
Abstract
The levels of brain-derived neurotrophic factor (BDNF) vary between different forebrain areas and show region-specific changes after cerebral ischemia. The present study explores the possibility that the levels of endogenous BDNF determine the susceptibility to ischemic neuronal death. To block BDNF activity the authors used the TrkB-Fc fusion protein, which was infused intraventriculary in rats during 1 week before and 1 week after 5 or 30 minutes of global forebrain ischemia. Ischemic damage was quantified in the striatum and hippocampal formation after 1 week of reperfusion using immunocytochemistry and stereological procedures. After the 30-minute insult, there was a significantly lower number of surviving CA4 pyramidal neurons, neuropeptide Y -immunoreactive dentate hilar neurons, and choline acetyltransferase- and TrkA-positive, cholinergic striatal interneurons in the TrkB-Fc-infused rats as compared to controls. In contrast, the TrkB-Fc treatment did not influence survival of CA1 or CA3 pyramidal neurons or striatal projection neurons. Also, after the mild ischemic insult (5 minutes), neuronal death in the CA1 region was similar in the TrkB-Fc-treated and control groups. These results indicate that endogenous BDNF can protect certain neuronal populations against ischemic damage. It is conceivable, though, that efficient neuroprotection after brain insults is dependent not only on this factor but on the concerted action of a large number of neurotrophic molecules.
Brain-derived neurotrophic factor (BDNF) is widely distributed in the adult rodent brain with the highest levels in the hippocampal formation (Ernfors et al., 1990; Nawa et al., 1995), The synthesis of BDNF is regulated in an activity-dependent manner by physiologic stimuli, and its biological effects are mediated through the high-affinity receptor, TrkB. The BDNF protein is transported retrogradely but probably mainly anterogradely in central neurons and can be released in the terminal regions (Altar and DiStefano, 1998). Recent experimental evidence indicates that BDNF can have acute effects on synaptic transmission and may be involved in the regulation of neuronal plasticity (McAllister et al., 1999). Various insults to the brain, such as epileptic seizures, cerebral ischemia, hypoglycemic coma, and traumatic injury induce changes of BDNF mRNA and protein levels in cortical and hippocampal neurons (Lindvall et al., 1994; Kokaia et al., 1996). The increased synthesis of BDNF triggered by insults in certain neuronal populations has been proposed to be a protective response, counteracting cell death (Lindvall et al., 1994). In support of a neuroprotective role of BDNF, exogenous BDNF can mitigate insult-induced neuronal death after administration both in vitro and in vivo. BDNF added to cultured hippocampal, cortical, or striatal neurons protects against hypoglycemic damage (Cheng and Mattson, 1994; Kokaia et al., 1994; Nakao et al., 1995). Intraventricular administration of BDNF ameliorates damage of CA1 pyramidal neurons after global forebrain ischemia (Beck et al., 1994) and reduces infarct size after middle cerebral artery occlusion (Schabitz et al., 1997). Recently, Wu and Pardridge (1999) reported that intravenous injections of BDNF, pegylated to optimize plasma pharmacokinetics and conjugated with the OX26 antibody to enable transport through the blood-brain barrier, protect against CA1 neuronal death caused by transient forebrain ischemia.
Whether the levels of endogenous BDNF also can influence the susceptibility to neuronal death after brain insults is less clear. Arguing in favor of this idea, addition of specific antibodies against BDNF leads to enhanced neuronal death in embryonic cortical cell cultures, indicating that these neurons are dependent on endogenous BDNF for survival (Ghosh et al., 1994). Lindholm et al. (1996) have reported reduced survival of cultured hippocampal neurons prepared from BDNF knockout mice. Furthermore, the regional BDNF protein levels both under basal conditions and after global forebrain ischemia in rats broadly correspond to the susceptibility of different neuronal populations to ischemic damage (Kokaia et al., 1996). Finally, hyperglycemia and hypercapnia, which aggravate ischemic damage in the CA3 and cingulate cortex, suppress BDNF gene expression in these regions (Uchino et al., 1997).
The main objective of the present study was to explore the possibility that the regional levels of endogenous BDNF can influence the magnitude of neuronal death after global forebrain ischemia in rats. To inhibit BDNF activity, we used the TrkB-Fc immunoadhesin (Shelton et al., 1995; Croll et al., 1998), which is a fusion protein combining the extracellular binding domain of TrkB with the Fc domain of a human immunoglobulin G (IgG). The TrkB-Fc specifically blocks the biological activity of BDNF on cultured peripheral neurons (Shelton et al., 1995), and has been used as a scavenger of endogenous BDNF in electrophysiologic studies on hippocampal slices (McAllister et al., 1999). Supporting that TrkB-Fc counteracts BDNF activity also in vivo, intraventricular infusion of TrkB-Fc was found to suppress the development of kindling epilepsy (Binder et al., 1999), similar to what has been observed in BDNF knockout mice (Kokaia et al., 1995). Also, blockade of endogenous BDNF in the visual cortex by TrkB-Fc immunoadhesin inhibited the formation of ocular dominance columns (Cabelli et al., 1997). In the present study, rats received intraventricular infusion of TrkB-Fc for 1 week before and 1 week after global forebrain ischemia, induced during 5 or 30 minutes by bilateral carotid artery occlusion and hypotension (Smith et al., 1984). The ischemic damage was quantified after 1 week of reperfusion using immunocytochemistry and stereologic procedures.
