
Abstract
Arterial hypertension is not only a major risk factor for cerebrovascular accidents, such as stroke and cerebral hemorrhage, but is also associated to milder forms of brain injury. One of the main causes of neurodegeneration is the increase in reactive oxygen species (ROS) that is also a common trait of hypertensive conditions, thus suggesting that such a mechanism could play a role even in the onset of hypertension-evoked brain injury. To investigate this issue, we have explored the effect of acute-induced hypertensive conditions on cerebral oxidative stress. To this aim, we have developed a mouse model of transverse aortic coarctation (TAC) between the two carotid arteries, which imposes acutely on the right brain hemisphere a dramatic increase in blood pressure. Our results show that hypertension acutely induced by aortic coarctation induces a breaking of the blood–brain barrier (BBB) and reactive astrocytosis through hyperperfusion, and evokes trigger factors of neurodegeneration such as oxidative stress and inflammation, similar to that observed in cerebral hypoperfusion. Moreover, the derived brain injury is mainly localized in selected brain areas controlling cognitive functions, such as the cortex and hippocampus, and could be a consequence of a defect in the BBB permeability. It is noteworthy to emphasize that, even if these latter events are not enough to produce ischemic/hemorrhagic injury, they are able to alter mechanisms fundamental for maintaining normal brain function, such as protein synthesis, which has a prominent role for memory formation and cortical plasticity.
Introduction
Arterial hypertension is not only a major risk factor for cerebrovascular accidents, such as stroke and cerebral hemorrhage, but is also associated to milder forms of brain injury, such as lacuna, focal brain damage, revealed only by magnetic resonance imaging (Del Bigio et al, 1999; Selvetella et al, 2003). These several types of cerebral damage are promoted by increased blood pressure levels, likely through different pathophysiological mechanisms. The brain injury in the presence of chronic hypertension has been mainly attributed to the involvement of a hypoxic/ischemic mechanism (de la Torre, 2002). In fact, hypertension induces vascular remodeling even in cerebral arteries, consisting in increased thickness of arterial wall accompanied by decreased lumen size, which favors chronic brain hypoperfusion. In contrast, the acute increase of blood pressure induces passive dilatation of cerebral arteries, which is accompanied by a dramatic increase in global cerebral blood flow (Mayhan, 2001). In this latter case, the impact of blood pressure on the cerebral blood flow is different from chronic hypertension, and it is still unclarified whether even acute blood pressure elevations could determine brain injury.
As regards the intrinsic causes of neuronal damage, one of the main events leading to neurodegeneration is the increase in reactive oxygen species (ROS) in the cerebral structures (Ischiropoulos and Beckman, 2003). Actually, an increased oxidative stress lowers the level of neuronal activity and, more important, causes the massive entrance of Ca2+ in neurons, thus activating proapoptotic mechanisms (Brown and Davis, 2002). Oxidative stress, however, is also a common trait of hypertensive conditions (Zalba et al, 2001), thus suggesting that such a mechanism could play a role even in the onset of hypertension-evoked brain injury (Zimmerman et al, 2004).
To investigate this latter issue, we have explored the effect of acute-induced hypertensive conditions on cerebral oxidative stress. To this aim, we have developed a mouse model of transverse aortic coarctation (TAC) between the two carotid arteries, which imposes acutely on the right brain hemisphere a dramatic increase in blood pressure.
Materials and methods
Animals
C57Bl/6 male mice 12 to 15 weeks old were used. Animals were kept under a constant 12-h light–dark cycle at a temperature of 22°C to 25°C. Standard chaw and water were provided ad libitum. All study protocol was in agreement with the guidelines for research in animals of our institution.
Transverse Aortic Constriction
Mice were anaesthetized with ketamine and xylazine. After anesthesia, mice were placed on thermal beds to prevent thermal dispersion, and respiration was assisted with a volume-cycled ventilator (Basile, Milano, Italia) connected to an 18-gauge cannula inserted into the trachea. Transverse aortic coarctation (TAC, n = 30) was performed. The transverse aorta between truncus anonymous and the left carotid artery was isolated and a 7.0 nylon suture ligature was placed around the aorta, with the two ends of the suture left outside the chest. A further group of mice underwent the same surgical procedures without realizing aortic stenosis (Sham, n = 20). A further group of TAC (n = 5) and sham (n = 3) mice were used to evaluate the hemodynamic impact of the TAC. The blood pressure gradient between the two carotids was measured by selective cannulation of the left and right carotid arteries after 1 and 7 days from the TAC.
Then the mice were weighed; their brains were excised and frozen in cool isopentane (−80°C). For immunocytochemical analysis mice were transcardially perfused, brains removed, and transferred to sucrose for cryoprotection as described previously (De Simoni et al, 2004).
Cerebral Perfusion Measurement
Blood flow was measured by laser Doppler flowmetry (Transonic BLF-21), using a flexible 0.5-mm fiberoptic probe (Transonic, Type M, 0.5 mm diameter) positioned on the skull (De Simoni et al, 2004). Measurements were performed on each hemisphere at the following coordinates: AP = −1 mm, L = ±1 mm; AP = −1 mm, L = ±3.5 mm; AP = −2 mm, L = ±2 mm; AP = −3.5 mm, L = ±1 mm; AP = −3.5 mm, L = ±3.5 mm. These coordinates have been chosen to explore brain perfusion on the whole hemisphere surface. Data are expressed as arbitrary perfusion units.
Evaluation of Blood–Brain Barrier Permeability
Brain injury might be coupled to alteration of blood–brain barrier (BBB) permeability. For this reason, we evaluated BBB integrity using albumin-fluorescein isothiocyanate FITC (Sigma), a dye that is able to cross BBB only when the latter is impaired. Mice were infused with albumin-FITC (10 mg/mL in PBS at 10 mL/kg) at the jugular vein level after 1 and 7 days from surgical procedures. At 12 mins after infusion, mice were decapitated, and the entire brain removed and frozen in cool isopentane (−80°C) was cut serially. The sections were observed with an × 20 magnification under an Axiophot2 fluorescence microscope (Zeiss) equipped with a FITC filter (Stamatovic et al, 2005).
