligodendrocytes), and vascular components (endothelial cells, pericytes, and smooth muscle cells). (a) Failure of nutrient supply after ischemia causes an immediately increase of glutamate in the parenchymal space that affects the neuronal tissue, and other components of the neurovascular unit. (b) At millimolar concentration range (excitotoxic levels), glutamate induces an over-excitation of NMDA receptors (NMDAr) and AMPA/KA receptors (AMPA/KAr) of neuronal cells and cellular components of the neurovascular unit.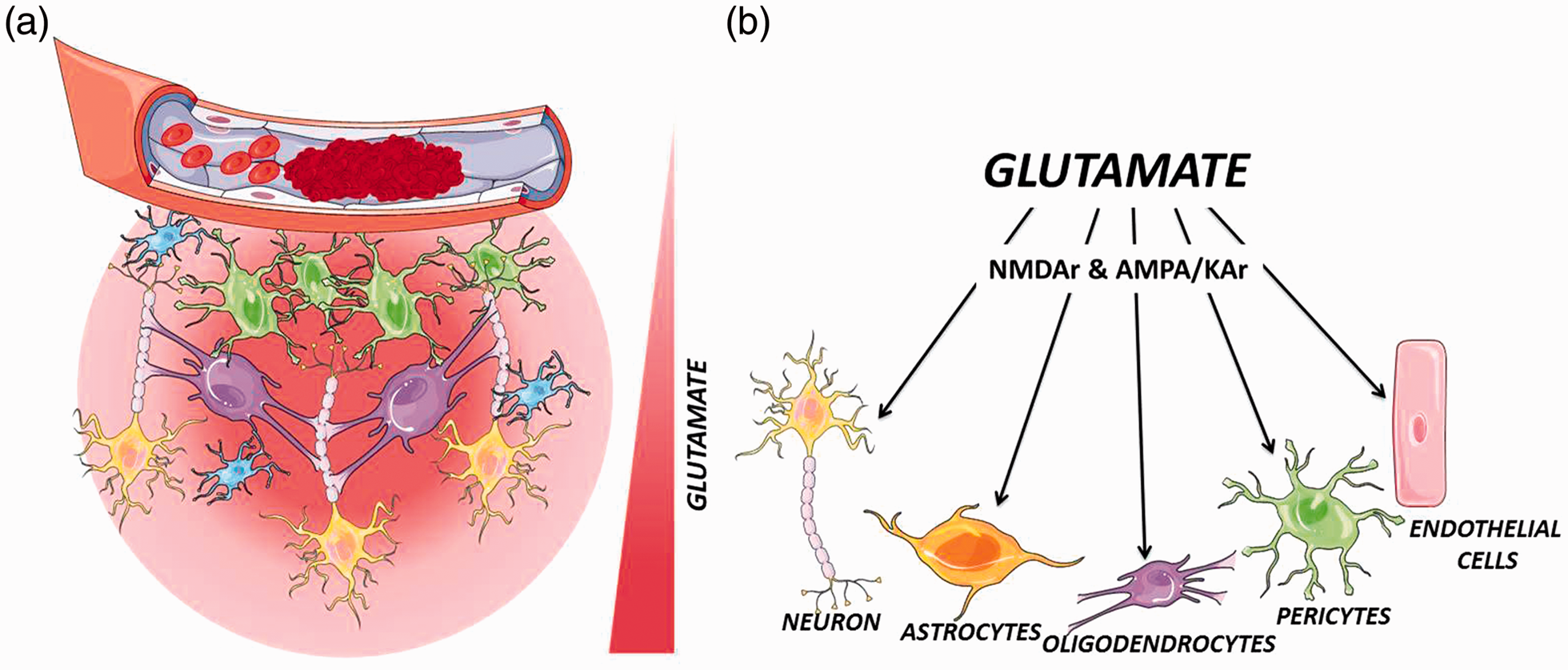
More than 15 years ago, studies in clinical patients, we reported that glutamate is critical for neuronal damage after ischemic stroke.32–37 Ischemic patients presented higher blood and cerebrospinal fluid (CSF) glutamate levels than control subjects at admission, 37 suggesting that glutamate concentrations above 200 μM in plasma acted as an important predictor of neuronal damage progression at 48 h, with a sensitivity of 85% and a specificity of 97%. 35 Moreover, high levels of glutamate in the plasma for at least 24 h were associated with early neurological deterioration, whereas in patients with stable ischemic stroke, glutamate levels dropped to normal values less than 6 h from onset. 35 All of these clinical data demonstrated for the first time the critical role of glutamate in stroke pathology and suggested a source of new strategies for the research of new protective therapies based on the inhibition of glutamate toxicity.