MATERIALS AND METHODS
Animals and experimental design
Twenty-nine male Wistar rats (Møllegaard Breeding and Research Center, Ejby, Denmark) weighing 260 to 300 g were housed under 12 hours light / 12 hours dark conditions with ad libitum access to food and water. All animals were first implanted with a cannula connected to an osmotic minipump delivering either TrkB-Fc (n = 14) or the Fc region of a human IgG (Hu-Fc; n = 15). One week later, 15 rats were subjected to 30 minutes (seven treated with TrkB-Fc and eight with Hu-Fc) and 14 rats to 5 minutes of global forebrain ischemia (seven infused with TrkB-Fc and seven with Hu-Fc). Throughout the experiment (i.e., during the ischemia, staining and nonstereologic cell counting) the investigator was blind to whether TrkB-Fc or Hu-Fc had been infused in the individual animal. After 1 week of recirculation, animals were killed and ischemic neuronal damage was quantified.
Cannula implantation and TrkB-Fc infusion
The rats were anesthetized with Equithesin (3 mL/kg intraperitoneally) and a cannula (Alzet brain infusion kit; ALZA, Palo Alto, CA, U.S.A.) implanted in the right lateral ventricle (0.5 mm caudal to bregma, 1.2 mm lateral from midline, and 3.5 mm ventral from the skull). The cannula was connected to an Alzet osmotic minipump (model 2002: 200 μL; infusion rate 0.5 μL/h; ALZA) that contained either TrkB-Fc or Hu-Fc (1 mg and 0.34 mg/pump, respectively; TrkB-Fc and Hu-Fc were gifts from Regeneron Phamarceuticals, Tarrytown, NY, U.S.A.) in 0.1 mol/L phosphate-buffered saline (PBS; pH 7.0–7.4).
Procedure for ischemia
Before the ischemic insult, the animals were fasted overnight with free access to water. The rats were anesthetized by inhalation of 3.5% halothane (Zeneca, Gothenburg, Sweden) in N2O:O2 (7:3), intubated, and then artificially ventilated with 1% to 2% halothane in N2O:O2 (7:3). The tail artery was cannulated for blood sampling and pressure recording, a tail vein for drug infusion, and a rectally placed thermometer measured body temperature, which was maintained around 37°C by a heating pad. Loose ligatures were placed around the isolated common carotid arteries, the jugular vein was cannulated, and electrodes were placed in the skull muscles. After surgery, 50 IU of heparin were administered, the halothane concentration was decreased to 0.5%, and vecuronium bromide (Organon Teknika B.V., Boxtel, Holland) was infused intravenously at 2 mg/h as muscle relaxant. A steady state period of 30 minutes followed during which physiologic parameters and electroencephalogram were monitored. Ischemia was induced by bilateral occlusion of the common carotid arteries, combined with hypotension (arterial blood pressure 40 to 50 mm Hg) evoked by withdrawal of 4 to 7 mL blood from the jugular vein with a 10-mL syringe (Smith et al., 1984). Circulation was restored after 5 or 30 minutes by reinfusion of the previously withdrawn blood and removal of the occluding claps. In the immediate recirculation period, sodium bicarbonate (0.5 mL intravenously, 50 mg/mL) was given to prevent systemic acidosis. After regaining spontaneous respiration, the animals were extubated. All animals had physiologic parameters within predetermined ranges and no significant differences were observed between the TrkB-Fc- and Hu-Fc-treated groups (Table 1).
Physiologic parameters in rats subjected to forebrain ischemia
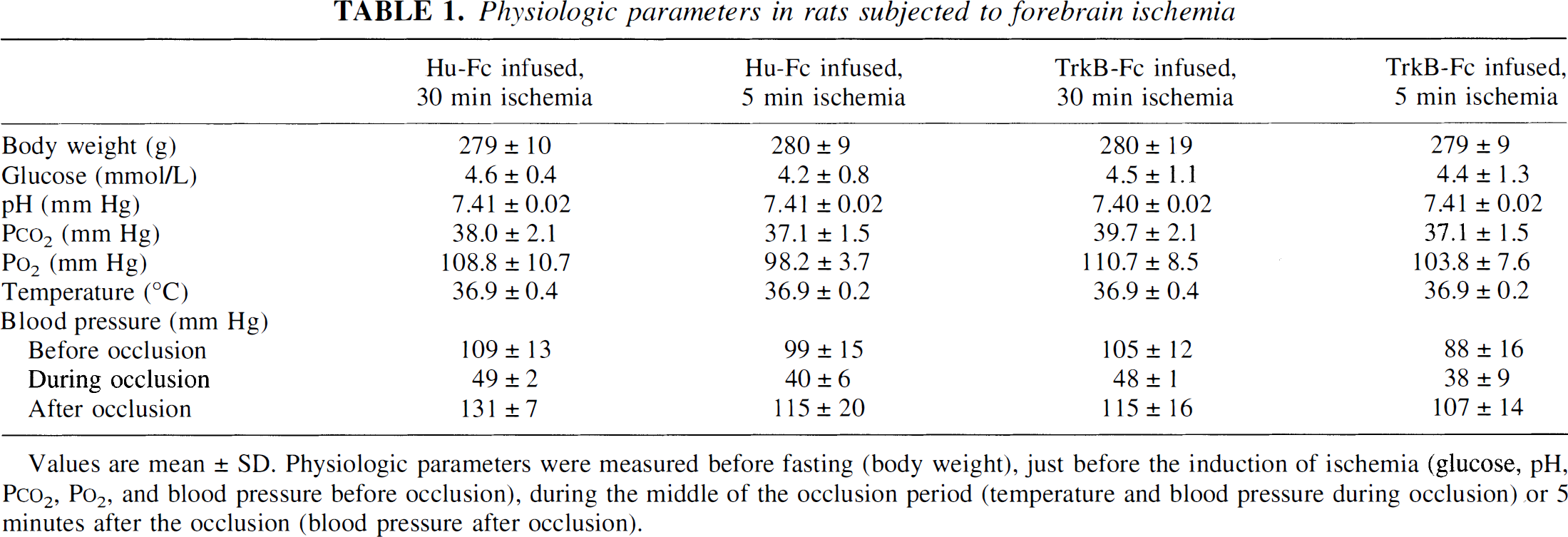
Values are mean ± SD. Physiologic parameters were measured before fasting (body weight), just before the induction of ischemia (glucose, pH, Pco2, Po2, and blood pressure before occlusion), during the middle of the occlusion period (temperature and blood pressure during occlusion) or 5 minutes after the occlusion (blood pressure after occlusion).
