
Abstract
In the present study, we investigate whether a long-term blockade of brain AT1 receptors in male Wistar rats before and after ischemic injury exerts neuroprotective effects and modulates apoptosis and inflammatory responses, which are associated with the post-ischemic progression of brain damage. The AT1 receptor antagonist irbesartan was continuously infused intracerebroventricularly using osmotic minipumps over a 5-day period before and for 3 or 7 days after middle cerebral artery occlusion (MCAO) for 90 minutes. Neurologic status was evaluated daily, starting 24 hours after MCAO. After MCAO (3 and 7 days), brains were removed for the measurement of infarct size and immunohistochemical evaluation of apoptosis and accumulation of reactive microglia and macrophages. Treatment with irbesartan before ischemia improved motor functions, whereas post-ischemic treatment improved sensory functions. Blockade of brain AT1 receptors reduced the infarct size on days 3 and 7 after MCAO. In the peri-infarct cortex, irbesartan treatment decreased the number of apoptotic cells on day 3 and attenuated the invasion of activated microglia and macrophages on days 3 and 7 after ischemia. Long-term blockade of brain AT1 receptors improves the recovery from cerebral ischemia. Antiapoptotic mechanisms and inhibition of post-ischemic inflammation are involved in the AT1 receptor blockade-induced neuroprotective effects in ischemic brain tissue.
The renin-angiotensin system (RAS) has been implicated in the pathogenesis of cardiovascular diseases and stroke. Angiotensin II (Ang II), the effector peptide of the RAS, exerts its actions through the activation of at least two receptor subtypes, referred to as the AT1 and the AT2 receptor (de Gasparo et al., 2000). A number of animal studies have demonstrated protective effects of high-affinity, non-peptide AT1 receptor antagonists against stroke (Inada et al., 1997; Stier et al., 1993; Von Lutterotti et al., 1992). Chronic systemic pretreatment with candesartan reduced the infarct size in both spontaneously hypertensive rats (SHR) and normotensive rats subjected to middle cerebral artery occlusion (MCAO) (Groth et al. 2003; Ito et al., 2002; Nishimura et al., 2000). Beneficial effects of an AT1 receptor deficiency during brain ischemic injury have also been demonstrated in mice with a genetic disruption of the AT1 receptor (Walther et al., 2002). Selective, long-lasting blockade of brain AT1 receptors before ischemic injury has been reported to improve the neurologic outcome of focal cerebral ischemia and to reduce the expression of AP-1 transcription factors c-Fos and c-Jun, which have been associated with programmed cell death (Dai et al., 1999). The effect of post-ischemic AT1 receptor inhibition on the recovery from ischemic injury has not yet been studied.
A local reduction or an arrest of blood supply leads to neuronal death in the ischemic core, which can be defined as the region of a profound adenosine triphosphate (ATP) depletion (Hara et al. 2001). Between the lethally damaged cells in the ischemic core and the normal brain tissue, the peri-infarct penumbra is located, a region in which energy state is initially preserved and the neurons can survive for several hours and days. A number of events, such as peri-infarct depolarization, occurring within hours after the onset of perfusion deficit, followed by more delayed post-ischemic inflammation and apoptosis, damage neurones and glial cells lethally and contribute to the progression and expansion of brain injury (Dirnagl et al., 1999, Hata et al. 2000b).
The effects of inhibition of angiotensin receptors in cerebral vessels upon the recovery from ischemic stroke have been intensively studied. On the other hand, little is known about the contribution of brain AT1 receptors in the initiation and regulation of processes leading to neuronal injury and damage after cerebral ischemia. In the present study, we investigate the effect of the inhibition of AT1 receptors in the brain upon the evolution of neurologic outcome, infarct size, and inflammation and apoptotic processes in peri-infarct cortical tissue after focal cerebral ischemia. To achieve an exclusive inhibition of brain AT1 receptors, the specific, high-affinity AT1 receptor antagonist irbesartan was continuously infused intracerebroventricularly (ICV) over a 5-day period before and for 3 (experiment 1) or 7 days (experiment 2) after MCAO with reperfusion. The used dose of the antagonist inhibited brain but not vascular AT1 receptors (Dai et al., 1999). The neurologic status of each rat was evaluated daily. Ongoing inflammatory processes were detected by staining brain slices with antibodies specific for activated microglia and macrophages. DNA fragmentation assessed with terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL), the cleavage fragment of the caspase 3 substrate, poly (ADP-ribose) polymerase (PARP), and activated caspase 3 in the peri-infarct parietal cortex were quantified as indicators of apoptosis.
METHODS
Male, normotensive Wistar rats weighing 200 g were obtained from Charles River (Sulzfeld, Germany). Animals were kept under controlled conditions with respect to temperature and humidity, and were housed on a 12 hours light/12 hours dark cycle, with free access to food and water.
Surgical methods
Implantation of osmotic minipumps.
Chloral hydrate (400 mg/kg) body weight) injected intraperitoneally was used as anesthetic for all surgical procedures. Osmotic minipumps (ALZET Model No. 2002) (Alza Corporation, Palo Alto, CA, U.S.A.), which continuously deliver dissolved substances at a rate of 0.5 μL/h, were filled with vehicle or irbesartan. The concentration of irbesartan in the solution was 4 mmol/L. The implantation of the osmotic minipumps was described in detail elsewhere (Dai et al., 1999). Osmotic minipumps were implanted 5 days before MCAO.
Implantation of the femoral artery catheter.
Three days after the implantation of ICV cannulae, a polyethylene catheter (PP-50) was inserted through the femoral artery into the abdominal aorta. The catheter was filled with heparinized saline, passed through a subcutaneous tunnel, and sealed at the back of the neck. The arterial catheter was used for blood withdrawals to determine blood parameters. pH, pCO2, pO2, and concentrations of lactate, glucose, sodium, potassium, and chloride were quantified using RADIOMETER ABL 700 SERIES (Radiometer Medical A/S, DK-2700 Brønshøj, Denmark) at the following time points: conscious rats; 30 minutes after the occlusion of middle cerebral artery; immediately after the onset of reperfusion; and 30 minutes thereafter.
Middle cerebral artery occlusion with reperfusion
In this study, the intraluminal occlusion of the middle cerebral artery (MCA) for 90 minutes with subsequent reperfusion was used (Koizumi et al., 1986). Regional cerebral blood flow (rCBF) was continuously monitored at one point (1 mm posterior to the bregma, 5 mm from the midline) on the surface of each hemisphere in the supply territory of the MCA before, during, and after MCAO by laser-Doppler-flowmetry (Periflux system 5000) (Schmid-Elsaesser et al., 1998). rCBF measurements were performed to assess the degree of MCAO. Abrupt reduction in rCBF by approximately 75 to 90% indicated a successful occlusion of the MCA. Rats in which the ipsilateral blood flow during ischemia was not reduced to less than 25% of baseline during the first 30 minutes of occlusion, or in which a premature increase in the ipsilateral blood flow was recorded, were excluded from the experiments. Body temperature was maintained at 37°C with a heating pad.
General procedures
Neurologic deficits.
Neurologic evaluations were performed by a blinded observer once a day starting 24 hours after the surgery and continuing until the end of each experiment, that is, 3 or 7 days. Two neurologic evaluation grading systems were used. The neurologic evaluation developed by Bederson and colleagues (1986) (grading scale 0–3) includes the assessment of the grade of the forelimb flexion contralateral to the injured hemisphere, resistance to lateral push, and observation of circling behavior. Rats with no observable neurologic deficits were graded 0. Rats with forelimb flexion and shoulder adduction were graded 1. Grade 2 was assigned to severely dysfunctional rats with decreased resistance to lateral push towards the paretic side. Rats displaying consistent circling behavior together with forelimb flexion and decreased resistance to lateral push were graded 3 (Bederson et al., 1986). The neurologic evaluation described by Garcia and colleagues (1995) consists of the following six tests: 1, spontaneous activity; 2, symmetry in the movement of four limbs; 3, forepaw outstretching; 4, climbing; 5, body proprioception; and 6, response to vibrissae touch. Severe impairments in each of the tests were graded 0 or 1, and no observable deficits were graded 3. The minimum neurologic score is 3, and the maximum is 18 (i.e., in intact rats).
Tissue processing
After MCAO (3 days, experiment 1, and 7 days, experiment 2), rats were deeply anesthetized and intracardially perfused with phosphate-buffered saline (PBS) (pH 7.4) followed by 4% paraformaldehyde. The brains were removed, postfixed overnight, and subsequently cryoprotected in 30% sucrose for 72 hours at 4°C. Coronal sections (40 μm) were cut in a cryostat from the level bregma +3.7 mm to the level bregma −6.7 mm. Every 20th slice was used for the determination of infarct size. Three consecutive slices obtained at the level of the anterior commissure (bregma ± 0) were used for the determination of apoptosis (staining for TUNEL, PARP p85 fragment and activated caspase 3) and the detection of activated microglia and macrophages.
Measurement of infarct volume
To measure the infarct area, 14 coronal brain sections from different levels of the brain (see previous sections) were stained with cresyl violet. Slice images were digitalized, and the area of infarct was determined on each slice (Leica QWin image analysis system). The infarct volume was calculated by multiplying the sum of the infarct areas with the distance between the sections. The volumes of the ipsilateral and contralateral hemispheres were measured, and the difference was used to calculate the edema volume. The correction of the infarct size for edema was calculated using the formula of Swanson and colleagues (1990).
Immunohistochemical detection of inflammation and apoptosis
For immunohistochemical analysis of activated microglia and macrophages, coronal free-floating brain slices (40 μm) were incubated with a primary antibody specific for activated microglia and macrophages (mouse anti-rat ED-1, 1:300, Serotec, Düsseldorf, Germany) for two nights at 4°C. The signal was enhanced with a secondary antibody (goat anti-mouse [1:300], Vector Laboratories Inc., Burlingame CA, U.S.A.), and a peroxidase was coupled using the avidin-biotin complex (ABC) method (Vectastain, Vector Laboratories Inc.). Immunolabeling was visualized using 3, 3'-diaminobenzidine as chromogen.
TUNEL staining was carried out using an apoptosis detection kit according to the manufacturer's instructions (Roche Diagnostics GmbH, Mannheim, Germany). Only strongly labeled TUNEL-positive cells were considered as apoptotic, whereas lightly stained cells suggesting necrosis were not evaluated.