Therefore, controlling the increase of extracellular glutamate may confer neuroprotection by terminating multiple downstream death signaling cascades at their converging upstream initiation point.
Previous strategies to reduce the glutamate excitotoxicity
Knowledge of the molecular mechanisms involved in glutamate excitotoxicity after cerebral ischemia allowed researchers to develop promising pharmacological strategies against this neurotoxic process. In the beginning, the major focus of research centered on NMDA receptors (NMDAr) antagonism. NMDAr provided a logical target for drug design because it represented a major gateway for the myriad of other downstream effects of glutamate excitotoxicity. Moreover, during this period, progress in protein biochemistry and small molecule design yielded a wealth of information regarding the structure and function of these receptors. 37
Several classes of NMDAr antagonists with different sites of action were developed: namely, the competitive NMDAr antagonist acting on glutamate or glycine binding sites, noncompetitive allosteric inhibitors acting at other extracellular sites, and NMDAr channel blockers acting on sites in the receptor channel pore. Though showing promise in animal studies, antagonist drugs, such as selfotel, gavestinel, and traxoprodil, have largely failed in randomized, controlled clinical trials in humans. A variety of reasons have been postulated to explain the lack of success for these NMDAr-targeting therapies. Many of these compounds lack sufficient brain penetrance while exhibiting significant dose-limiting side effects.27,38,39 The adverse event profiles included hallucinations, agitations, catatonia, peripheral sensory loss, nausea, and elevation in blood pressure. 40 Moreover, in the case of acute processes such as stroke or traumatic brain injuries, glutamate excitotoxicity is thought to cause harm within a narrow timeframe after which the neurotransmitter reassumes its normal function. Therefore, the use of agents acting on NMDAr, a major receptor of glutamate, may have not only missed the window for therapeutic efficacy but also led to undesired side effects from prolonged receptor blockades.27,41,42
Research has continued on NMDAr antagonism despite initial disappointments. However, studies in the last two decades have expanded beyond the NMDAr with newer experiments seeking ways to control the upstream glutamate concentration as well as downstream protein signals. 27
Beyond glutamate antagonist: Blood to brain glutamate grabbing
High glutamate concentrations at the synaptic cleft are rapidly (up to 1000-fold) reduced by the action of glutamate transporters present primarily on astrocytes surrounding the nerve terminal to prevent glutamate excitotoxicity.23,27 An unfavorable gradient exists between blood (40–60 μM) and brain (1–10 μM) glutamate concentrations. In addition to astrocytes, EAATs on the antiluminal membrane act to accumulate the excess extracellular glutamate into the endothelial cells.
26
When the endothelial glutamate concentration becomes higher than the blood glutamate concentration, glutamate is transported into the blood by means of facilitated diffusion, a mechanism that facilitates blood excretion of glutamate from the brain.
27
Based on this mechanism, a decrease of blood glutamate levels by means of glutamate scavengers or grabbers leads to a larger glutamate gradient between the brain and blood, facilitating the lowering of extracellular glutamate in the brain, and reducing the toxic effects of this neurotransmitter. Therefore, manipulating this mechanism may have potential neuroprotective effects after stroke (Figure 3).