Color pictures (24-bit) were acquired and analyzed using a digital camera system coupled to an imaging software (Spot, Diagnostic Instruments, Sterling Heights, MI, USA) under a constant exposure time, gain and offset, chosen as to increase the threshold for fluorescence. Albumin extravasation was evaluated measuring selectively the green fluorescence intensity corrected for background fluorescence, and expressed as fluorescence arbitrary units (FAU). All images were taken in the first 30 secs of light exposure, when no fluorescence decay was detected in preliminary studies.
Measurement of Superoxide in the Brain by Dihydroethidium
To evaluate the impact on cerebral oxidative stress in our animal model, we analyzed brain slices from several cerebral districts by different approaches. Analysis of superoxide production was assessed by dihydroethidium (DHE) dyeing as described previously (Vecchione et al, 2005; Dayal et al, 2004). In particular, DHE is able to detect directly superoxide production, since it evokes a red fluorescent signal after binding of dihydroethidium to these oxygen-free radicals.
The entire brain was removed and frozen in cool isopentane (−80°C) and cut serially with a Jung CM1900 Cryostat (Leica) in 20-μm-thick sections. Sections were placed onto polylysinated microscope slides, incubated at room temperature for 30 mins with DHE (Sigma; 10 μmol/L) and protected from light. Images were acquired and fluorescence intensity was measured as described above.
Measurement of Peroxynitrite in the Brain by Immunohistochemistry
Immunohistochemistry with antinitrotyrosine antibodies recognizes the reaction products of oxygen-free radicals with nitric oxide, such as peroxynitrite, on tyrosine residues of proteins. Brain sections contiguous to those treated with DHE were placed onto polylysinated microscope slides, fixed at room temperature in 4% paraformaldehyde in PBS, blocked with 0.1% BSA in PBS for 1 h, incubated with antinitrotyrosine antibodies (5 μg/mL, Transduction Laboratories, Rockville, MD, USA) overnight at 4°C, and with anti-IgG TRITC conjugated for 1 h at room temperature. Images were acquired and fluorescence intensity was measured as above.
Evaluation of Nicotinamide Adenine Dinucleotide Phosphate (NAD(P)H) Oxidase Enzymatic Activity
Oxidative stress is the final event derived from activation of several enzymes. It is well known that, in brain tissues, ROS derive mainly from NAD(P)H oxidase enzymatic activity (Shimohama et al, 2000). Thus, we set to evaluate NAD(P)H oxidase enzymatic activity in the left and right brain hemispheres from 1 and 7 days TAC mice and in sham mice. NAD(P)H oxidase activity was measured using a lucigenin assay as described previously (Li et al, 2002). The brain was cut into two hemispheres and lysed in a buffer containing protease inhibitors (20 mmol/L, pH 7.0, monobasic potassium phosphate, 1 mmol/L EGTA, 10 μmol/L aprotinin, leupeptin, pepstatin, 0.5 mmol/L phenylmethylsulfonyl fluoride). Lysate protein was collated and resuspended in TRIS-sucrose buffer, and the protein content was measured by Bradford method. NAD(P)H oxidase activity was performed in a 50 mmol/L phosphate buffer (pH 7.0) containing 1 mmol/L EGTA, 150 mmol/L sucrose, 5 μmol/L lucigenin as the electron acceptor, and NAD(P)H (100 μmol/L) as the substrate (final volume 2 mL). The reaction was started by the addition of 100 μg of protein. Photon emission was measured every 30 secs for 30 mins in a scintillation counter. Background counts were determined by protein-free incubations and subtracted from protein readings. Results were expressed cpm/(mg protein) min.
Immunocytochemistry
Astrocytes have been evaluated by glial fibrillary acidic protein (GFAP) immunostaining. Coronal sections (20-μm thick), prepared from perfused brains, were rinsed for 1 h in 0.3% Triton X-100 and 10% normal goat serum (NGS) in PBS 0.01 mol/L, pH 7.4. Then, they were incubated at 4°C overnight with GFAP (mouse monoclonal, 1:2000, Boehringher Mannheim, Mannheim, Germany) for astrocytes. Then, sections were incubated with biotinylated secondary antibody (goat anti-mouse, Vector Laboratories, USA) for 1 h, washed and incubated in avidin–biotin-peroxidase (Vectastain ABC kit, Vector Laboratories, USA). After reacting with 3′-3-diaminobenzidine (DAB, Sigma, Munich, Germany) in Tris-HCl-buffered saline (TBS), pH 7.4, and 0.01% H2O2, the sections were washed, mounted onto gelatin-coated slides, dried, dehydrated through graded alcohol, fixed in xylene, and coverslipped using a distyrene plasticizer xylene (DPX) mountant (BDH, Poole, UK) before light microscopy analysis.
Messenger RNA Expression of Inflammatory Cytokines by Real-Time PCR
Total RNA was extracted from the right and left hippocampi of sham-operated and TAC mice according to the acid guanidium–phenol–chloroform method (De Simoni et al, 2000). Complementary DNA was synthesized as follows: 2 μg total RNA from each sample was reverse transcribed with random hexamer primers using Multi Scribe reverse transcriptase (TaqMan Reverse transcription reagents, Applied Biosystems, Foster City, CA, USA). The following thermal cycling protocol for reverse transcription was used: 10 mins at 25°C for incubation, 30 mins at 48°C for reverse transcription, and then 5 mins at 95°C for inactivation. Real-time PCR was performed using a GeneAmp 5700 Sequence detection System (Applied Biosystems, Foster City, CA, USA) as described previously (Bergamaschini et al, 2004). In all, 50 ng of cDNA and gene-specific primers (200 nmol/L final concentration) were added to Master Mix SYBR Green I Dye, TaqMan DNA polymerase, deoxyribonucleotide triphosphate (dNTPs) with deoxyuridine triphosphate (dUTP) and optimal buffer components (Applied Biosystems, Foster City, CA, USA), and subjected to PCR amplification in a total volume of 25 μL. The PCR protocol included one cycle at 50°C for 2 mins, one cycle at 95°C for 10 mins, and 40 cycles at 95°C for 15 secs and 60°C for 1 min. For each experimental group, five RNA samples were used. Real-time PCR was conducted in triplicate with each RNA sample. The amplified transcripts were quantified using the comparative cycle threshold method (Applied Biosystems users bulletin 2). Primer-optimizing procedures and validation experiments (data not shown) were performed according to the manufacturer's instructions to show that efficiencies of target and reference were equal. β-Actin was used as reference gene and relative gene expression levels were determined according to the manufacturer's ρρCt method (Applied Biosystems). Primers were designed using Primer Express software (Applied Biosystems Foster City, CA, USA), based on the following GenBank accession numbers: NM007393 (β-actin), NM008361 (interleukin (IL)-1β), and NM013693 (tumor necrosis factor (TNF)-α).