To evaluate PARP p85 fragment generation, free-floating brain sections were incubated with primary antibody overnight at 4°C (rabbit anti-rat PARP p85 fragment polyclonal antibody, 1:100, Promega GmbH, Mannheim, Germany), followed by biotinylated secondary antibody (goat-anti rabbit, Vector Laboratories Inc.) and the avidin-biotin complex method (Vectastain, Vector Laboratories Inc.) as described in previous sections.
Brain sections to be stained for activated caspase 3 were incubated with a primary antibody against caspase-3 (rabbit anti-rat cleaved caspase 3 antibody, 17kDa, Cell Signalling Technology, dilution 1:300) overnight at 4°C followed by biotinylated secondary antibody (anti-rabbit, Vector Laboratories Inc) and the avidin-biotin complex peroxidase method (Vectastain, Vector Laboratories, Inc) as described in previous sections.
Treatments and experimental design
Using osmotic minipumps, vehicle (control group) and irbesartan at a dose of 2 nmol/hour were continuously infused over a 5-day period. On day 6, the MCA was occluded for 90 minutes in all rats. Without interruption, ICV infusions of vehicle and the AT1 receptor antagonist were continued for 3 (experiment 1; vehicle: n = 10, irbesartan: n = 10) and 7 (experiment 2; vehicle n = 14, irbesartan. n = 16) consecutive days after MCAO. The first evaluation of neurologic outcome was performed 24 hours after the onset of reperfusion, and then daily until day 3 or 7.
Experiment 1.
On day 3 after focal cerebral ischemia, rats treated with vehicle (n = 10) and rats treated with irbesartan (n = 10) were perfused intracardially with 4% paraformaldehyde under deep anesthesia. The brains were removed and used for the measurement of infarct volume (all brains). Brain section from five brains in each group were used for immunohistochemical detection of TUNEL, PARP p85 fragment, and activated microglia.
Experiment 2.
The brains (vehicle, n = 12; irbesartan, n = 10) were removed on day 7 after MCAO with reperfusion and processed as described in the previous section.
The same protocol (Experiment 1) was used to study the activation of the executive caspase 3 in the boundary zone of the infarct core after AT1 receptor blockade in the brain. Rats were treated ICV with vehicle (n = 4) or irbesartan (n = 4) as described in the protocol of Experiment 1. On the third day after focal cerebral ischemia, brains were removed and cut in a cryostat, and coronal sections at the level of the anterior commissure were used for immunohistochemical detection of activated caspase 3.
Morphometric studies
Coronal sections through the brain at the level of the anterior commissure (bregma ± 0) were used for the immunohistochemical analysis of apoptosis and inflammation. Three random and non-overlapping areas (0.125 mm2 per area) were chosen in the boundary zone of the ischemic core in the frontoparietal cortex, as described in detail previously (Xu et al., 2003). The quantification of positively stained cells was carried out using Leica image analyzing software (Leica Qwin).
Statistical analyses
All values are expressed as mean ± SD. Comparisons of neurologic outcomes in vehicle- and irbesartan - treated rats (7 days) were analyzed by the repeated measures of analysis of variance with two independent groups of subjects followed by a post-hoc Bonferroni test for pairwise comparisons at individual time points. The effects of vehicle or irbesartan treatment on neurologic deficits over the time were analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni test. Comparisons of infarct volumes, apoptosis (staining for TUNEL and PARP p85 fragment), and activated microglia on days 3 and 7 in the vehicle- and irbesartan - treated rats were performed by ANOVA followed by post-hoc Bonferroni test. Student's t-test for unpaired samples was used to compare the data on caspase 3 staining between the vehicle- and irbesartan-treated groups on day 3 after MCAO. Statistical significance was accepted at P>0.05.
RESULTS
Physiologic variables in blood
The values of pH, pCO2, and pO2 and the blood concentrations of lactate, glucose, and electrolytes were not significantly different between the vehicle- and the irbesartan-treated groups at any time point (Table 1).
Arterial blood oxygen, carbon dioxide, and pH values and glucose, lactate, sodium, potassium, and chloride concentrations before, during, and after middle cerebral artery occlusion in rats treated intracerebroventricularly with vehicle or irbesartan
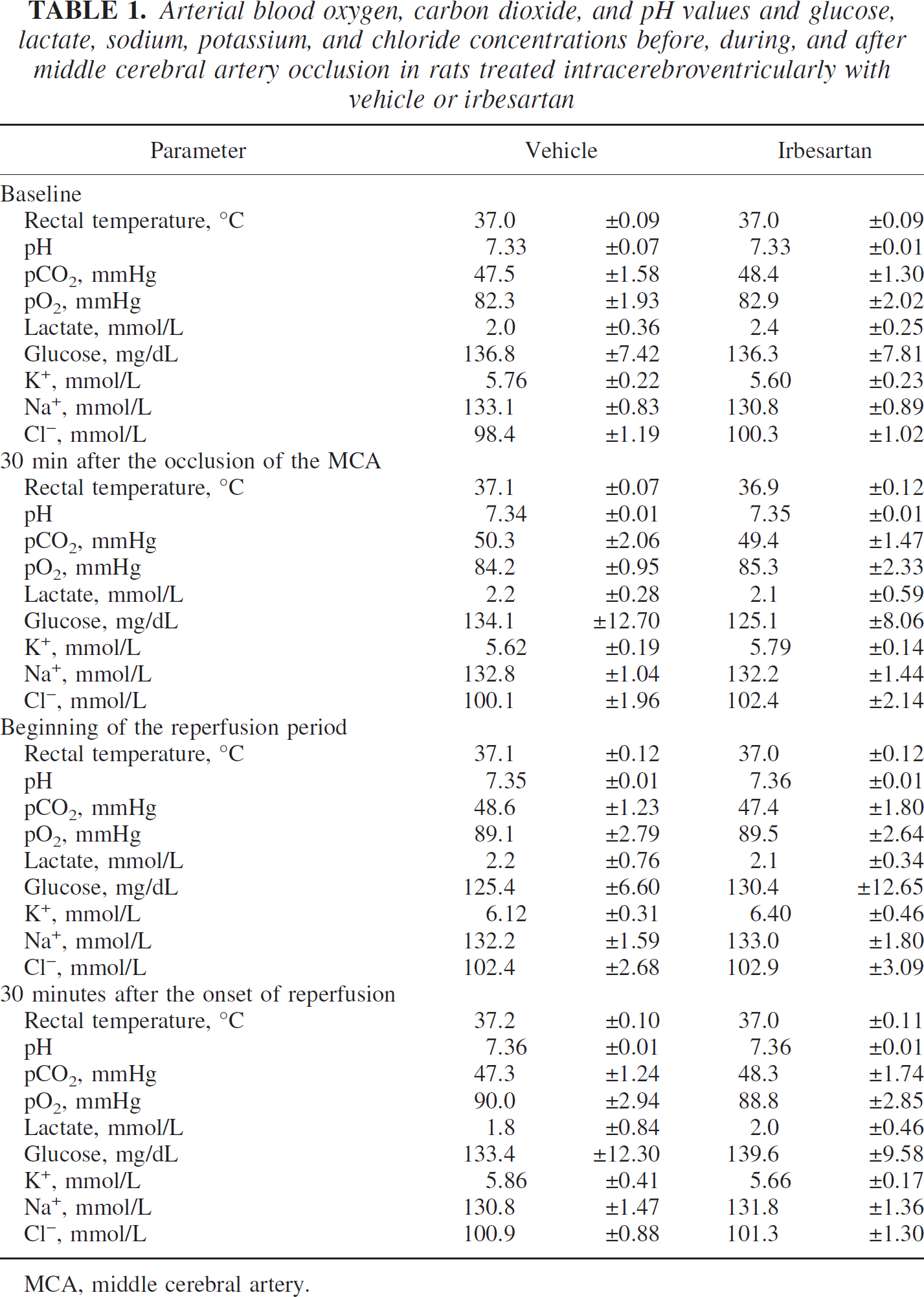
MCA, middle cerebral artery.
Regional cerebral blood flow
A reduction of rCBF to less than 25% of baseline indicates a successful occlusion of the middle cerebral artery; therefore, only rats with a reduction of rCBF greater than 75% were included into the study. Rats with subarachnoid hemorrhage were excluded from the protocol. rCBF reductions during MCAO and during reperfusion were identical in both groups of rats (Fig. 1). Contralateral rCBF fluctuated at approximately 100% of baseline during the ischemia period. After the withdrawal of the filament, ipsilateral blood flow was restored to approximately 70% to 80% of baseline in both groups of rats (Fig. 1).
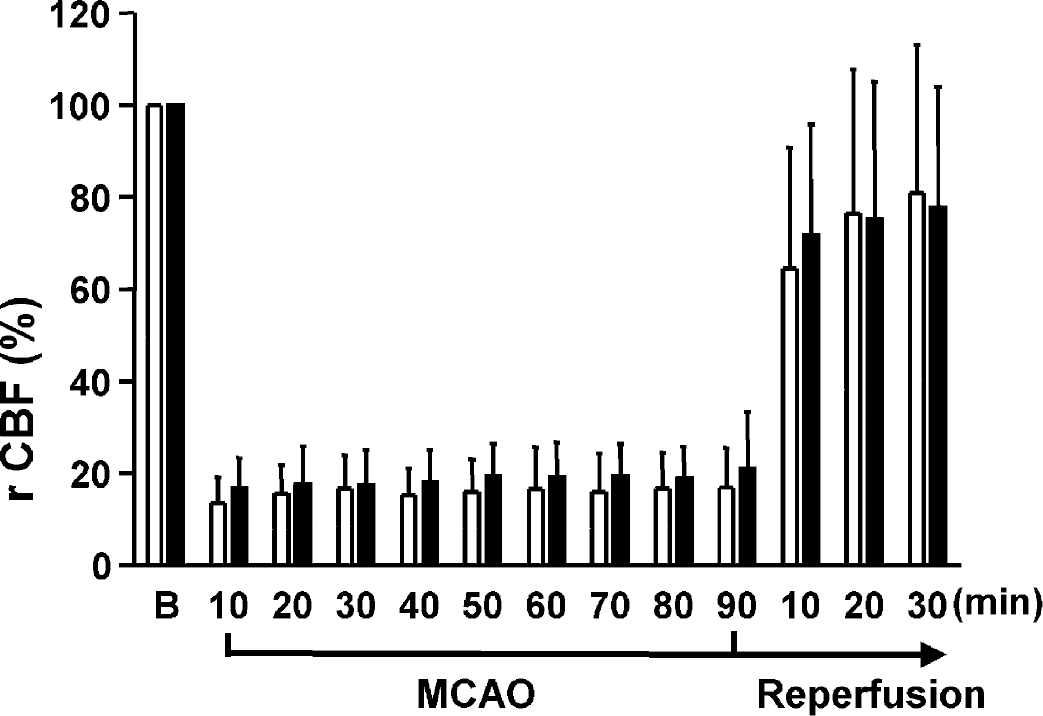
Changes in the rCBF in the zone of ischemia during occlusion of the middle cerebral artery for 90 minutes and during the reperfusion period in rats treated intracerebroventricularly with vehicle (empty columns) or irbesartan (solid columns) on 5 consecutive days before the induction of ischemia. CBF values (the means ± SD) are expressed as the percentage of baseline values recorded before occlusion of the middle cerebral artery. No significant differences in cerebral blood flow between the vehicle- group and the irbesartan-treated groups were detected. CBF, cerebral blood flow; rCBF, regional CBF.