Schematic representation of the diffusion of glutamate from the brain to the blood. Under physiological conditions, the brain–blood barrier (BBB) acts as a semi-permeable membrane preventing the diffusion of glutamate from the blood to the brain. During ischemia, glutamate concentrations in the brain rise to levels 10 times higher than normal, leading to increased blood levels of glutamate as well. Lowering blood glutamate levels leads to a reduction of extracellular levels in the brain.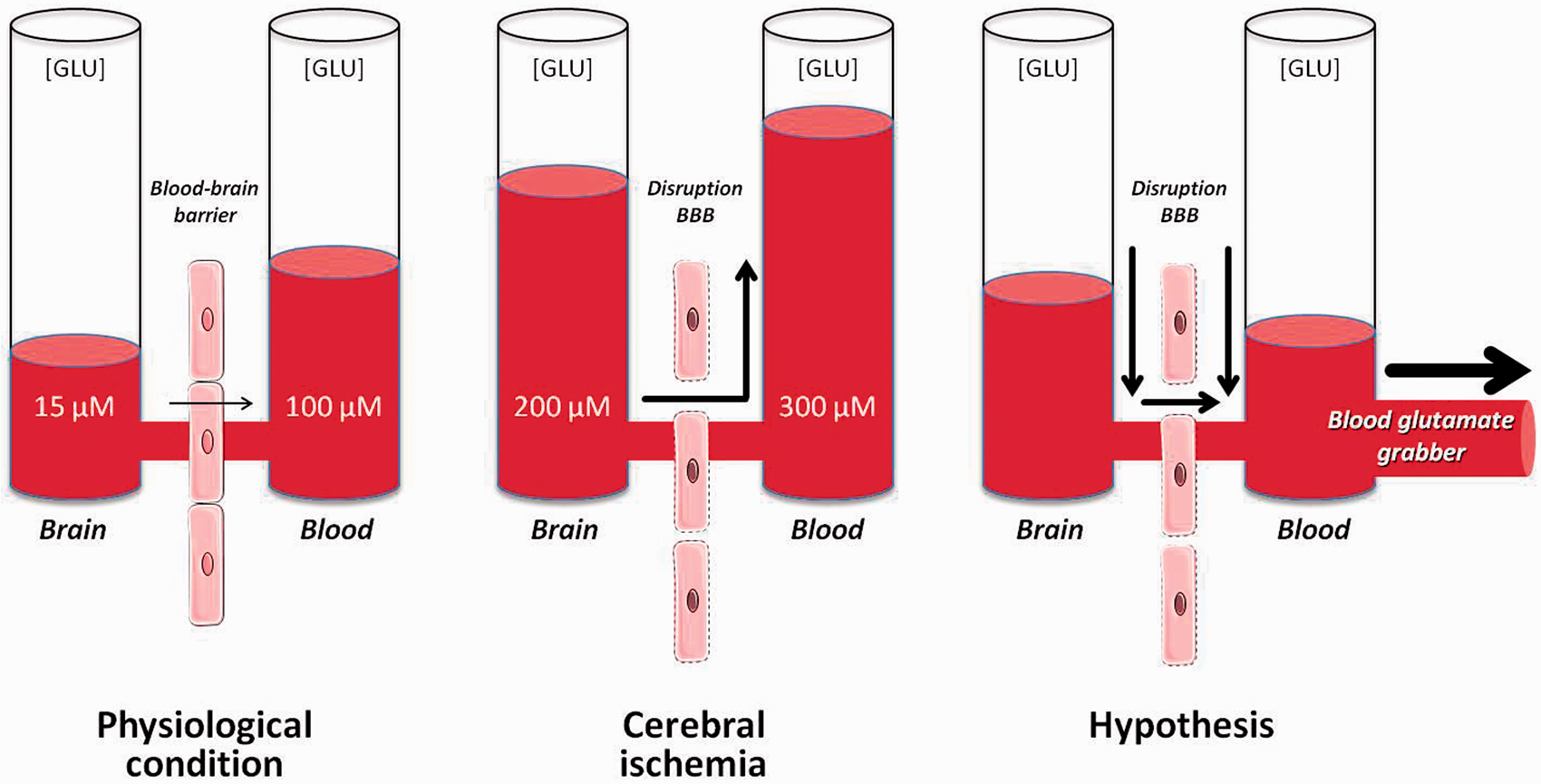
The term scavenger is commonly used to describe biological processes based on the removal of waste material to maintain an adequate ecosystem (e.g. free radical scavengers). Glutamate is an essential amino acid in the function of the CNS, and reducing the excess levels of glutamate to maintain homeostasis of this neurotransmitter is critical. In this regard, the term grabbing glutamate seems more accurate than glutamate scavenger.