Immunocytochemistry
Animals were deeply anesthetized with chloral hydrate (400 mg/kg intraperitoneally) and transcardially perfused with 0.9% NaCl followed by ice-cold 4% paraformaldehyde in 0.1 mol/L PBS. The brains were removed and postfixed in the same fixative overnight before being transferred to a 20% sucrose solution in PBS. All brains were cut in 40-μm thick coronal sections on a freezing microtome and stored in a cryoprotectant solution at −20°C. Immunocytochemical staining was performed on free-floating sections using antibodies against neuronal-specific antigen (NeuN; 1:100; Chemicon), dopamine-and cyclic AMP-regulated phosphoprotein with a molecular weight of 32 kd (DARPP-32; 1:20000; a gift from Dr. P. Greengard, Rockefeller University, New York, NY, U.S.A.), choline acetyltransferase (ChAT; 1:750; Chemicon), TrkA ectodomain (TrkA; 1:5000; a gift from Dr L. F. Reichardt, University of California, San Francisco, CA, U.S.A.), Fc-specific human IgG (Hu-IgG; 1:10000; Sigma), neuropeptide Y (NPY; 1:1000; Incstar, Stillwater, MN, U.S.A.) and parvalbumin (1:1000; a gift from Dr. P. Emson, The Babraham Institute, Cambridge, U.K.). Briefly, sections were rinsed and endogenous peroxidase activity was blocked by 3% hydrogen peroxide and 10% methanol in potassium-phosphate buffered saline (KPBS; pH 7.0–7.4), followed by preincubation in 5% appropriate serum in 0.25% Triton X-100 in KPBS (T-KPBS) for at least 1 hour. The sections were then incubated overnight (for NeuN, DARPP-32, TrkA, Hu-IgG, NPY and parvalbumin staining) or 68 hours (for ChAT staining) with the primary antibody in 1 % serum in T-KPBS in 4°C (for NeuN, DARPP-32, ChAT, TrkA and Hu-IgG staining) or room temperature (for NPY and parvalbumin staining). After rinsing, the sections were incubated with the appropriate biotinylated secondary antibody (horse anti-mouse, 1:200, Vector Laboratories, Burlingame, CA, U.S.A., for NeuN, DARPP-32 and ChAT staining; rabbit anti-goat, 1:200, Vector Laboratories for Hu-IgG staining; and goat anti-rabbit, 1:200, Vector Laboratories for TrkA, NPY and parvalbumin staining) in 2% serum in T-KPBS for 90 minutes. The sections were then rinsed and incubated in avidinbiotin-peroxidase complex (Elite ABC kit, Vector Laboratories) for 1 hour, followed by reaction with diaminobenzidine (0.5 mg/mL) and hydrogen peroxide (including nickel chloride for intensification of staining for NeuN, DARPP-32, ChAT, TrkA and Hu-IgG staining).
Fluoro-Jade staining
Sections from perfused brains (see above) were washed three times during 10 minutes in KPBS, mounted on Super plus slides and dried overnight. The slides were immersed in absolute ethanol for 3 minutes followed by I minute in 70% ethanol and 1 minute in distilled water. After incubation in 0.06% potassium permanganate for 15 minutes, the slides were rinsed in distilled water for 1 minute and transferred to the Fluoro-Jade solution (10 mL 0.01 % Fluoro-Jade (Histo-Chem Inc., Jefferson, AR, U.S.A.) in 90 mL 0.1 % acetic acid), which was agitated for 30 minutes. After three 1-minute washes, the slides were dried, immersed in xylene for 10 minutes, and cover-slipped.
Cell counting
Stereo logic analyses were performed according to the optical fractionator method using Olympus BH-2 microscope, CCD-IRIS color video camera and CAST-GRID software (Olympus, Albertslund, Denmark). The number of DARPP-32-, ChAT-and TrkA-positive neurons in the striatum was quantified at six coronal levels located 1.5, 0.7, and 0.2 mm anterior and 0.3, 0.8, and 1.3 mm posterior to bregma, respectively, according to the atlas of Paxinos and Watson (1997). The striatum was divided into three areas (dorsolateral, dorsomedial, and ventral) with the help of a grid system using 1.25 × objective, and counting was then performed with 40 × objective for DARPP-32 staining and 10x objective for ChAT and TrkA staining. The number of NeuN-positive neurons in the CA1, CA3, and CA4 pyramidal layers of the hippocampus was quantified at three or four levels located between 2.8 and 4.2 mm posterior to bregma using 100× objective. Total numbers of DARPP-32- and NeuN-positive neurons were calculated using the optical fractionator formula (West et al., 1991). Also ChAT- and TrkA-positive neurons were counted using the optical fractionator method. However, the numbers of these cells were too low to allow for the use of the fractionator formula to calculate total cell number (Gundersen and Jensen, 1987). Therefore, the counting of ChAT- and TrkA-positive neurons in both TrkB-Fc- and Hu-Fc-treated groups were performed with the same stereologic settings. Because neither the area for counting nor the thickness of the analyzed sections (measured using the CAST-GRID software) differed between the groups (data not shown), the numbers of cells counted in the different areas could be compared. The low numbers of parvalbumin- and NPY-positive neurons in the dentate gyrus hilus made stereologic analysis impossible. These neurons were instead counted manually at four levels, located between 2.8 and 4.2 mm posterior to bregma, using 20× objective and the numbers for each animal were added together.