Neurologic outcome after focal cerebral ischemia
The neurologic evaluation system developed by Bederson and colleagues (1986) tests motor impairments. ICV pretreatment with the AT1 receptor antagonist improved reflex and motor functions 24 hours after MCAO, as published previously (Dai et al., 1999). The significant difference in neurologic scores between the irbesartanand the vehicle-treated groups could be detected until the end of the experiment (F = 10.017; P>0.01). Surprisingly, no further improvement in motor functions was observed in the irbesartan-group in the post-ischemic period, although the treatment with the AT1 receptor antagonist continued without interruption. (Fig. 2). The assessment of neurologic impairments using the neurologic examination developed by Garcia and colleagues (1995) yielded different results. On the initial 2 days after MCAO, the neurologic outcomes of both groups of rats were not significantly different (day 1, P = 0.309; day 2, P = 0.051) (Fig. 2). Beginning on day 3, rats treated with irbesartan displayed less functional impairments than rats treated with vehicle (F = 9.088; P>0.01). The neurologic outcome of irbesartan-treated rats steadily improved, and the scores on days 6 and 7 were significantly better than the score on day 1 after MCAO (F6,105 = 2.2907; P>0.05) (Fig. 2). The observed discrepancies in the development of neurologic deficits obviously resulted from differences in the evaluation systems used for the assessment of neurologic impairments. The neurologic evaluation grading system developed by Bederson and colleagues (1986) tests simple reflex and motor impairments, whereas the one developed by Garcia (1995) quantifies, in addition to motor impairments, spontaneous activity and sensory functions. Therefore, we separately analyzed the evolution of reflex and motor functions (symmetry of the movement of four limbs and forepaw outstretching) and sensory deficits (body proprioception and vibrissae touch) after focal cerebral ischemia by the evaluation grading system developed by Garcia and colleagues (1995). An improvement of both motor deficits, the forepaw outstretching (F = 6.6310; P>0.05) and the symmetry of movement of four limbs (F = 17.260; P>0.001), was recorded in the irbesartan-treated group of rats, beginning on day 2 after MCAO and lasting over the whole observation period. In line with the results obtained with the neurologic evaluation grading system described by Bederson and colleagues (1986), post-ischemic treatment with irbesartan did not further improve motor functions (Fig. 3). Although the neurologic scores of sensory tests did not differ between the vehicle- and the irbesartan-treated groups over the time after MCAO (body proprioception, F = 0.015, P = 0.90; response to vibrissae touch, F = 0.167, P = 0.16), the neurologic scores of both sensory deficits steadily improved in rats treated with irbesartan (body proprioception, F6,105 = 4.210, P>0.001; vibrissae touch, F6,105 = 3.064, P>0.01). On days 6 and 7, these rats showed less sensory impairments than on the first day after focal cerebral ischemia. Sensory deficits in rats treated with vehicle remained unchanged during the entire observation period, and no improvement was recorded (Fig. 3).
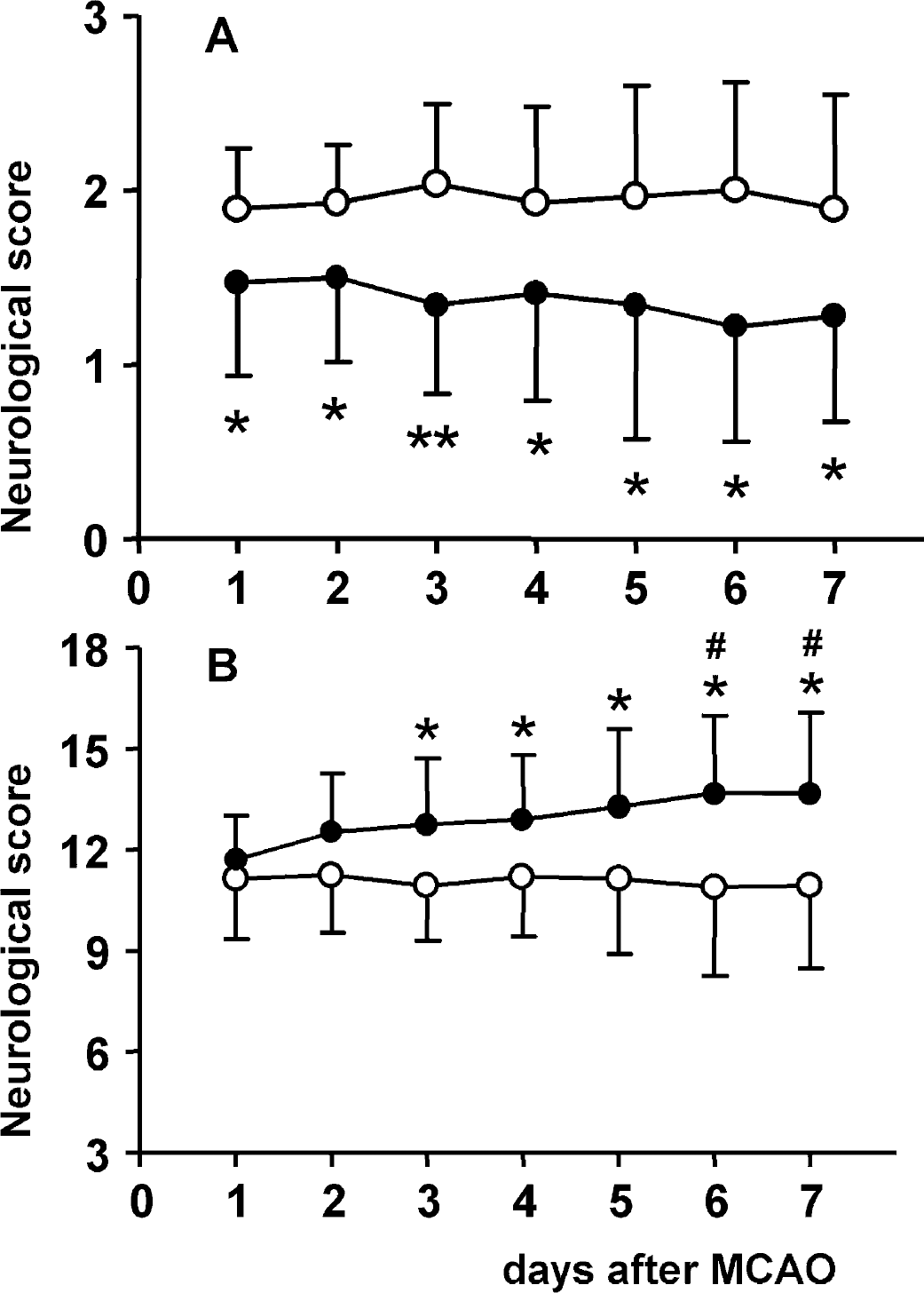
Neurologic scores in normotensive rats treated ICV with vehicle (empty circles, n = 14) or with irbesartan (2 nmol/h) (solid circles, n = 16). (A) Neurologic evaluation grading system 0–3 developed by Bederson and colleagues (1985). Rats displaying better neurologic outcomes receive lower neurologic scores. (B) Neurologic evaluation grading system 3–18 developed by Garcia and colleagues (1995). Rats with better neurologic outcomes receive higher neurologic scores. Neurologic scores are expressed as means ± SD . *P>0.05, **P>0.01 statistical comparisons with the vehicle-treated group at the given time point; #P>0.05 statistical comparison with day 1 after MCAO, calculated with repeated measures of analysis of variance followed by a post-hoc Bonferroni test. ICV, intracerebroventricularly; MCAO, middle cerebral artery occlusion.