To demonstrate this glutamate grabbing hypothesis the blood-resident enzyme glutamate-oxaloacetate transaminase (GOT), which transforms glutamate into α-ketoglutarate and aspartate in the presence of oxaloacetate (OxAc) (Figure 4), was used. This enzyme, when OxAc is artificially increased shifts the equilibrium of the reaction to the right side, thereby decreasing glutamate levels in blood.
43
Experimental animals were injected with radioactive glutamate into the lateral ventricles, and the addition of OxAc induced a decrease in blood glutamate levels followed by an increase in the diffusion of radioactive brain glutamate into the blood.
41
Similar effects were observed in other studies using two microdialysis probes, where the first one infused and the other one collected glutamate; OxAc treatment reduced the rate of radioactive glutamate collection by the second probe.
44
Alternatively, malate pretreatment, a GOT blocker, has also demonstrated inhibition of the OxAc-dependent lowering of blood glutamate, confirming that the effect of OxAc or GOT on blood glutamate levels was mediated by blood glutamate lowering.
45
The neuroprotective effect of oxaloacetate is due to the decrease in blood glutamate levels as a result of the activation of a blood-resident enzyme glutamate-oxaloacetate transaminase (GOT). The latter enzyme causes a reversible reaction in which glutamate reacts with oxaloacetate to transfer an amino group, transforming glutamate into α-ketoglutarate and oxaloacetate into aspartate. Artificially increasing oxaloacetate concentration shifts the reaction to the right, decreasing glutamate levels in the blood.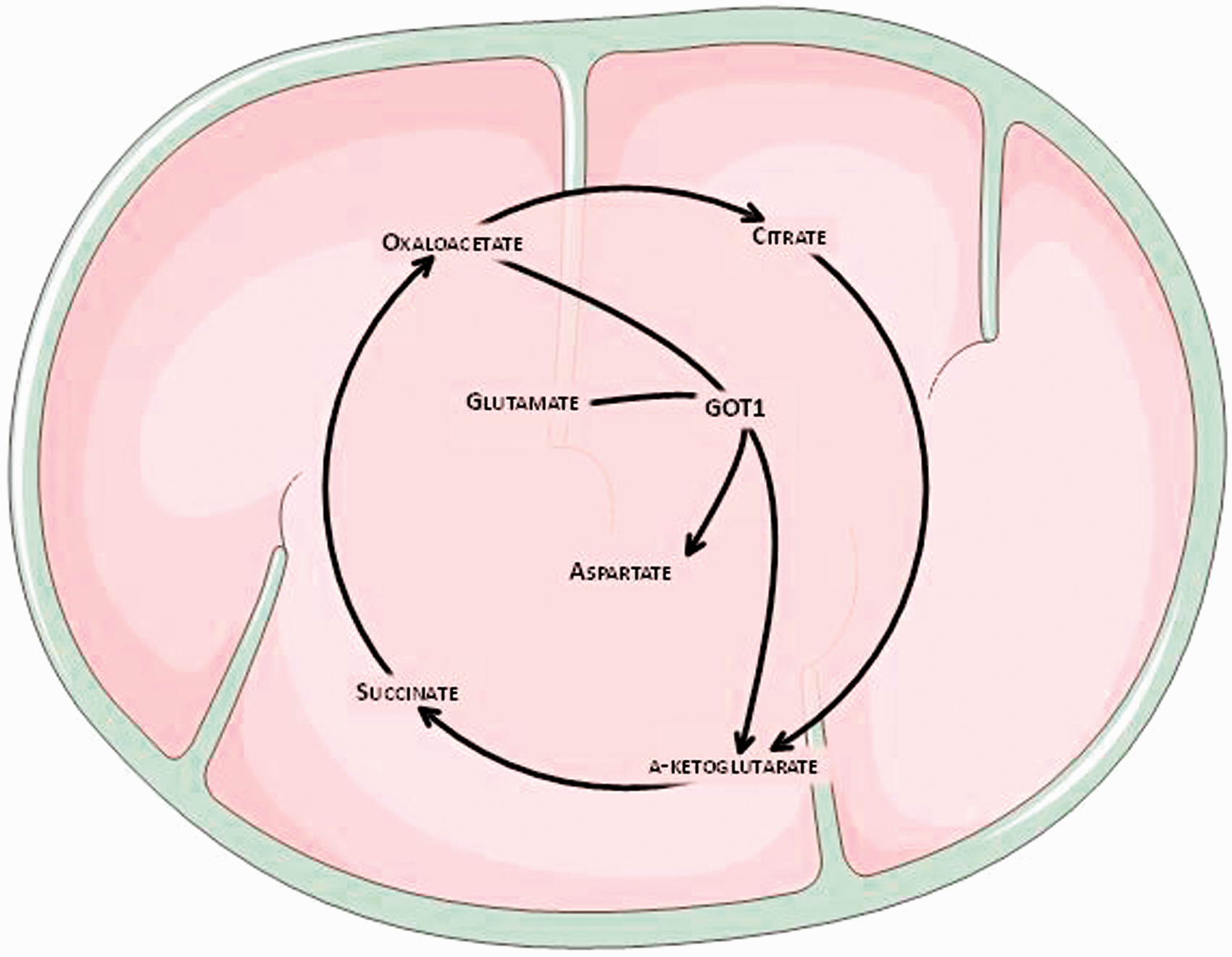
Based on this mechanism, when glutamate levels in brain fluids are elevated OxAc treatment causes a decrease in blood glutamate levels; this leads to a larger glutamate gradient between the brain and blood, which facilitates a decrease in extracellular levels of brain glutamate reflected in a reduction of ischemic damage.43,44