Statistical analysis
Evaluation of differences in physiologic parameters or number of immunopositive cells between groups was performed using unpaired t-test.
RESULTS
Distribution of ischemic damage and penetration of TrkB-Fc
To determine whether blockade of BDNF activity can influence neuronal death after global cerebral ischemia, cell counting was performed in those forebrain regions which showed both ischemic damage and penetration of TrkB-Fc or Hu-Fc. Using Fluoro-Jade, a marker for degenerating neurons (Schmued et al., 1997), we observed that at 1 week after the 30-minute ischemic insult there were many stained neurons in the dorsal and dorsolateral striatum, and more caudally, also, in the medial striatum, whereas only single neurons were stained in more ventral areas. The thalamic reticular nucleus contained many Fluoro-lade-positive neurons and in the piriform cortex and olfactory tubercle clusters of degenerating neurons were observed. In the hippocampal formation, the majority of neurons in the subiculum and the CA1 pyramidal layer were stained, while only few neurons were found in the CA3 and CA4 pyramidal layers and in the dentate gyrus hilus. Scattered degenerating neurons were detected in the parietal, cingulate, and retrosplenial cortices.
The penetration of TrkB-Fc and Hu-Fc into the brain parenchyma from the infusion site in the right lateral ventricle was analyzed using immunocytochemistry with a Fc-specific anti-human IgG antibody. Figure 1B shows the distribution of TrkB-Fc and control Hu-Fc as observed at the same two coronal levels in rats subjected to 30 minutes of ischemia. Both TrkB-Fc and Hu-Fc were detected bilaterally in the dorsomedial striatum. The staining on the right side (i.e., ipsilateral to the infusion) was more intense and had a wider distribution, reaching also the dorsolateral striatum. In the more ventral part of the striatum, the staining was variable. The septal region was intensely stained. Both TrkB-Fc and Hu-Fc were detected in the rostral part of the hippocampus, in most rats bilaterally, but with a higher staining intensity and wider distribution on the right side (Fig. 1A). There was also penetration of TrkB-Fc and Hu-Fc into the cerebral cortex, mostly in the cingulate and frontal cortices close to the cannula tract. The distributional pattern of TrkB-Fc and Hu-Fc was similar in the rats subjected to 5-minute ischemia as observed in the 30-minute group (data not shown). Two of the Hu-Fc-treated rats subjected to 30-minute ischemia were excluded from further analysis due to lack of Hu-IgG staining in the brain parenchyma, probably caused by misplacement of the cannula. These animals are not shown in Fig. 1B. In one rat from the TrkB-Fc-infused group subjected to 30 minutes of ischemia, cells were only counted in the striatum (shown in Fig. IB) due to poor Hu-IgG staining in the hippocampus.
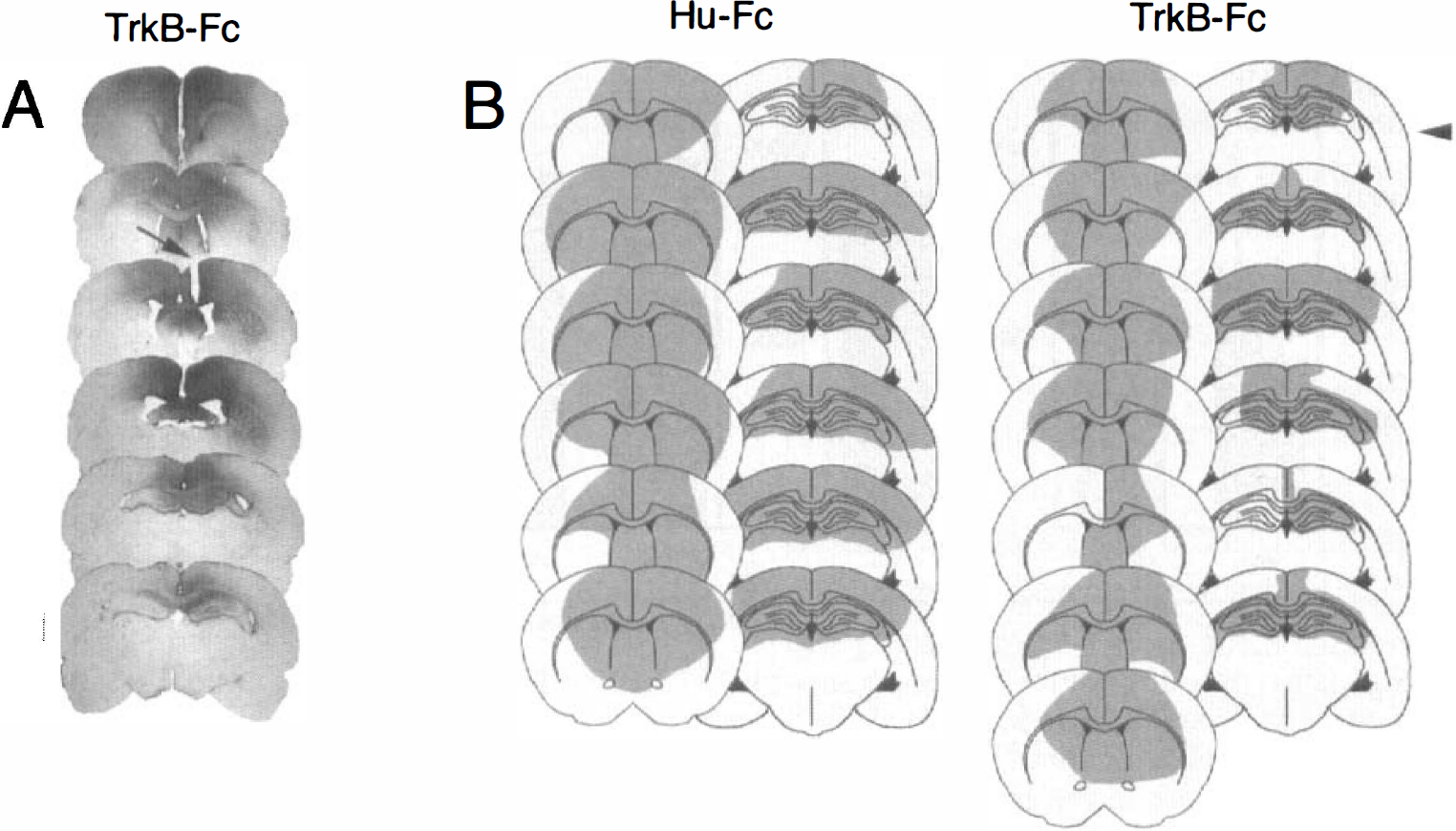
Penetration of Hu-Fc and TrkB-Fc as assessed by Hu-lgG immunostaining in rats subjected to 30 minutes of ischemia.