Detection of Leukocytes in Brain Tissues
Circulating leukocytes were marked using Rhodamine 6G (0.3 mg/kg intravenous, Sigma) as described previously (Ishikawa et al, 2004). Briefly, the dye was injected intravenously 12 h before performing TAC. At 1 and 7 days after TAC, brains were excised, cooled in isopentane and cut into slices. Rhodamine 6G-associated fluorescence in the brain and spleen, used as positive control, was visualized by epiillumination at 510 to 560 nm, using a 590-nm emission filter.
Statistical Analysis
All results are expressed as mean ± s.e.m. Data were compared by two-way ANOVA, followed by post hoc simultaneous multiple comparisons with Bonferroni analysis. A two-tailed value of P < 0.05 was considered significant.
Results
Transverse Aortic Coarctation Induces an Increase in Cerebral Perfusion in the Right Hemisphere
Transverse Aortic Coarctation induced a significant increase in blood pressure in the right carotid as compared with the left one (160 ± 12 versus 80 ± 8 mm Hg, P < 0.05). This resulted in a significant increase in cerebral perfusion in the right hemisphere and a concomitant reduction of perfusion in the left hemisphere 1 day after TAC. These alterations of blood flow were still present 7 days after TAC (Figure 1).
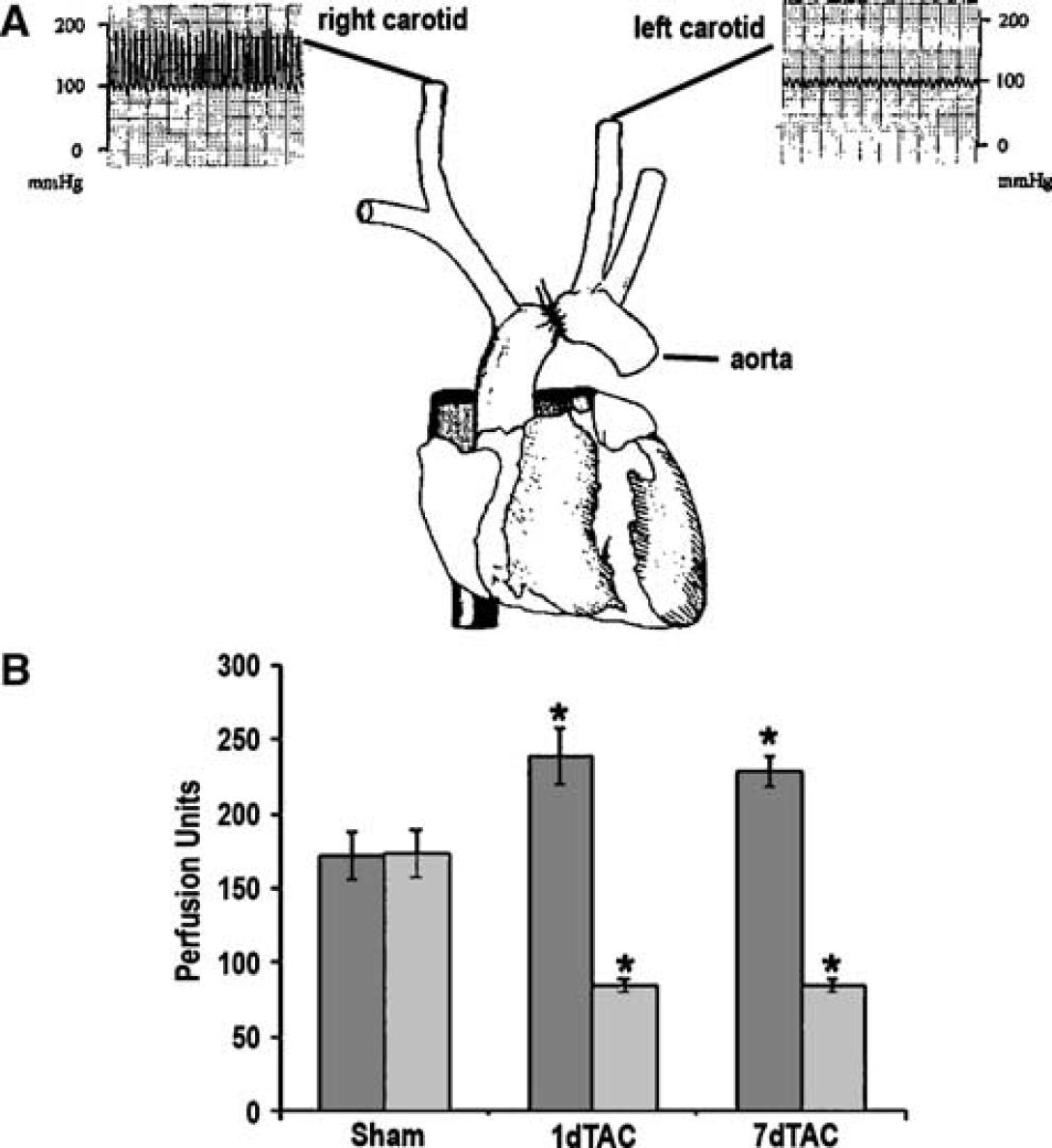
(
Transverse Aortic Coarctation Induces Alterations of the Blood–Brain Barrier Permeability
At 1 and 7 days after surgery, the signal due to albumin extravasation was increased in selected right and left brain areas, such as the hippocampus and cortex of TAC, compared with sham-operated mice, indicating a damage to the BBB in these areas. This effect was similar in both hemispheres. Blood–brain barrier at striatal level was unaffected by TAC (Figure 2).