The first evidence of the neuroprotective effect with OxAc in cerebral ischemia was observed in rats submitted to photothrombotic lesions. 46 This effect was subsequently probed in a model of ischemia induced by the transient occlusion of the middle cerebral artery (MCAO). 47 After a dose–response study on the effect OxAc on blood glutamate lowering, and under the STAIR guidelines, 48 a bolus intravenous injection of OxAc (effective dose used 3.5 mg/100 g, animal weight) was provided 90 min after occlusion, leading to decreased blood glutamate levels, followed by a decrease in infarct volume and edema after ischemia. These effects were associated with a reduction in motor deficit. To confirm that the neuroprotective effect was due to a decrease in brain glutamate levels, spectroscopic analysis revealed that the increase in brain glutamate observed in control animals after MCAO was clearly reduced in animals treated with OxAc. These results were also validated by other independent laboratories. 49
To further demonstrate the clinical relevance of these preclinical results as potential therapeutic strategies, high blood levels of GOT were later hypothesized to be correlated with lower blood glutamate levels and subsequently with a better functional outcome. To test this hypothesis, two independent clinical and observational studies were performed, in which the primary end point was functional outcome at three months.50,51 In these studies, patients with good outcomes showed lower glutamate levels and higher GOT levels in blood samples collected at admission. A significant inverse correlation was observed between GOT and glutamate levels. The favorable effect on functional outcome was also supported by reduced lesion volumes. These results postulated that GOT could metabolize blood glutamate and induce a glutamate gradient from brain parenchyma to the circulation. These clinical findings represented the first clinical evidence of the neuroprotective effect of blood glutamate grabbing mechanisms in ischemic stroke patients and supported the potential applicability of OxAc or GOT as future treatments for acute ischemic stroke.