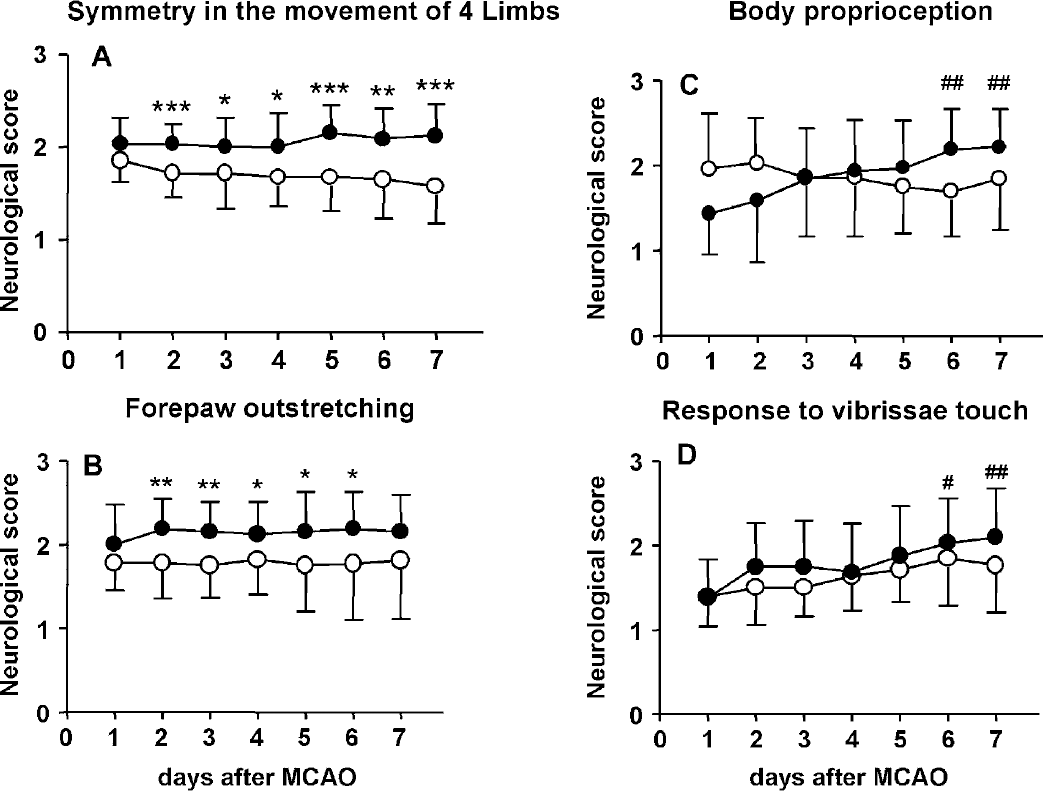
Neurologic scores in normotensive rats treated ICV with vehicle (empty circles, n = 14) or with irbesartan (2 nmol/h) (solid circles, n = 16). (A and B) Neurologic scores of motor deficits. (C and D) Neurologic scores of sensory deficits. Neurologic impairments were assessed by the evaluation grading system developed by Garcia and colleagues (1995). Rats with no observable neurologic deficits are graded 3, and severely affected rats are graded 0 or 1. Rats with better neurologic outcomes receive higher neurologic scores. Neurologic scores are expressed as means ± SD. *P>0.05, **P>0.01, ***P>0.001, statistical comparisons with the vehicle-treated group at the given time point; #P>0.05, #P>0.01, statistical comparison with day 1 after MCAO, calculated with repeated measures of analysis of variance followed by a post-hoc Bonferroni test. ICV, intracerebroventricularly; MCAO, middle cerebral artery occlusion.
Infarct volume
The size of the ischemic injury was determined by cresyl violet staining 3 and 7 days after MCAO with reperfusion. The cytoarchitecture of the cortex was normal in the contralateral hemisphere. Three days after focal cerebral ischemia, severe unilateral injury was clearly detected as area of pallor, which was sharply demarcated from the adjacent tissue. The ischemic lesion covered the territory of the middle cerebral artery, comprising parts of the frontoparietal and frontotemporal cortex and the lateral and medial segments of the caudate putamen (Fig. 4, upper panel A). Irbesartan treatment significantly reduced the infarct volume by approximately 42% (Fig. 4, upper panel B and lower panel). Seven days after transient MCAO, the infarct area consisted of cavities of different sizes, necrotic zones detected as area of pallor, and extensive, inhomogenous accumulations of cells throughout the ischemic core and peripheral regions. The accumulated cells often formed nodes with varying densities; only a few cells could be detected in the area of pallor (Fig. 4, upper panel). The sizes of ischemic lesions on days 3 and 7 after MCAO were approximately 200 mm3. The total volume of injury (cortical plus subcortical areas) in irbesartan-treated rats with either survival period was significantly reduced (F3,38 = 26.086, P>0.001) (Fig. 4, lower panel). Irbesartan also decreased brain edema 3 days after ischemic injury compared with vehicle-treated rats (vehicle, 45.20 ± 4.86 mm3; irbesartan, 20.44 ± 3.50 mm3; P>0.05).
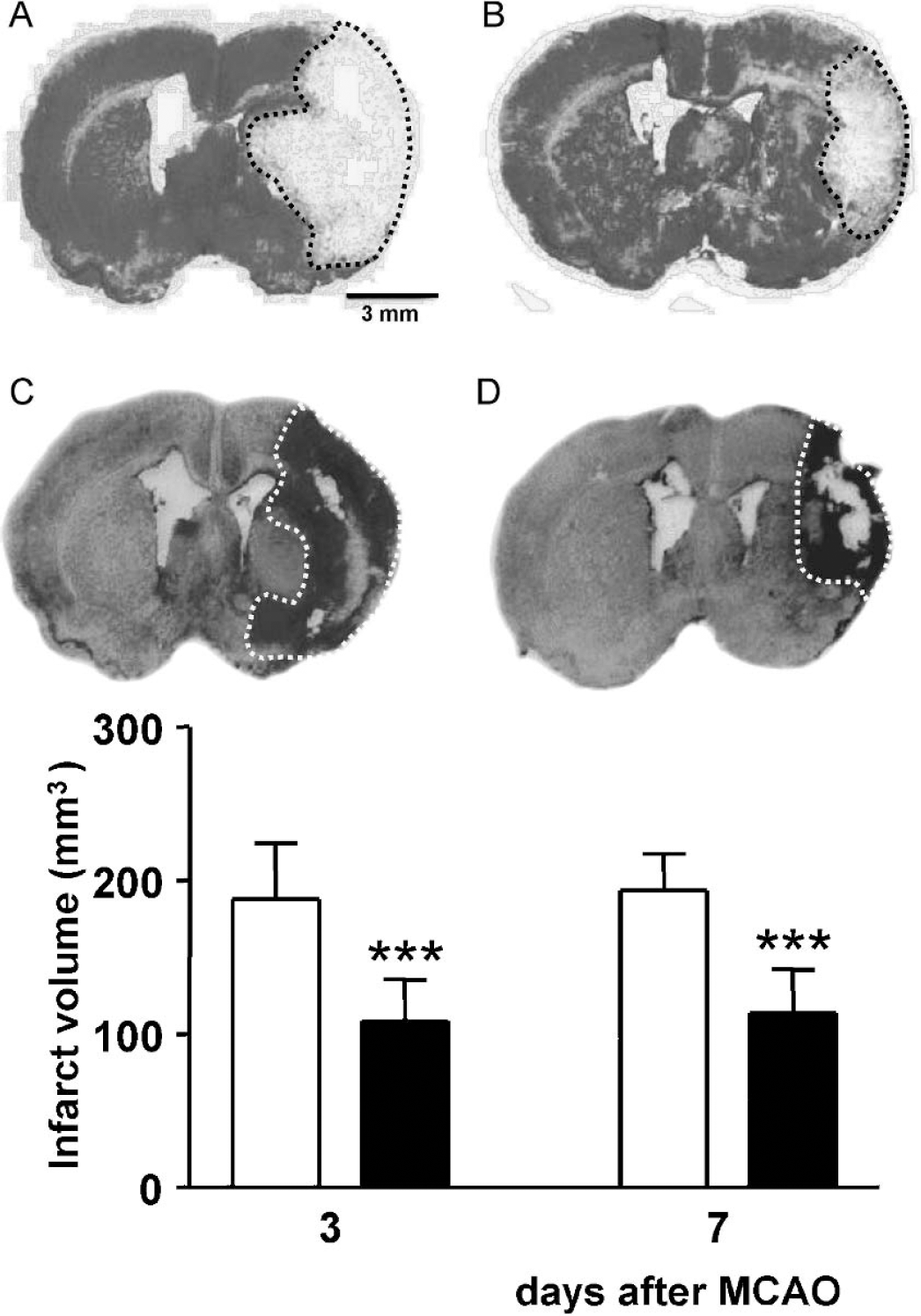
Upper panel: topography and size of cerebral infarct in rats exposed to MCAO with reperfusion. A (vehicle) and B (irbesartan): cerebral infarct after 90 minutes of MCAO/3 days of reperfusion. C (vehicle) and D (irbesartan): cerebral infarct after 90 minutes of MCAO/7 days of reperfusion. Figures represent typical serial 40-μm thick brain sections stained with cresyl violet. Lower panel: Effect of intracerebroventricular treatment with vehicle (empty columns) and irbesartan (solid columns) upon the size of the total infarct volume 3 days (vehicle: n = 10; irbesartan: n = 10) and 7 days (vehicle: n = 12; irbesartan: n = 10) after MCAO for 90 minutes. Results are expressed as means ± SD. Statistical comparisons to the vehicle-treated group: ***P>0.001, calculated with ANOVA followed by a post-hoc Bonferroni test. MCAO, middle cerebral artery occlusion.
Immunohistochemical detection of apoptosis
Apoptotic processes were evaluated using immunohistochemical staining for TUNEL and PARP p85 cleavage fragment. Three days after MCAO with reperfusion, TUNEL-positive cells were abundant throughout the peri-infarct areas of the parietal cortex. TUNEL staining was predominantly localized in cells displaying morphologic features of degenerating neurons. There were significantly fewer TUNEL-positive cells in rats treated with irbesartan than in those treated with vehicle (P>0.05) (Figs. 5 and 6). On day 7, TUNEL-positive cells were detected predominantly in areas infiltrated by inflammatory cells throughout the ischemic core. The density of TUNEL-positive cells in the boundary zone to the infarct core did not differ between the vehicle treated- and irbesartan-treated rats (Fig. 5). No TUNEL staining was observed in the contralateral hemisphere in any experimental group.