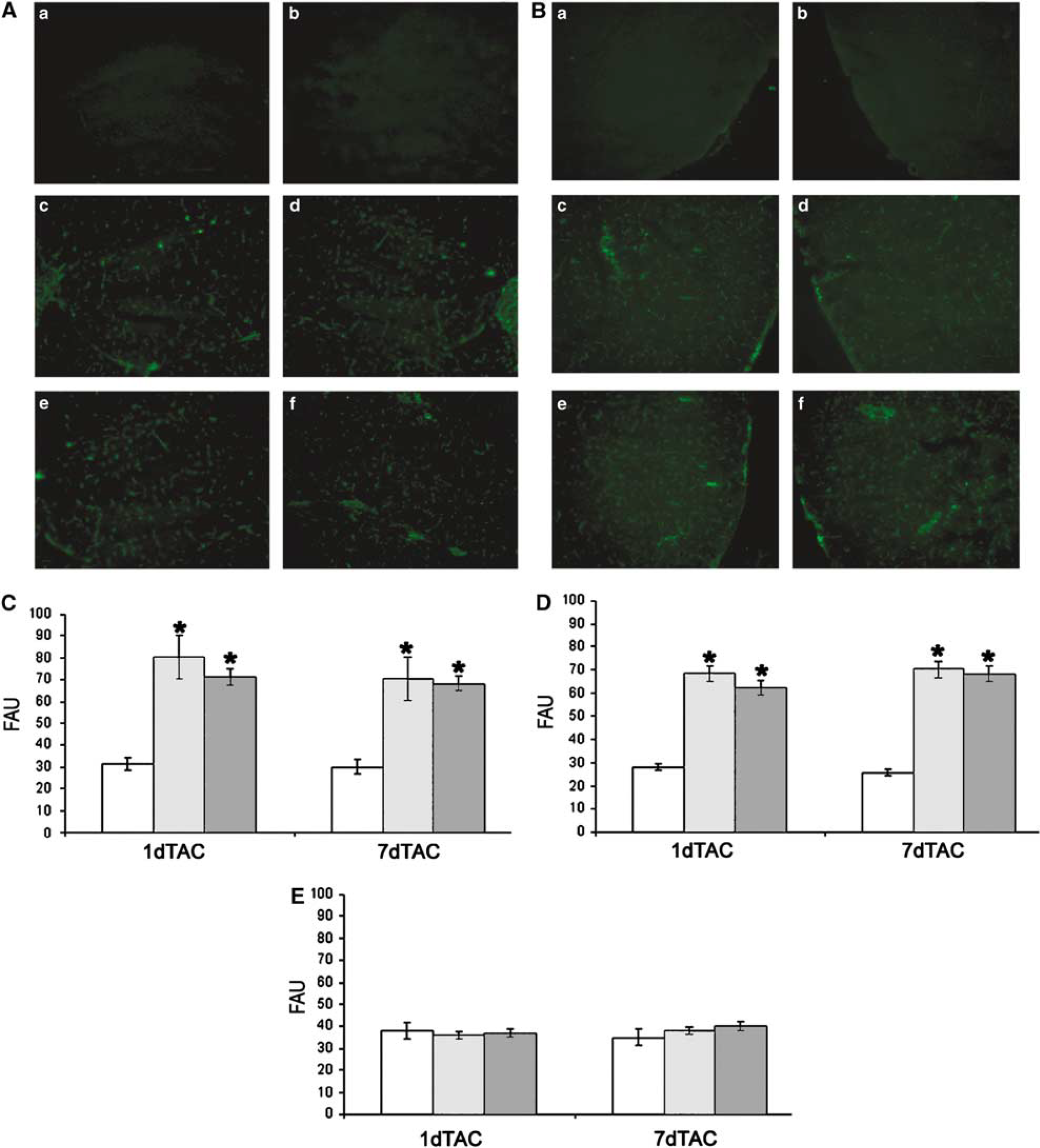
High-power micrograph of albumin-FITC extravasation through BBB in the hippocampus (
Transverse Aortic Coarctation Induces Oxidative Stress in Selected Brain Areas
At 1 day after TAC, the hippocampus and cortex showed an increase in DHE fluorescent signal as compared with that observed in sham mice, thus reflecting an increased superoxide production in both brain areas subjected to TAC. This effect was present also at 7 days from TAC. Notably, DHE fluorescence signal increased to a similar extent in both cerebral hemispheres. In contrast, other brain areas, such as the striatum, were unaffected by TAC (Figure 3). The analysis of brain nitrotyrosines showed similar results. Their presence was revealed in the hippocampus and cortex 1 and 7 days after TAC and not in the striatum (Figure 4). The increase in superoxide production and the presence of nitrotyrosines suggested that TAC is able to induce an augmented oxidative stress in selected brain areas such as the cortex and hippocampus.