Quantification of ischemic damage after TrkB-Fc infusion
The number of neurons in the CA1 and CA3 pyramidal layers was counted using stereologic procedures in sections stained for the neuron-specific marker NeuN. In animals subjected to 30 minutes of ischemia, there was no difference in the number of CA1 pyramidal cells between TrkB-Fc- and Hu-Fc-treated groups, either ipsilateral (Figs. 2 and 3) or contralateral to the infusion (Fig. 2). We speculated that the lack of aggravation of damage in the CA1 region by the TrkB-Fc infusion in the 30-minute ischemia group might be due to that the insult-induced neuronal loss was already close to maximal and further enhancement would not be possible to detect. To test this hypothesis, we subjected animals to 5 minutes of forebrain ischemia which leads to modest loss of CA1 pyramidal neurons (unpublished observation). As shown in Fig. 2, the number of surviving neurons in the CA1 pyramidal layer was about 12-fold higher after 5 minutes as compared to 30 minutes of ischemia. However, also after this mild ischemic insult, no difference in CAl cell numbers was detected between TrkB-Fc- and Hu-Fc-treated rats. The number of CA3 pyramidal neurons, which were counted only ipsilateral to the infusion due to lack of consistent Fc-immunoreactivity contralaterally, was also unchanged in the TrkB-Fc- as compared to the Hu-Fc-infused animals (Fig. 2).
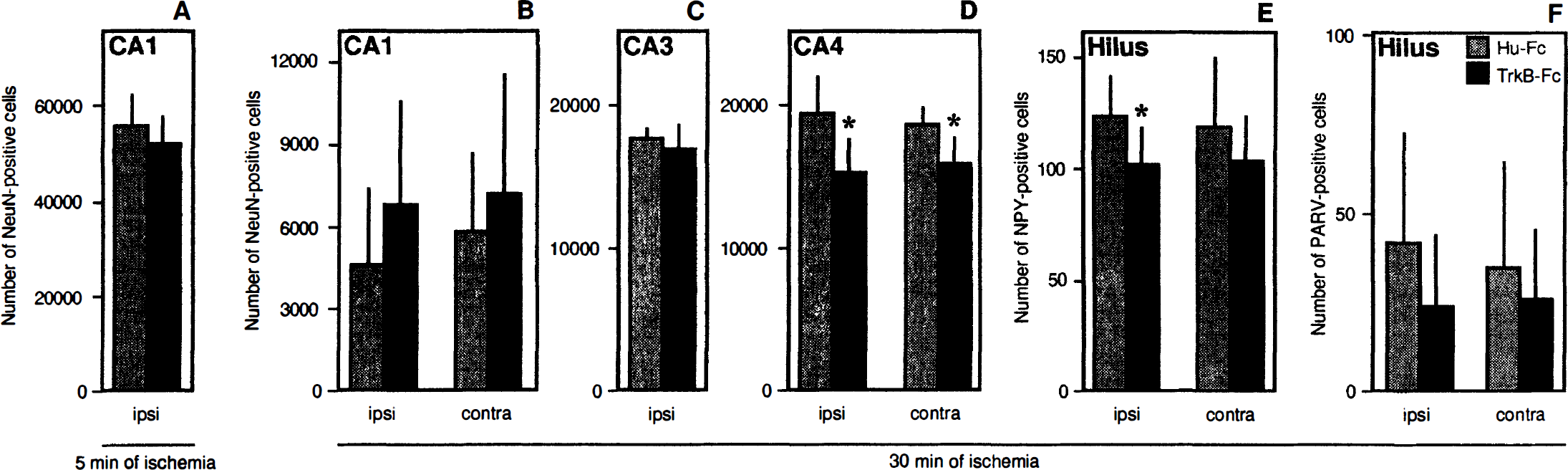
Number of NeuN-positive neurons within the hippocampal CA1 (
The number of NeuN-positive CA4 pyramidal neurons and NPY- and parvalbumin-immunoreactive interneurons was quantified in the dentate gyrus hilus (Fig. 2). One rat from the TrkB-Fc group was not analyzed on the side contralateral to the infusion due to poor penetration in the dentate gyrus hilar region. After 30 minutes of ischemia, the TrkB-Fc-treated group had significantly fewer NeuN-positive neurons as compared to the Hu-Fc-treated group in the CA4 pyramidal layer on both sides (21 % and 15% reduction ipsilateral and contralateral to the infusion, respectively) (Figs. 2 and 3). Also, the number of NPY-immunopositive neurons was significantly lower (about 18%) in the TrkB-Fc-treated group ipsilateral to the infusion (Fig. 2). There was a similar trend for a reduction of parvalbumin-immunoreactive hilar neurons, but the difference between TrkB-Fc- and Hu-Fc-infused rats was not statistically significant.