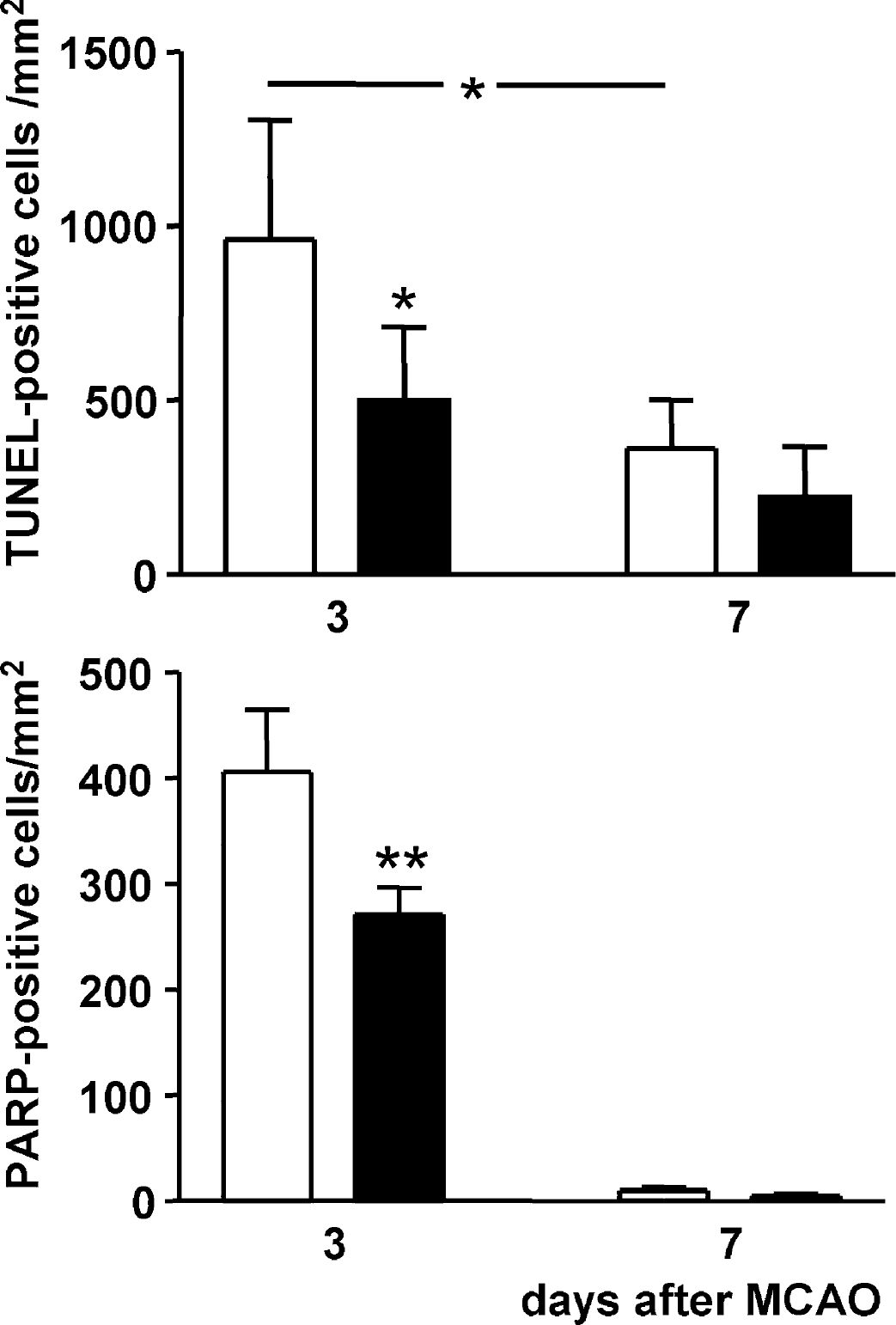
Effect of intracerebroventricular treatment with vehicle (empty columns, n = 5) and irbesartan (solid columns, n = 5) upon the number of cells that stained positive for TUNEL (upper panel) or for PARP P85 fragment (lower panel) in the parietal cortex ipsilateral to the occlusion, 3 and 7 days after MCAO for 90 minutes. Results are expressed as means ± SD. Statistical comparisons with the vehicle-treated group: *P>0.05; **P>0.01, calculated with ANOVA followed by a post-hoc Bonferroni test. MCAO, middle cerebral artery occlusion.
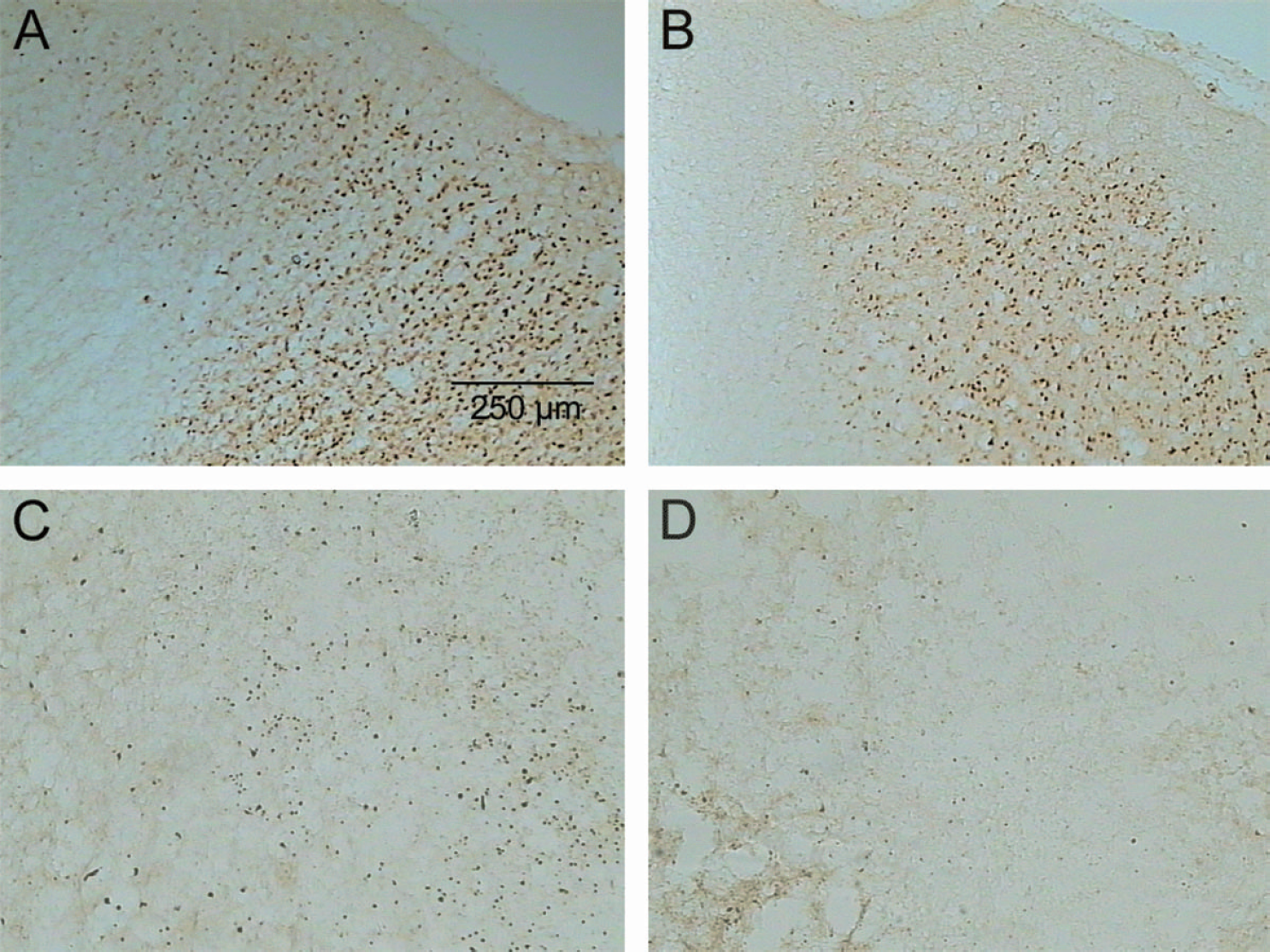
Immunohistochemical staining for TUNEL-positive cells in the parietal cortex ipsilateral to the occlusion of rats treated intracerebroventricularly with vehicle (A and C) and irbesartan (B and D), 3 days (A and B) and 7 days (C and D) after MCAO for 90 minutes. Irbesartan reduced the number of TUNEL-positive cells (B and D). MCAO, middle cerebral artery occlusion.
A large number of cells that stained positive for PARP p85 cleavage fragment appeared 3 days after ischemic injury. The density of PARP p85 cleavage fragment-positive cells in the peri-infarct parietal cortex was lower than that of TUNEL-positive cells. Morphometric analysis revealed a 33% reduction in the number of PARP p85 cleavage fragment-positive cells in the cortical tissue adjacent to the ischemic core in rats treated with irbesartan relative to rats treated with vehicle (Figs. 5 and 7).
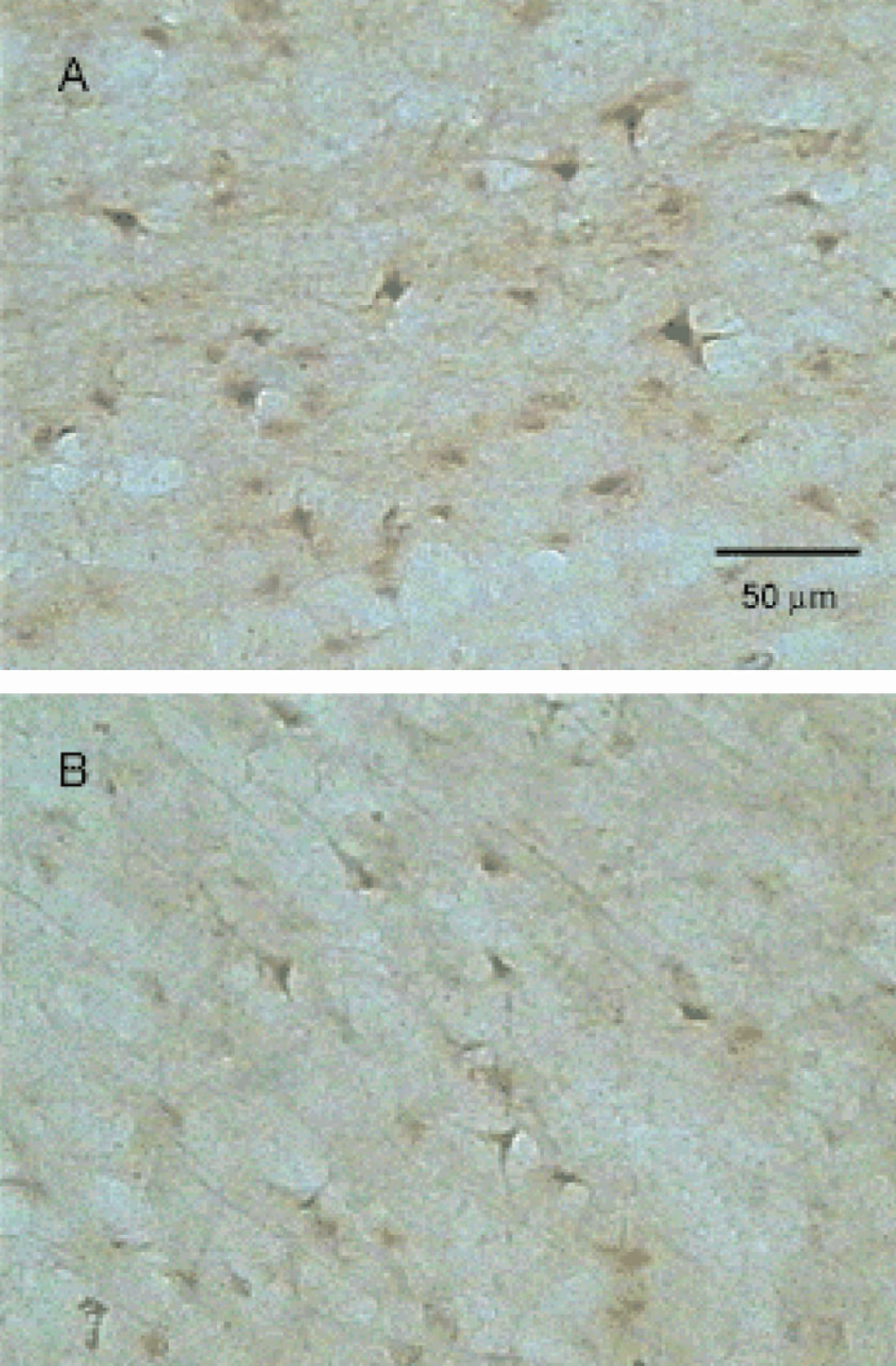
Immunohistochemical staining for PARP P85 fragment-positive cells in the parietal cortex ipsilateral to the occlusion of rats treated intracerebroventricularly with vehicle (A) and irbesartan (B) 3 days after MCAO for 90 minutes. The density and intensity of PARP P85 fragment-positive cells is lower in irbesartan-treated rats. MCAO, middle cerebral artery occlusion.