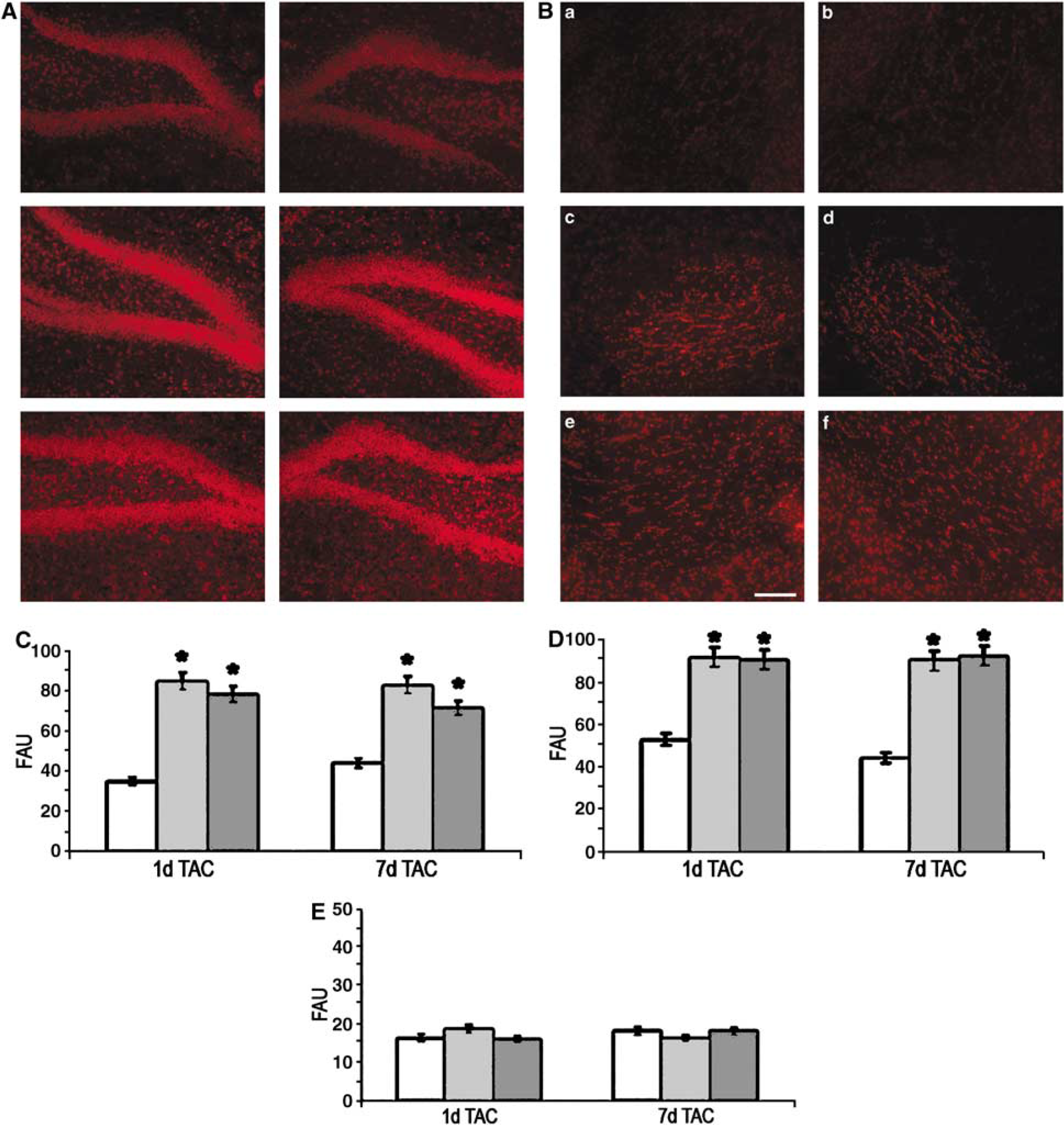
High-power micrograph of the histochemistry by DHE of the hippocampus (
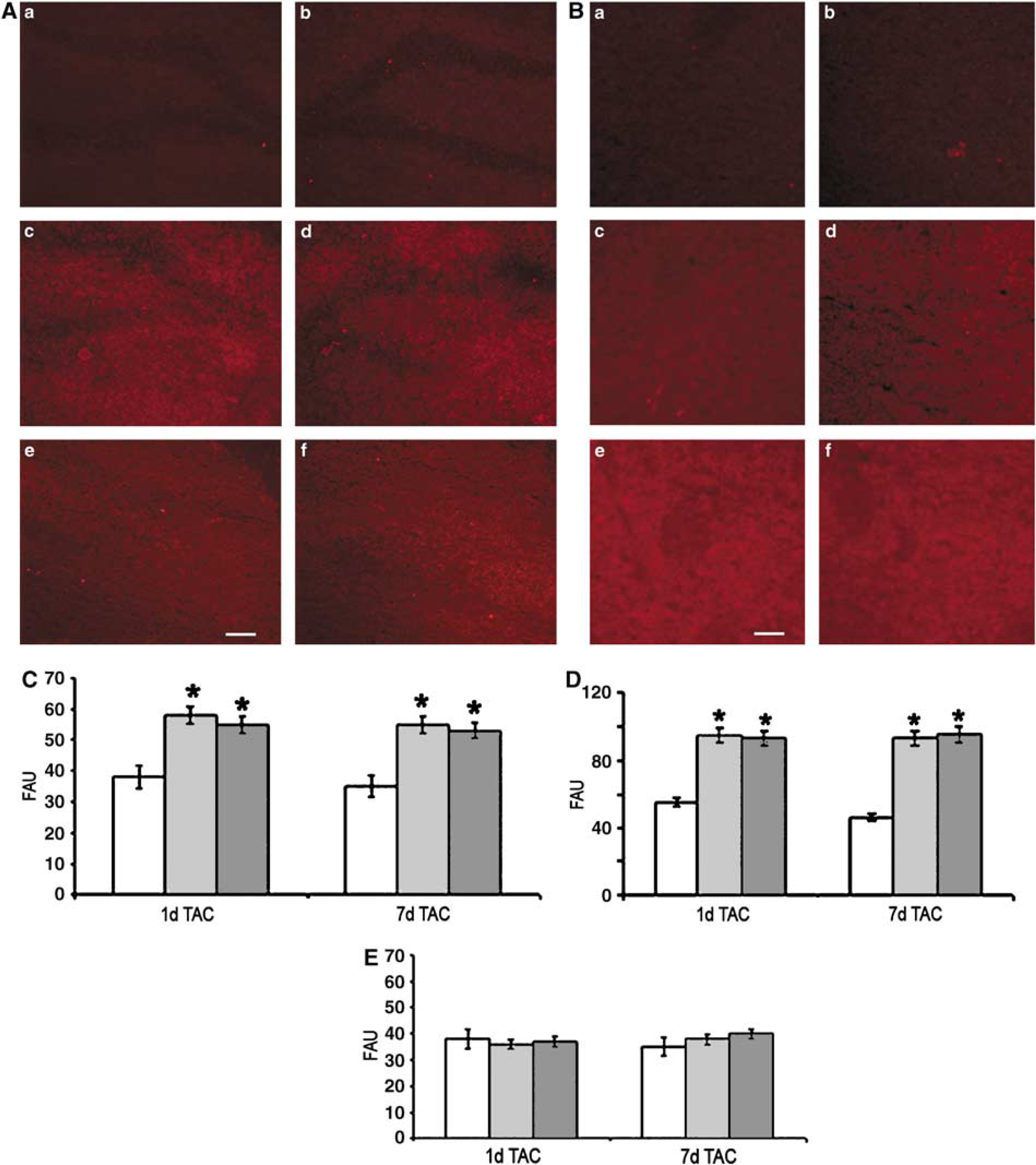
High-power micrograph of the immunohistochemistry by antinitrotyrosine of the hippocampus (
Transverse Aortic Coarctation Induces the Activation of NAD(P)H Oxidase and of Proinflammatory Cytokines
As shown in Figure 5, NAD(P)H oxidase activity was increased in both hemispheres 1 day after surgery in TAC compared with sham-operated mice, and continued to be still significantly higher in the right hemisphere, while it became similar to sham in the left hemisphere, after 7 days from TAC.