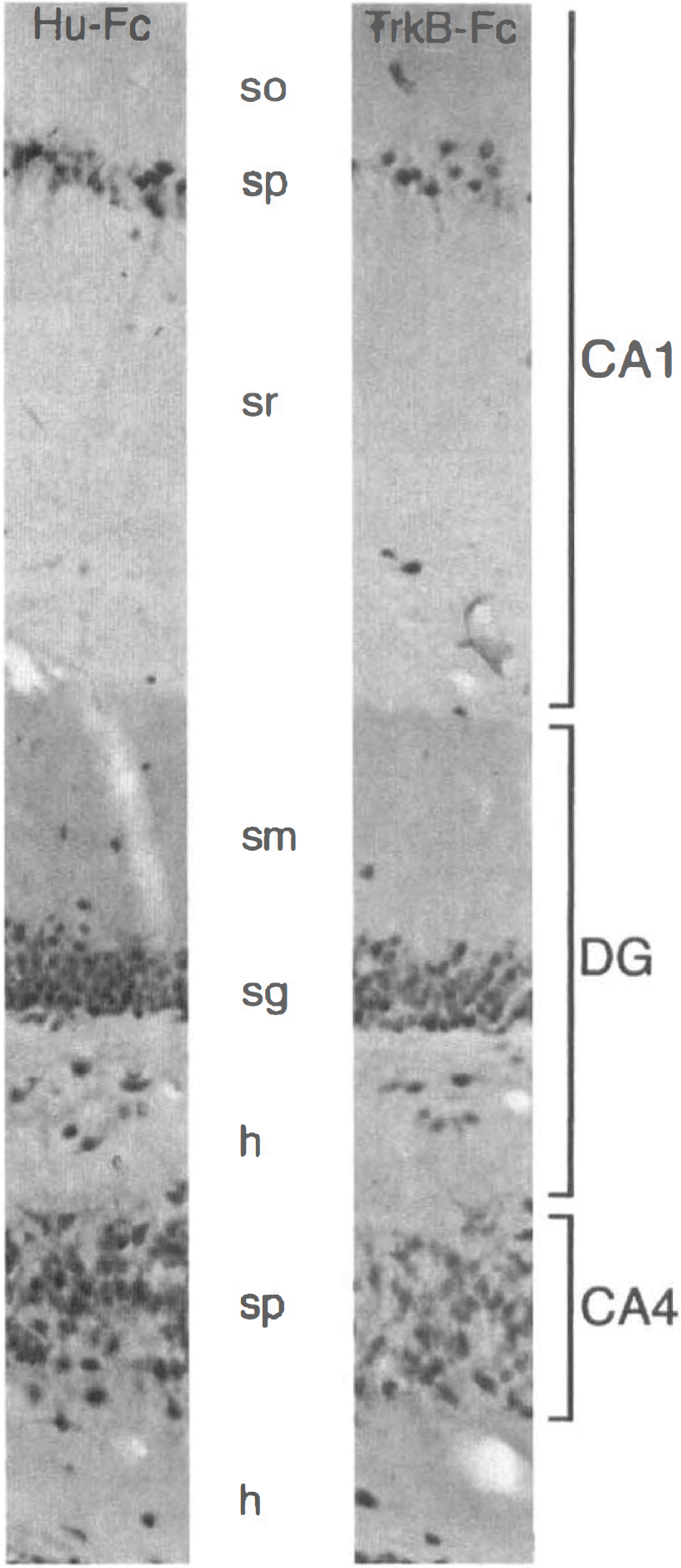
Photomicrographs showing neuron-specific NeuN staining in the hippocampal formation, ipsilateral to the infusion of Hu-Fc and TrkB-Fc, respectively, at 1 week after 30 minutes of ischemia. Note the similar loss of neurons in the CA1 pyramidal layer, and the more pronounced reduction of neurons in the CA4 pyramidal layer and in the dentate gyrus hilus of the TrkB-Fc rat as compared to the Hu-Fc rat. so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; sm, stratum moleculare; sg, stratum granulosum; h, hilus.
In the striatum, we first quantified the number of projection neurons, identified by their immunoreactivity to DARPP-32, in the dorsolateral, dorsomedial, and ventral parts as defined in Fig. 4D. These medium-sized spiny neurons are GABAergic and constitute more than 90% of all neurons in the striatum. The projection neurons in the dorsolateral part are the most sensitive ones to ischemic damage (Chesselet et al., 1990; Kokaia et al., 1998), and, in agreement there was a lack of DARPP-32-stained neurons in this area after the 30-minute ischemic insult. As shown in Fig. 4, no significant difference between the TrkB-Fc- and Hu-Fc-treated groups in the number of DARPP-32-positive neurons could be detected in any striatal region either ipsilateral or contralateral to the infusion.