On day 7 after ischemic injury, only a few PARP p85-positive cells were found throughout the entire ischemic area (Fig. 5).
Irbesartan treatment significantly reduced the number of cells positively stained for TUNEL or PARP p85 cleavage fragment in the boundary zone of the infarct core 3 days after MCAO. Positively stained cells for TUNEL or PARP p85 fragment need not, however, exclusively represent apoptotic cells. Therefore, coronal sections obtained from an additional experiment were stained for activated caspase 3.
Activation of this execution caspase is termed as the hallmark of apoptosis (Huppertz et al., 1999). Three days after MCAO with reperfusion, the boundary zone of the infarct core in the parietal cortex revealed significantly fewer activated caspase 3-positive cells (−27%) in rats treated with irbesartan than in those treated with vehicle (t = 3.157; P>0.05) (Figs. 8 and 9).
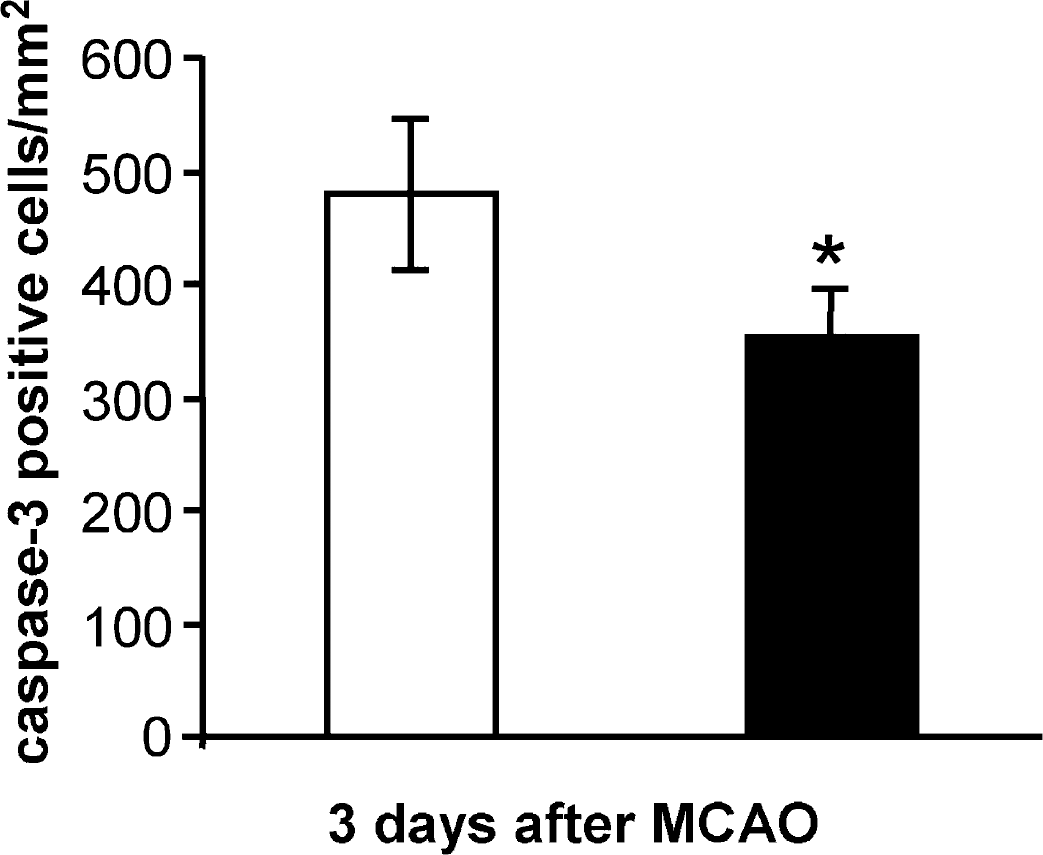
Effect of intracerebroventricular treatment with vehicle (empty columns, n = 4) and irbesartan (solid columns, n = 4) upon the number of cells positively stained for activated caspase 3 in the parietal cortex ipsilateral to the occlusion, 3 days after MCAO for 90 minutes. Results are expressed as means ± SD. Statistical comparisons with the vehicle-treated group: *P>0.05, calculated with Student's t-test for unpaired samples. MCAO, middle cerebral artery occlusion.
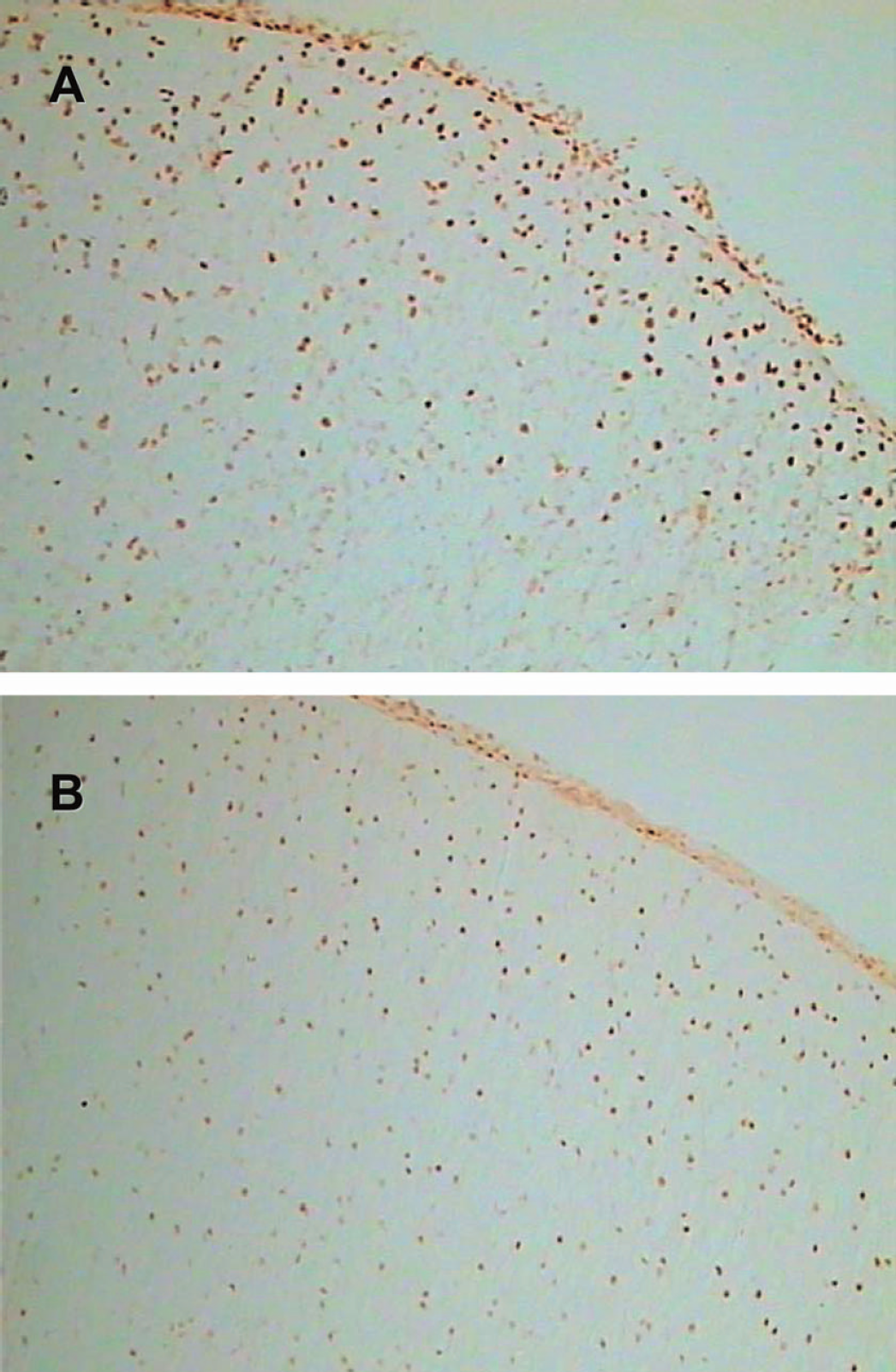
Immunohistochemical staining for activated caspase 3-positive cells in the parietal cortex ipsilateral to the occlusion of rats treated intracerebroventricularly with vehicle (A) and irbesartan (B) 3 days after MCAO for 90 minutes. Irbesartan reduced the number of cells positively stained for activated caspase 3. MCAO, middle cerebral artery occlusion.