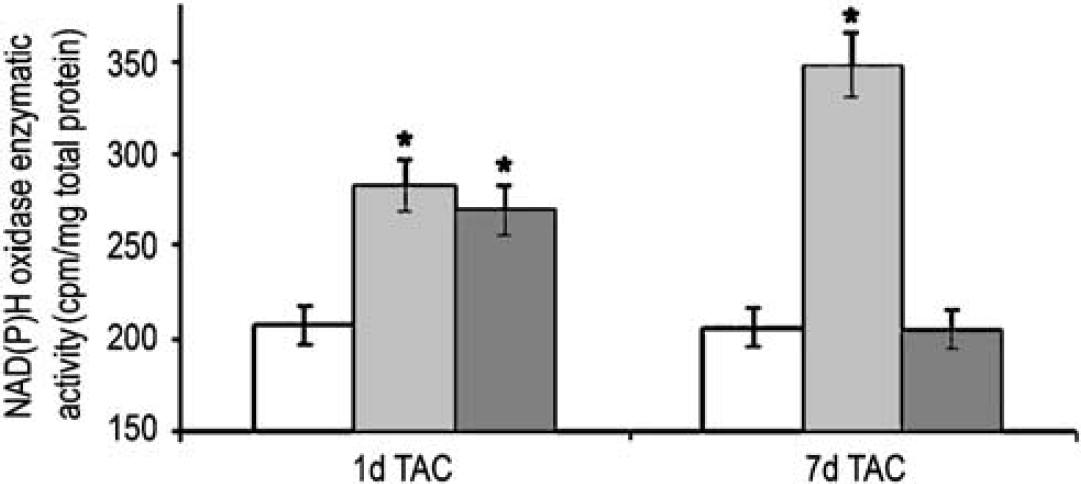
Quantitation of the NAD(P)H oxidase enzymatic activity in sham (white bar), or TAC mice: right (light gray bar) and left (dark gray bar) hemispheres. *P < 0.05 versus sham; n = 5.
Since oxidative stress is associated with inflammation (Lo et al, 2003), we evaluated the mRNA expression of inflammatory cytokines IL-1β and TNF-α. An increase in IL-1β and TNF-α mRNA expression was detected 1 and 7 days after TAC in both the right and left hippocampi (Figure 6). No leukocyte recruitment from circulation was observed after 1 and 7 days from TAC (data not shown).
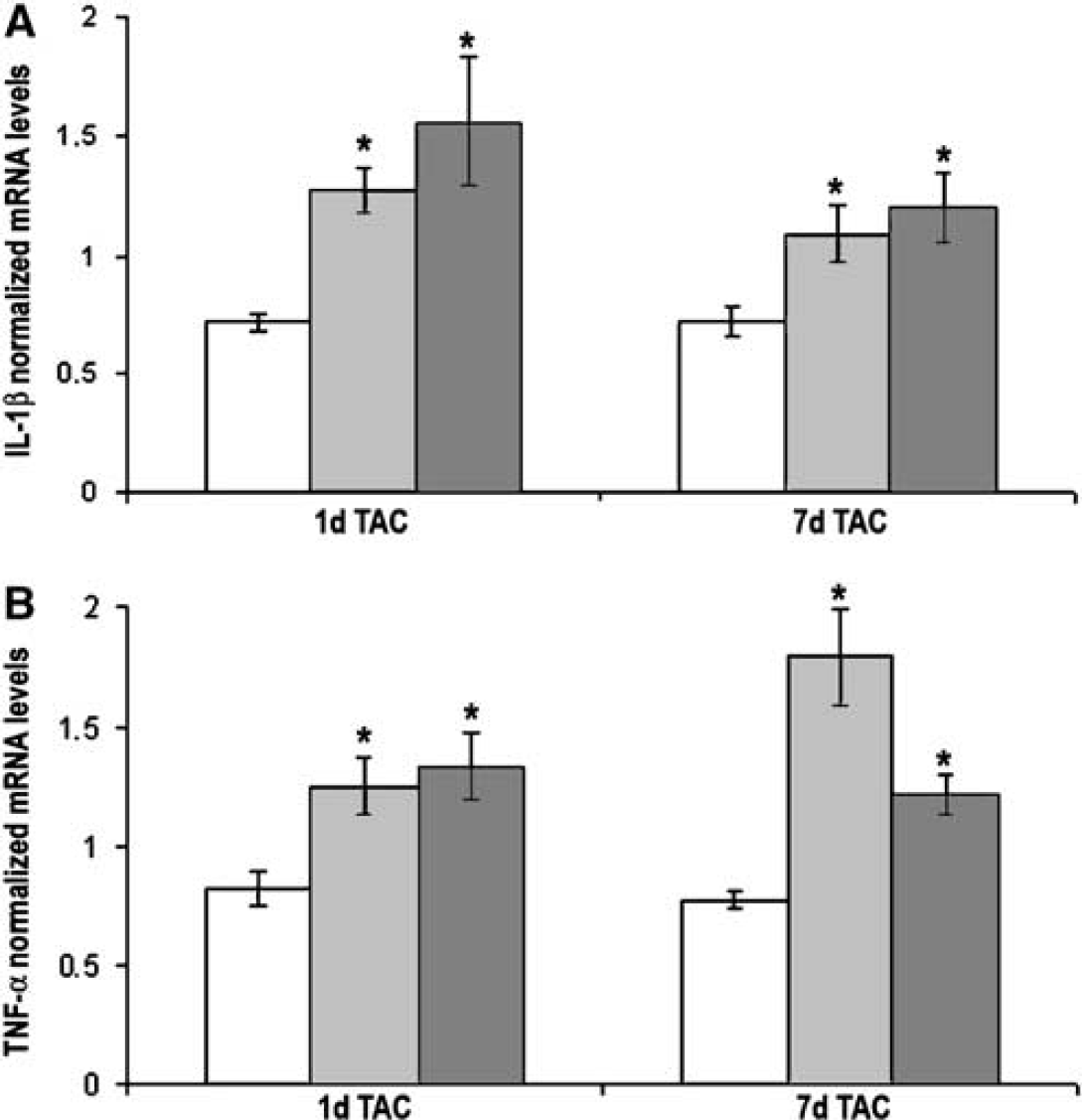
Interleukin-1β (
Transverse Aortic Coarctation Induces Reactive Astrogliosis
At 1 day after surgery, an increase in the immunoreactivity of GFAP-positive cells was apparent in the hippocampus and cortex (Figure 7) of TAC compared with sham-operated mice. Transverse aortic coarctation induced a clearcut activation of these cells whose phenotypic changes from the quiescent to the activated state became apparent by the enlargement of cell bodies and thickening of processes. While in the hippocampus cells showing an activated phenotype were widespread throughout the brain area, in the cortex distinct clusters of activated GFAP-positive cells were present. These features could be observed in both hemispheres.