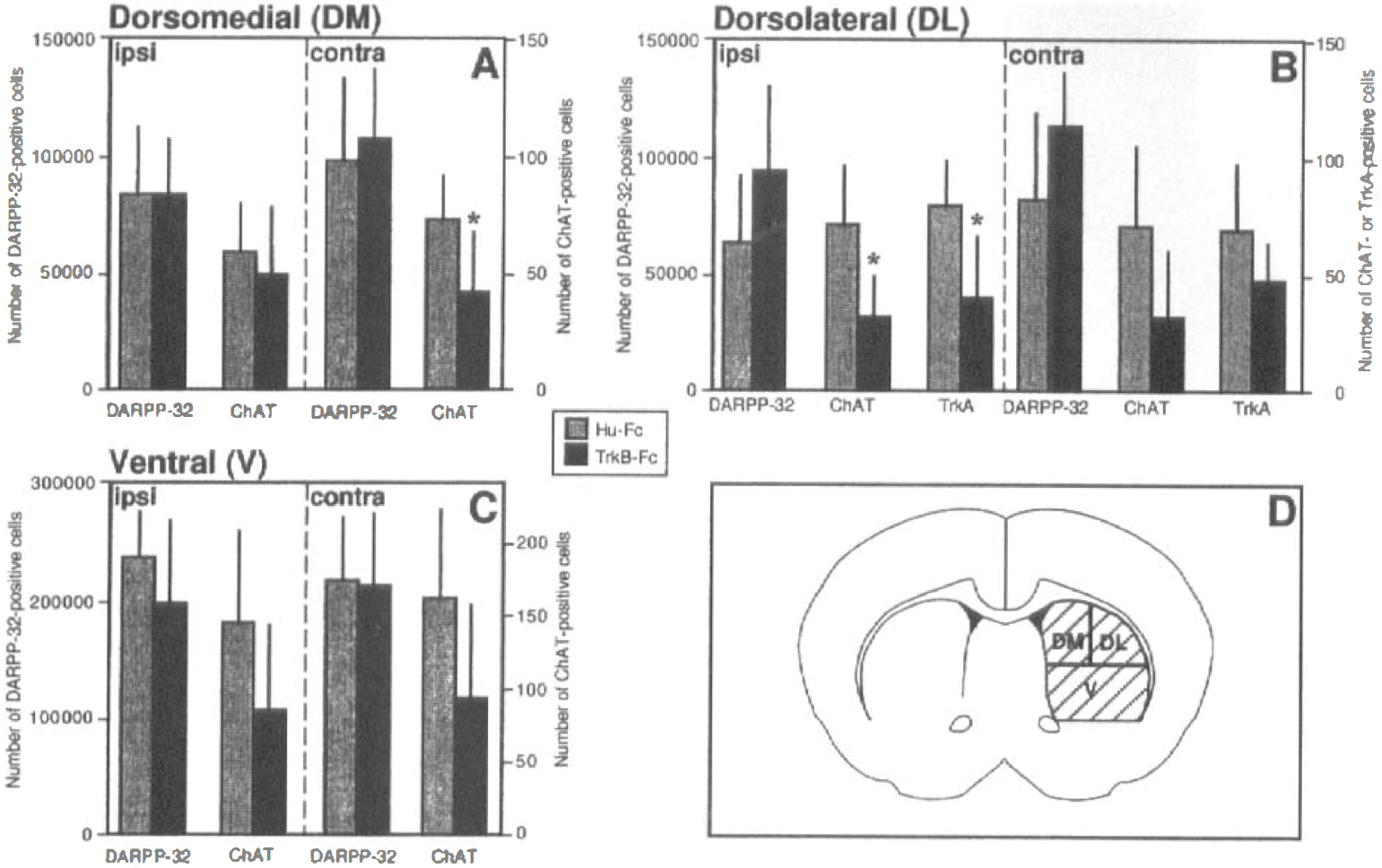
Number of OARPP-32-, ChAT-, and TrkA-immunopositive neurons in the dorsomedial (DM;
In contrast to the projection neurons, striatal cholinergic interneurons are resistant to ischemic damage (Chesselet et al., 1990; Kokaia et al., 1998). We counted the number of cholinergic neurons, stained using a ChAT antibody, in the different parts of the striatum (Fig. 4). In the Hu-Fc-treated rats, the number and distribution of ChAT-immunoreactive neurons agreed closely with previous reports (Kokaia et al., 1998). The TrkB-Fc-infused animals, on the other hand, showed a marked reduction of the number of cholinergic neurons (Figs. 4 and 5). There was a clear trend in all counted areas but the most dramatic, statistically significant cell loss (56% reduction) was observed in the dorsolateral striatum, ipsilateral to the infusion. To explore the possibility that the reduction of ChAT-positive neurons was merely due to downregulation of this enzyme, we also counted the number of striatal neurons expressing the high-affinity nerve growth factor (NGF) receptor, TrkA. This receptor is coexpressed with ChAT in striatal cholinergic interneurons (Sobreviela et al., 1994). The same cell loss, shown by quantification of ChAT-positive neurons, was observed using TrkA immunocytochemistry (Figs. 4 and 5).