Inflammatory response
Transient occlusion of the MCA induced an inflammatory response. Three days after MCAO with reperfusion, extensive accumulation of ED-1 positive cells (activated microglia and macrophages) occurred at the periphery of the ischemic core and, to a minor extent, in the ischemic core. Activated microglial cells of amoeboid type, identified by their enlarged size and stout processes, were most frequently observed in the boundary zone of the infarct (Fig. 10). Seven days after MCAO with reperfusion, a substantial number of ED-1 immunoreactive cells infiltrated the core and a large amount of these cells was seen in the whole ischemic area and the inner boundary zone of the cerebral cortex. Most of the ED-1 positive cells were localized in the areas that displayed a distinct degree of hypercellularity as evidenced in the corresponding section stained with cresyl violet. Compared with day 3 after MCAO, a marked increase in the numbers of reactive microglial cells and macrophages was also observed at the border of the infarct core in the vehicle-treated group of rats (P>0.01). A semiquantitative analysis of the numbers of ED-1 positive cells in this area is shown in Fig. 11. In both groups of rats treated with the AT1 receptor antagonist, the density of reactive microglial cells and macrophages in the penumbra of the parietal cortex was significantly reduced (F3,16 = 19,663, P>0.001).
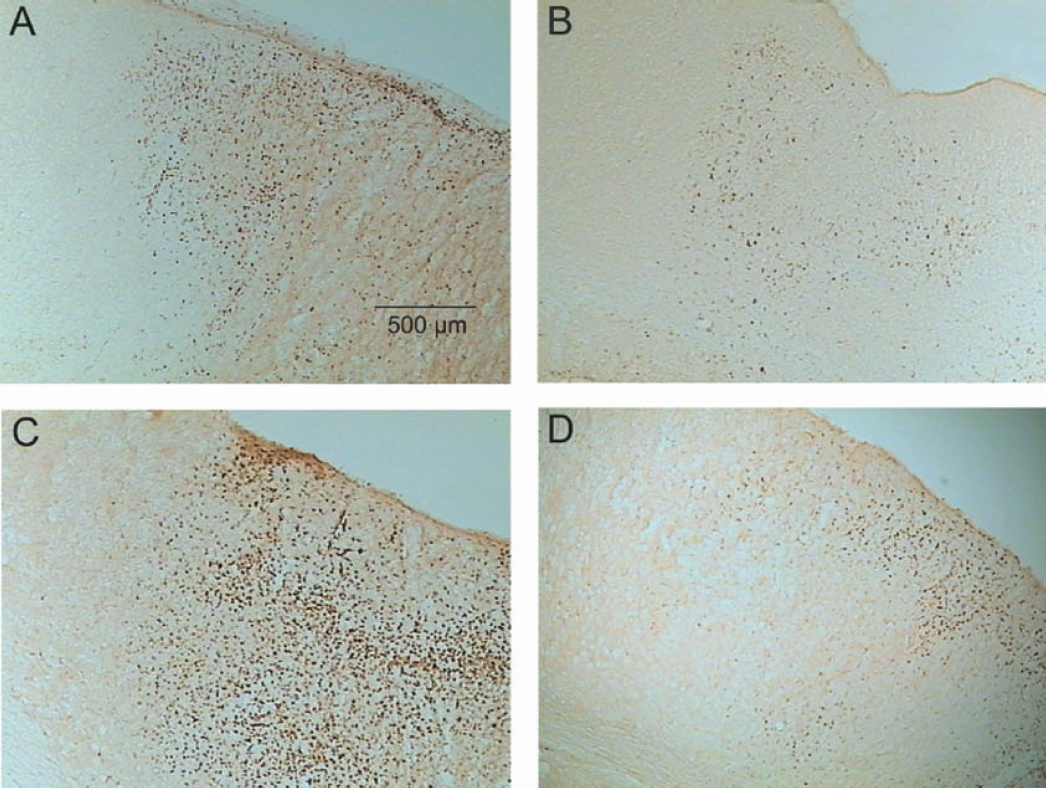
Immunohistochemical staining for ED-1-positive cells (activated microglia and macrophages) in the parietal cortex ipsilateral to the occlusion of rats treated intracerebroventricularly with vehicle (A and C) and irbesartan (B and D), 3 days (A and B) and 7 days (C and D) after MCAO for 90 minutes. The magnitude of ED-1 immunoreactivity is higher 7 days after MCAO than it is after 3 days. The density of ED-1-positive cells is lower in irbesartan-treated rats (B and D). MCAO, middle cerebral artery occlusion.
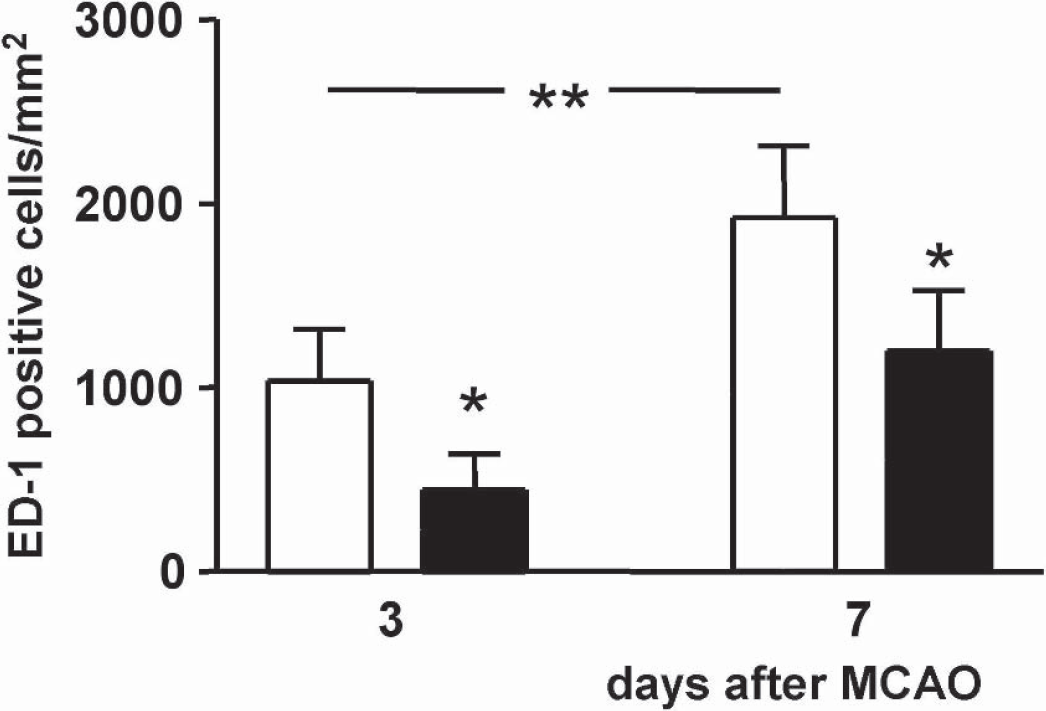
Effect of intracerebroventricular treatment with vehicle (empty columns, n = 5) and irbesartan (solid columns, n = 5) upon the number of activated microglia and macrophages (cells that stained positive for ED-1) in the parietal cortex ipsilateral to the occlusion, 3 and 7 days after MCAO for 90 minutes. The density of ED-1-positive cells is higher 7 days after MCAO than it is after 3 days. Results are expressed as means ± SD. Statistical comparisons with the vehicle-treated group: *P>0.05, calculated with ANOVA followed by a post-hoc Bonferroni test. MCAO, middle cerebral artery occlusion.
DISCUSSION
Beneficial effects of systemically administered AT1 receptor antagonists in ischemic stroke have been demonstrated in a number of animal studies. AT1 receptor antagonists can improve the recovery from ischemic stroke by the prevention of the decrease in blood flow in the marginal zone of ischemia because of increased capacity of cerebral arteries to dilate when cerebral blood flow decreases (Ito et al., 2002; Nishimura et al., 2000). However, AT1 receptor antagonists cross the blood-brain barrier and may improve the recovery from stroke by blocking the pathophysiologic processes and biochemical and metabolic events at the ischemic cascade level, such as the expression of various inducible transcription factors (Dai et al., 1999). Thus the beneficial effects of systemically administered AT1 receptor antagonists in stroke result from blockade of both peripheral and central AT1 receptors. In the present study, we sought to investigate the effect of a selective inhibition of brain AT1 receptors on post-ischemic inflammation and apoptosis. To achieve a steady-state blockade of brain AT1 receptors, the selective AT1 receptor antagonist irbesartan was continuously infused ICV over a 5-day period before and during 3 or 7 consecutive days after MCAO with reperfusion. We have reported previously that irbesartan infused ICV at the given dose interacts exclusively with central AT1 receptors because the antagonist did not affect basal mean arterial pressure nor did it reduce the blood pressure pressor response to a low dose of intravenously injected Ang II (Dai et al., 1999).
Chronic ICV infusion of irbesartan reduced infarct volume and improved neurologic outcome of focal cerebral ischemia. We report for the first time that long-term blockade of AT1 receptors in the brain inhibits apoptosis and the accumulation of reactive microglia and macrophages in the boundary zone of the infarct core. Unilateral occlusion of the MCA in rats induces necrosis mainly in the frontal sensorimotor cortices and in the caudal-putamen region. The ischemia-induced functional impairments comprise a wide range of motor and sensorimotor deficits, including partial paralysis, locomotor activity, and lack of coordination, which closely resemble those observed after focal occlusion of the MCA in humans (Hunter et al., 1998).
ICV pretreatment with the AT1 receptor antagonist improved only the recovery of motor functions, which were already better 24 hours (forelimb movements and resistance to lateral push) or 2 days (symmetry of movement of four limbs and forepaw outstretching) after MCAO and remained improved during the entire postischemic period. AT1 receptor blockade in the postischemic period did not produce any further benefit. In contrast to the motor impairments, pretreatment with the AT1 receptor antagonist before ischemia had no effect upon the grade of sensory impairments, such as body proprioception and vibrissae touch, but irbesartan treatment in the post-ischemic period improved the recovery of both sensory deficits. Obviously, the evolution of motor and sensory impairments after cerebral ischemia may considerably differ upon pharmacologic intervention (Hunter et al., 1998). The present findings stress the importance of a separate evaluation of motor and sensory functions in the assessment of the functional benefit of potential neuroprotective agents. Long-term blockade of central AT1 receptors before ischemic insult may considerably improve motor dysfunction after ischemic insult, which represents the major focus in the stroke rehabilitation. In addition, a sustained and long-term blockade of AT1 receptors in the brain after cerebral ischemia may reduce sensory deficits and alleviate cognitive and emotional impairments, as AT1 receptor antagonists have been demonstrated to improve cognitive function in hypertensive patients (Gard, 2002).