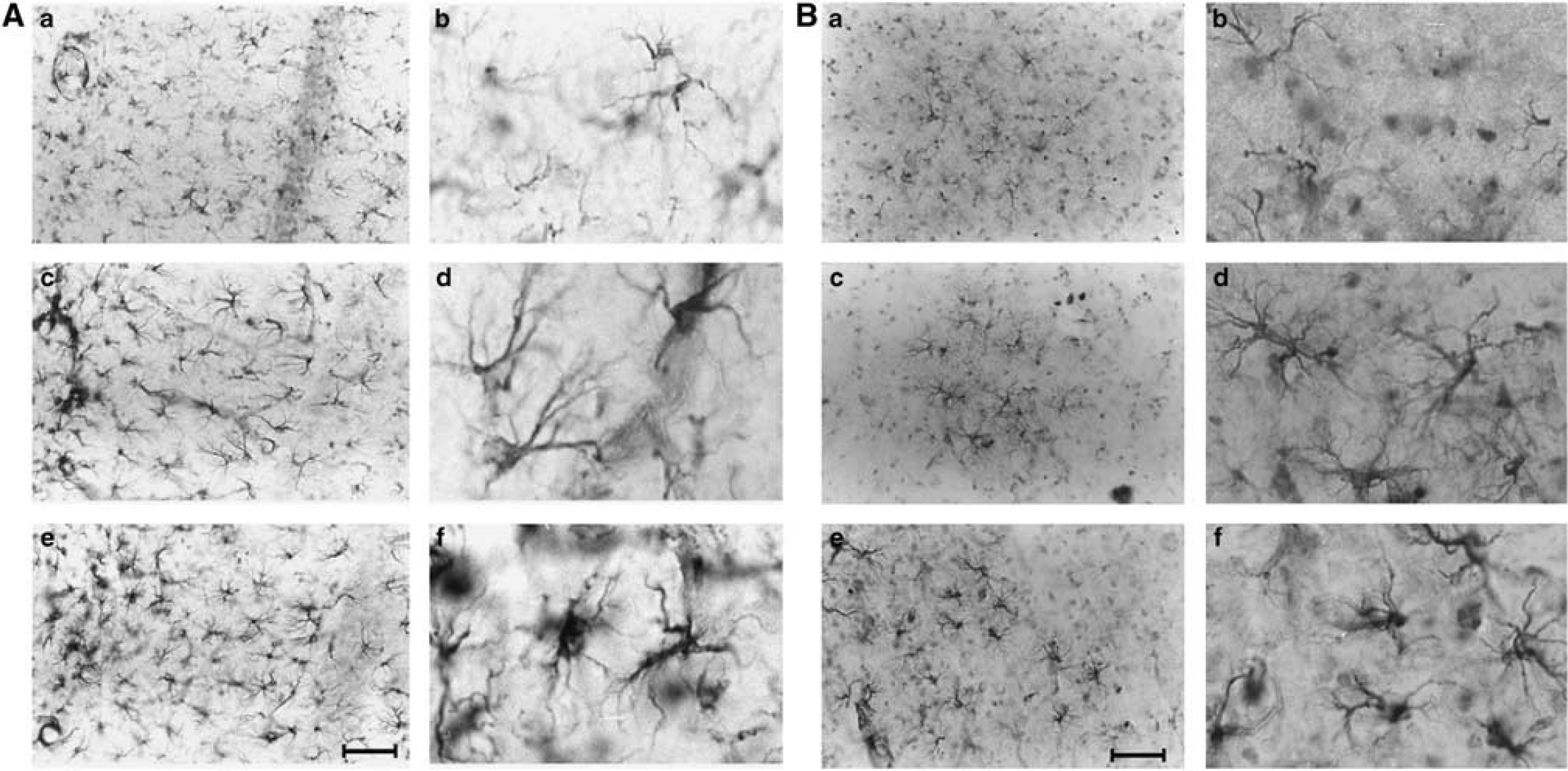
Representative photomicrographs (n = 5) of the right hippocampal (
Discussion
Our results show for the first time that hypertension acutely induced by aortic coarctation evokes a breaking of the BBB associated to oxidative stress, and inflammation in selected brain areas such as the hippocampus and cortex.