New results from independent clinical laboratories have shown a strong relationship between plasma glutamate and GOT levels at admission and the development of post-stroke depression 52 with functional outcome in acute ischemic stroke within three months, 53 confirming once again the efficacy of the mechanism.
Regarding the use of OxAc as a serum glutamate-grabbing treatment, some limitations may be associated with the high doses likely to be necessary in patients to achieve the same effects as those observed in experimental animals. However, although to date, no consistent data exist in humans confirming this toxic effect, a study published in the 1960s 54 showed that OxAc was used to treat diabetic patients (from 200 to 1000 mg per day in three divided doses given orally), and no effects on liver function or blood acetone and cholesterol levels was reported at the doses administered.
The protective effects of OxAc have also been discussed. 43 Because this molecule participates in energy metabolism and provides antioxidant protection to cells subjected to stress, such as hydrogen peroxide, the protective role of OxAc has been suggested to be related to other mechanisms apart from serum glutamate reduction. Studies in experimental animals showed that the lowering of blood glutamate levels induced by OxAc was abolished in the presence of excess glutamate in the serum or when it was administered in combination with malate (a GOT inhibitor), 44 which effectively demonstrates its serum glutamate-grabbing action.
GOT could also be used as another potential blood glutamate grabber. As such, human recombinant GOT1 (rGOT1) in the rat model of ischemia induced by transient MCAO 55 caused a reduction in serum and brain glutamate levels, resulting in a reduction in infarct volume and sensorimotor deficit. Dose–response study showed that an rGOT1 dose of 12.88 µg per 100 g (animal weight) caused a maximum blood glutamate reduction. In order to determine whether the effect of the enzyme could be potentiated by OxAc (co-substrate of the enzymatic reaction), the dose of rGOT1 (12.88 µg/100 g) was supplemented with a non-effective dose of OxAc (1.5 mg/100 g), observing that the protective effect of rGOT1 was more evident with this new combination, indicating that the combined administration of rGOT1 and OxAc may be the more efficient serum glutamate-grabbing approach.
The administration of an endogenous serum enzyme such as GOT1 as a new protective treatment against ischemia is an interesting strategy because the administration of rGOT1 is unlikely to induce toxic effects in humans as the levels of this enzyme vary among healthy human subjects (7–45 U/l), and has been shown to increase >10-fold in patients with liver damage. 56
The therapeutic synergistic effect observed between OxAc and GOT can be attributed to the fact that the endogenous OxAc concentration can become a limiting factor to the enzymatic reaction when GOT activity is increased after treatment. Therefore, by attending to the rapid and maintained reduction in serum glutamate concentration, treatments based on the combined administration of rGOT1 supplemented with a low concentration of OxAc were concluded to be optimal to reach the maximum protective effect in ischemia. Notably, the low dose of OxAc is sufficient to increase the glutamate-grabbing activity of rGOT1, which reduces the potential complications associated with a high dose of this molecule as previously described.
Other blood glutamate lowering strategies
Pyruvate
In addition to the use of OxAc as a blood glutamate grabber, pyruvate has also been described as an effective drug able to reduce blood glutamate levels. Similar to OxAc, pyruvate induces an activation of the blood-resident enzyme glutamate-pyruvate transaminase (GPT), an enzyme that catalyzes the reversible transformation of pyruvate and glutamate to alanine and α-ketoglutarate; therefore, artificially increasing pyruvate shifts the equilibrium of the reaction to the right side, thereby decreasing glutamate levels in the blood.