Long-term irbesartan treatment reduced the volume of brain infarct and tissue swelling after focal cerebral ischemia. The similar reductions in infarct volume on days 3 (−42%) and 7 (−41%) after MCAO indicate that blockade of brain AT1 receptors after day 3 did not provide any additional benefit with respect to the size of the ischemic injury. Nishimura and colleagues (2000) report upon a reduction in infarct size after temporal or permanent MCAO in SHR pretreated systemically with the high-affinity AT1 receptor antagonist candesartan. Cerebral vessels of these rats show hypertrophy and increased resistance, which may affect blood flow to ischemic tissue during MCAO. Pretreatment with candesartan protected SHR against cerebral ischemia by normalizing the cerebrovascular regulation and the reduction of the media thickness of the MCA, resulting in an increased arterial compliance and improvement of the collateral blood flow to the marginal zone of the ischemia (Ito et al., 2002; Nishimura et al., 2000). Such mechanisms cannot be implicated in the reduction of infarct size in our experimental setting because (1) cerebral arteries in normotensive rats have not undergone pathologic remodeling and can preserve cerebral blood flow at both high and low perfusion pressures, and (2) irbesartan infused ICV at the dose that was used did not interact with vascular AT1 receptors (Dai et al., 1999). Instead, the underlying mechanisms may involve an inhibition of Ca2+ entry into neuronal cells during ischemic insult. Overstimulation of glutamate receptors during hypoxia-ischemia leads to an excessive Na+ and Ca2+ entry into neurons resulting in cell body swelling and necrosis. In cultured cortical neurons, intense excitotoxic insults produced necrosis, whereas milder insults induced apoptosis (Choi, 1996). Stimulation of AT1 receptors also increases intracellular free Ca2+, which, together with the excessive, glutamate receptor-mediated Ca2+ entry, may augment excitotoxic necrosis. The finding of a much smaller infarct size but a larger penumbral area in AT1 receptor knock-out mice compared with wild-type mice after MCAO is in line with this assumption (Walther et al., 2002).
Apoptosis is the predominant mechanism of neuronal cell death at later time points after ischemic injury and, together with inflammation, promotes the gradual expansion of the ischemic lesion. Typical biochemical events occurring during apoptosis comprise, among others, the production of reactive oxygen radicals and peroxynitrite resulting in damage of proteins, lipids, activation of caspases known as the executioners of the apoptotic process, cleavage of poly (ADP-ribose) polymerase by activated caspase 3, and fragmentation and degradation of DNA (Huppertz et al., 1999; Mattson et al., 2000). To assess the effects of irbesartan therapy on apoptotic processes after focal cerebral ischemia, we used the TUNEL-assay and staining for PARP p85 cleavage fragment. Both methods reliably identify apoptotic cells (Huppertz et al., 1999).
The peri-infarct penumbra is the brain region in which most of the neuronal cells undergo apoptosis due to peri-infarct depolarization. Apoptosis occurs early, within minutes and hours after the onset of perfusion deficit, whereas inflammation processes damage neurons lethally within hours and days after ischemic injury. The penumbra and the ischemic core can be well defined at early time points after ischemic injury by measuring, for example, the protein synthesis and the regional tissue content of ATP. Suppression of protein synthesis and ATP depletion characterize the infarct core, whereas the penumbra can be defined as the region in which energy state is initially preserved but the protein synthesis is suppressed. Thus, the final size of infarct after transient focal cerebral ischemia corresponds to the region of early inhibition of protein synthesis (Hata et al., 2000a,b).
Because our experimental and methodologic approach does not allow for definition of the penumbra-region, cells that stained positively for apoptosis and inflammatory cells were quantified in the parietal cortex, on the border between the infarcted tissue and tissue with minimal morphologic signs of injury. Irbesartan treatment significantly reduced the number of cells that stained positively for TUNEL and PARP p85 cleavage fragment in the peri-infarct cortical region on day 3 after MCAO. On day 7 after ischemic injury, only a few PARP p85-positive cells were found throughout this cortical area. Immunohistochemical detection of the PARP p85 cleavage fragment is a more specific marker for apoptosis than the TUNEL-assay. PARP, an enzyme involved in DNA repair, is the best characterized substrate for the execution caspase 3. PARP is cleaved by caspase 3 to generate the characteristic 85 and 24 kDa fragments (Huppertz et al., 1999). In the present study, PARP was increasingly cleaved to its p85 cleavage fragment in the peri-infarct cortical region, but whether degradation of PARP is an event linked solely to apoptotic cell death remains to be elucidated (Chen et al., 1998; Paschen et al., 2000). On the contrary, activation of the execution caspase 3 indicates an irreversible progression of apoptotic cascade (Huppertz et al., 1999). A number of cells that stained positively for the activated caspase 3 were found in the cortical tissue adjacent to the infarct core. Consistent with the data obtained with staining for TUNEL and PARP p85 cleavage fragment, caspase 3- positive cells were decreased in irbesartan-treated rats on day 3 after MCAO. Our results clearly demonstrate that blockade of AT1 receptors in the brain significantly inhibited apoptosis events 3 days after ischemic insult; however, they do not allow for the exact characterization of the underlying mechanisms. Stimulation of neuronal AT1 receptors results in an increase in intracellular Ca2+ and activation of protein kinase C(de Gasparo et al., 2000). The AT1 receptor-mediated activation of protein kinase C has been reported to trigger apoptosis in endothelial cells (Li et al., 1999). Ang II acting upon AT1 receptors initiates apoptosis in neonatal myocytes via an elevation of cytosolic calcium and the stimulation of calcium-dependent endogenous endonuclease. Selective blockade of AT2 receptors failed to affect the AT1-receptor initiated myocyte apoptosis (Cigola et al., 1997). Increased levels of intracellular calcium also play a central role in the initiation of neuronal apoptosis (Mattson et al., 2000). Inhibition of the rise in intracellular Ca2+ levels may, therefore, represent one of the mechanisms that is responsible for the suppression of neuronal apoptosis after AT1 receptor blockade.
The antiapoptotic effects of the AT1 receptor antagonist may result from the activation of AT2 receptors in the brain. AT2 receptors have been associated with neuronal differentiation, regeneration, and tissue repair (for review see Culman et al., 2001). Kagiyama and colleagues (2003) report upon increased expression of AT2 receptors and levels of Ang II in the ipsilateral and contralateral ventral cortices after transient MCAO. When AT1 receptors are inhibited, Ang II can increasingly interact with AT2 receptors and initiate regeneration of neurons in the ischemic penumbra. On the other hand, activation of this receptor subtype can also promote apoptosis in neuronal tissue, especially when neurons become so severely damaged that they are beyond rescue. The proapoptotic events mediated by the AT2 receptor include activation of tyrosine phosphatases and inactivation of MAP kinase (ERK1/2), which results in Bcl-2 protein dephosphorylation (inactivation of an antiapoptotic factor) and upregulation of the proapoptotic Bax protein (Horiuchi et al., 1997, 1998). An attempt has been made to link the increased expression of AT2 receptors with excessive apoptosis leading to the loss of neurons in the ischemic tissue (Kagiyama et al., 2003). These findings exemplify that the role of AT2 receptors in neuronal apoptosis after cerebral ischemia is far from being understood.
In addition, the observed reduction of apoptosis need not be directly mediated by angiotensin receptors. Inhibition of AT1 receptors may alter the expression of a number of proinflammatory genes, resulting in suppression of inflammatory processes and, consequently, reduced apoptosis (see next sections).
Cerebral ischemia activates proinflammatory genes and the synthesis of mediators of inflammation in the ischemic tissue. Accumulating evidence has shown that post-ischemic inflammation leads to delayed loss of neurons and considerably contributes to the expansion of brain injury (Feuerstein et al., 1998). Injured brain cells produce various cytokines such as interleukin-1β and tumor necrosis factor α, which promote the transformation of quiescent microglia into reactive microglia and enhance the expression of adhesion molecules on the endothelial cells, resulting in the migration of granulocytes and macrophages into the ischemic brain parenchyma. Reactive microglia and invading macrophages represent the principal factors responsible for neuronal death in damaged neuronal tissue (Dirnagl et al., 1999; Mabuchi et al., 2000; Morioka et al., 1993). The present findings demonstrate that long-term treatment with irbesartan significantly inhibited the accumulation of reactive microglia and macrophages in the cortical tissue surrounding the ischemic core. According to findings noted by Dr. Blume (unpublished data, 2001), microglial cells in cell culture do not express angiotensin receptors. On the other hand, Ang II acting on AT1 receptors has been reported to upregulate peroxide production in macrophages and to increase oxidative stress (Yanagitani et al., 1999). In addition, Ang II increases lipooxigenase activity in macrophages through an AT1 receptor-mediated mechanism, and the peptide is also able to stimulate leukocyte migration (Elferink and de Koster, 1997; Scheidegger et al., 1997). The AT1 receptor antagonist-induced suppression of the accumulation of inflammatory cells may reduce the damage to potentially still-viable tissue and enhance functional recovery from stroke. This view is indirectly supported by data showing smaller infarct volume in neutropenic animals than in normal controls (for review see Pantoni et al., 1998).
CONCLUSION
We observe that long-term blockade of brain AT1 receptors before and after focal cerebral ischemia improves motor functions and alleviates sensory deficits. Inhibition of apoptosis and inflammatory processes in the ischemic tissue may substantially contribute to the reduction of infarct volume. The present findings demonstrate that inhibition of AT1 receptors in the brain can effectively reduce the magnitude of ischemic injury to the brain parenchyma and improve the recovery from ischemic insult.
Footnotes
Acknowledgment:
The authors thank Mr. Jan Brdon for his excellent technical assistance.