In this study, we have firstly characterized the impact of coarctation of the aortic arch between the two carotid arteries on cerebral blood flow (CBF). In particular, using this strategy, we have realized a murine model of increase of CBF in the right brain hemisphere, with a concomitant reduction of the CBF in the contralateral one. Thus, in the hemisphere where blood pressure is acutely increased, a hyperperfusion is realized, whereas the decrease in blood pressure downstream the aortic coarctation generates a hypoperfusion in the left hemisphere. Our model allowed us to study concomitantly, in the same animal, the effects of hyperperfusion and hypoperfusion. Moreover, beside hypertension, this particular experimental model could have other analogues in human pathology, such as epileptic seizures (Van Paesschen et al, 2003; Ho et al, 2000) and the left unilateral carotid stenosis (Stiver and Ogilvy, 2002; McCabe et al, 1999). Interestingly, the analysis of the impact of aortic coarctation on the BBB revealed an increased permeability at hippocampal and cortical levels in both hemispheres, thus suggesting that hyperperfusion as well as hypoperfusion are able to alter BBB permeability. Blood–brain barrier function depends on the so-called ‘neurovascular unit’ composed of neurons, astrocytes, and, more important, endothelial cells of microvessels (Mayhan, 2001). Endothelial cells of cerebral vessels are devoid of fenestrations and transendothelial channels (Matter and Balda, 2003). Thus, for the most part, the BBB prevents the movement of substances into brain tissues (Wolburg and Lippoldt, 2002). Our results are in agreement with other previous studies showing that mechanical stress induced by acute increase in the intracranial blood pressure is able to provoke a breaking of the BBB through the impairment of the tight junctions between adjacent endothelial cells (Rapoport, 2000).
However, it has been reported that the reduction of blood flow to cerebral microvessels, according to the critically attained threshold of cerebral hypoper fusion (CATCH) hypothesis, determines a failure in the delivery of oxygen to the endothelial cells of cerebral microvessels, altering intracellular homeostasis, with consequent alteration of the function of the tight junction that controls BBB permeability (de la Torre, 2000).
In the same brain areas where BBB damage was observed, a marked reactive astrogliosis was also present. These cells showed the typical activated morphology with enlarged cell bodies and highly ramified thick processes. Astrocytes, that are readily activated by injury, are major players in the establishment and maintenance of the BBB function (del Zoppo and Mabuchi, 2003; Dietrich et al, 2003). By their endfeet processes that surround brain vessel endothelial cells, they control the migration of inflammatory cells and act as buffers for extravasated fluids and molecules that may gain access to the brain parenchyma.
When the overall control of BBB function is lost, possible toxic substances may gain access to the brain and induce activation of oxidative pathways, with an increase in ROS production (Eisenblatter et al, 2003; Del Maestro et al, 1981). In fact, in this model, we have observed a significant increase in oxidative stress at hippocampal and cortical levels, in both hemispheres, thus indicating that oxidative stress could be a consequence of BBB damage induced by hyperperfusion as well as hypoperfusion. Moreover, free radicals can further alter endothelial membrane permeability to various molecules through peroxidation of membrane lipid (Mayhan, 1998), enhancing further BBB dysfunction.
One of the main oxidative pathways involved in the production of ROS is represented by NAD(P)H oxidase (Ishikawa et al, 2004; Kazama et al, 2004). On this issue, we analyzed the impact of hyperperfusion and hypoperfusion on the activation of NAD(P)H oxidase in cerebral tissue and its contribution to oxidative stress. We observed that NAD(P)H oxidase enzymatic activity is significantly increased in both hemispheres after 1 day from TAC. More interestingly, it is still high in the right hyperperfused hemisphere after 7 days from TAC, while it returns to the basal levels in the left hypoperfused hemisphere. Our data are supported by a recent study that ascribes NAD(P)H oxidase among the main sources of superoxide in brain tissues (Patel et al, 2005). Indeed, the presence of oxidative stress in the left hemisphere even after 7 days from TAC, in which NAD(P)H oxidase enzymatic activity is reduced, prompted us to analyze the involvement of other trigger factors that can explain the imbalance in cerebral oxidative homeostasis. Actually, neuroinflammatory processes, induced in part by proinflammatory cytokines, yield enhanced ROS production that has neurotoxic properties (Barone and Feurstein, 1999; de Vries et al, 1997). For this reason, we have analyzed also the expression of the cytokines that are mainly involved in the inflammatory component of cerebrovascular disease: IL-1β and TNF-α (Giulian et al, 1986). Interestingly, we observed an increase in mRNA levels of the two cytokines in both hemispheres after 1 and 7 days from TAC. More important, cytokine mRNA levels are still higher in the left hemisphere after 7 days from TAC. To understand whether the inflammatory cytokines observed in brain tissues from TAC mice were the result of local release or of the circulating leukocyte recruitment and extravasation, we have marked circulating leukocytes. No extravasation of leukocytes into brain tissues at the time of hypertension we have evaluated indicates that the proinflammatory cytokines we observed are from local production and release. However, our results showed the activation of the astrogliosis, which has been clearly shown as among the most powerful inducers of IL-1β and TNF-α. Thus, altogether, these data suggest that alterations in cerebral microcirculation, through a disruption of the BBB, are able to induce the activation of redundant oxidative and inflammatory pathways, which in turn condition the brain oxidative stress.
Neurons are highly susceptible to oxidative stress. Excessive ROS, in fact, lead to the destruction of cellular components including lipids, proteins, and DNA, and ultimately cell death via apoptosis or necrosis (Lo et al, 2003). In our experimental model and at the time points considered in our study, we could not find evidence of cell death as evaluated by cresyl violet or fluoro jade stainings, or by neurofilament H expression (data not shown). We propose that this condition of inflammatory and oxidative stress state may represent a risk factor for neuronal death, and/or lead to functional deficits via protein damage.
In conclusion, our data show that acute blood pressure increase, through hyperperfusion, evokes trigger factors of neurodegeneration such as oxidative stress and inflammation, similar to that observed in cerebral hypoperfusion. The derived brain injury is mainly localized in selected brain areas controlling cognitive functions such as the cortex and hippocampus, and could be a consequence of a defect in the BBB permeability. It is noteworthy to emphasize that, even if these latter events are not enough to produce ischemic/hemorrhagic injury, they are able to alter the mechanisms fundamental for maintaining normal brain function, such as protein synthesis, which has a prominent role for memory formation and cortical plasticity (Serrano and Klann, 2004; Scharf et al, 2002).