The efficacy of this drug has been tested in stroke.57,58 Similar to previous studies with GOT, GPT levels were associated with lower levels of blood glutamate, better functional outcome, reduced infarct volume, and lower percentages of early neurological deterioration, although this association was stronger for GOT than GPT levels. 50 Similarly, in experimental studies, OxAc presented stronger protective effects than pyruvate.57,58
Hemodialysis and peritoneal dialysis
Another interesting and potentially useful method for reducing blood glutamate concentrations is the use of dialysis to filter the blood and remove excess glutamate. Compared with healthy controls, patients with end-stage renal failure on hemodialysis had higher concentrations of blood glutamate. 59 During hemodialysis (especially in the first hour), glutamate concentrations decreased regardless of the size of filter pores, blood flow rate, or gender. Similarly, peritoneal dialysis resulted in decreases in blood glutamate with a corresponding increase of glutamate in the dialysis solution. 60 In a rat model of stroke, the reduction of blood glutamate levels observed with peritoneal dialysis was associated with a decrease in infarct area. 61 These studies provided, by means of an alternative way, that blood glutamate reduction is an effective protective strategy and may also be used as evidence that hemodialysis and peritoneal dialysis may be an effective modality in reducing blood glutamate concentration. This hypothesis is currently being tested in clinical trials in phase IIa (EudraCT Number: 2012-000791-42).
Analogs of OxAc: Repositioning of drugs against glutamate toxicity
All preclinical studies on different models and the clinical observational analysis reported guarantee the therapeutic efficacy of the reduction of blood glutamate as well as the glutamate grabber drugs; however, translation to clinical practice has critical steps before their use in humans. These critical steps are those necessary to develop a clinical trial, which will involve high financial support and risk of investment for the sponsors interested in the study. In addition, the repeated failure of protective drugs against glutamate excitotoxicity in clinical trials has reduced the trust of pharmaceutical companies and other sponsors in stroke studies.
Aiming to demonstrate the clinical efficacy of glutamate grabbers in humans and reduce the risk of investment in the study the pharmacological strategy known as drug repositioning represents an interesting alternative to find new grabbing drugs.62,63 This allowed researchers to search drugs already known and used for other pathologies and in which the clinical phase I and phase II were already completed, reducing the risk of investment for sponsors in case the clinical study result failed.
Following this pharmacological strategy, a new clinical study (proof of concept) with a new grabbing drug is currently ongoing (EudraCT Number: 2014-003123-22).
Limitations of glutamate grabbers
From a clinical point of view, glutamate grabbers may have some important limitations. In acute pathologies such as stroke, glutamate increases immediately after neuronal damage, inducing a rapid effect, and therefore exhibits a short therapeutic window. OxAc or GOT showed a therapeutic window of no longer than 3 to 4 h after insult 55 ; therefore, the efficacy of this protective mechanism depends on the time-point of drug administration. However, in a recently published model of brain hemorrhage, glutamate grabbers were shown to decrease the blood glutamate levels but did not affect the hemorrhagic hematoma, 64 suggesting that this treatment can be given as early as possible, without neuroimaging, in case of suspected stroke, which may potentially increase the number of patients treated within the therapeutic window.
Conclusions
The critical role of glutamate excitotoxicity has been long described as a key molecular cause of neuronal injury from stroke. At present, we have a better understanding of the mechanisms by which elevated glutamate ultimately leads to cell death; unfortunately all clinical trials that have been primarily based on the inhibition of glutamate excitotoxicity through glutamate antagonist to date have failed. Currently, many experimental evidences in different models of diseases use blood glutamate grabbers as effective treatments against neuronal damage induced by glutamate excitotoxicity. Future clinical trials (some of them in progress) will allow to know the application of this novel therapeutic strategy.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: partially supported by grants from Instituto de Salud Carlos III (PI13/00292; PI14/01879). T Sobrino (CP12/03121) and F Campos (CP14/00154) are recipients of a research contract from Miguel Servet Program of Instituto de Salud Carlos III.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.