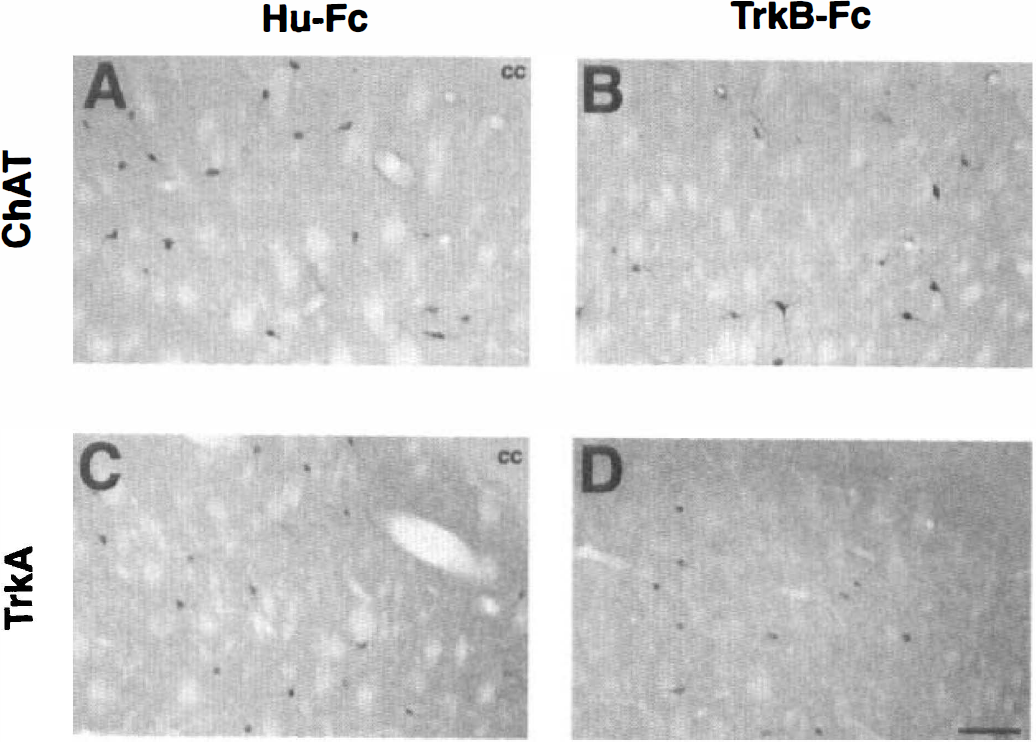
Photomicrographs showing ChAT- (
To explore the possibility that the differences in cell numbers, observed using immunocytochemical markers, were due to downregulation of the respective proteins and not neuronal death, we performed cresyl violet staining. In the regions where we could detect clear loss of DARPP-32- or NeuN-immunostained neurons, i.e., the striatum (DARPP-32 and NeuN) and the CA1 pyramidal layer (NeuN), the cresyl violet staining showed unequivocal signs of dead neurons with shrunk, condensed, and darkly stained cell bodies (data not shown). This finding strongly supports the interpretation that the loss of NeuN- and DARPP-32-immunostaining represents neuronal death and not downregulation of protein synthesis due to the ischemia and/or TrkB-Fc infusion.
Moreover, in a parallel study, in which we compared TrkB-Fc– and Hu-Fc–infused nonischemic animals, we could not detect any effect of the TrkB-Fc treatment on neuronal survival or on immunoreactivity for NeuN, DARPP-32, ChAT, NPY, and parvalbumin (data not shown). In agreement, Croll et al. (1998) and Binder et al. (1999) also did not observe any significant cell loss after intrahippocampal and intraventricular infusion of TrkB-Fc.
DISCUSSION
The functional implications of the regional differences in endogenous levels of BDNF and of the changes in the expression of this factor after cerebral ischemia are poorly understood. In the present study, we tested the hypothesis of a neuroprotective action using an experimental strategy intended to attenuate or block BDNF signaling by administration of a scavenger of endogenous BDNF. The results presented here indicate that blockade of endogenous BDNF activity by intraventricular infusion of TrkB-Fc can lead to aggravated neuronal death after global forebrain ischemia in rats. Specifically, as compared to Hu-Fc-exposed controls, the TrkB-Fc-treated animals subjected to 30 minutes of ischemia had significantly lower numbers of CA4 pyramidal neurons, NPY-immunopositive hilar neurons, and ChAT- and TrkA-positive striatal interneurons. Cell counting was performed using unbiased stereologic procedures in two regions, striatum and hippocampal formation, which showed good penetration of both TrkB-Fc and Hu-Fc. Based on previous in vitro studies (Shelton et al., 1995), it seems highly likely that the continuous TrkB-Fc infusion during 2 weeks lead to blockade of BDNF activity. The magnitude of this blockade probably varied between different parts of the striatum and hippocampus because the immunocytochemical analysis indicated a gradient in the penetration of TrkB-Fc within these areas. It also seems possible that low levels of TrkB-Fc, not clearly detectable with the method used here, gave rise to reduced BDNF activity in wider areas than those depicted in Fig. 1. Intracerebral infusion of TrkB-Fc will scavenge not only BDNF but also other ligands which bind to the TrkB receptor, i.e., neurotrophin-4/5 (NT-4/5) and neurotrophin-3 (NT-3). However, the levels of NT-4/5 in the hippocampal formation and striatum are very low (Timmusk et al., 1993) and NT-3 interacts mainly with the TrkC receptor (Lamballe et al., 1991). Therefore, the effect of the TrkB-Fc infusion observed here is most likely due to scavenging of endogenous BDNF. Recently, Croll et al. (1998) described that, when co-infused with TrkB-Fc, the distribution of exogenous BDNF in the rat forebrain was greatly increased. However, it is highly unlikely that the present findings were due to TrkB-Fc enhancing the distribution and activity of endogenous BDNF. First, such a mechanism has not been shown to occur either in vitro or in vivo (Croll et al., 1998). Second, the observation of reduced cell numbers in TrkB-Fc-treated rats would then imply that BDNF has toxic effects on these neuronal populations, which is inconsistent with available literature (Beck et al., 1994; Wu and Pardridge, 1999).
In the striatum, no difference in the survival of DARPP-32-immunopositive projection neurons was observed between the two experimental groups after 30 minutes of ischemia. In contrast, there was, on the side of the infusion, a 56% reduction of the number of ChAT-positive interneurons in the dorsolateral striatum of the TrkB-Fc-treated rats as compared to the Hu-Fc-infused group. A similar reduction of cell number was observed using an antibody against TrkA, which is coexpressed with ChAT in cholinergic interneurons (Sobreviela et al., 1994). There is no evidence that BDNF regulates the expression of these markers in the striatum (Jones et al., 1994), therefore, it is conceivable that the difference between the groups reflects a lower number of surviving cholinergic interneurons in the TrkB-Fc-treated rats. Our findings argue against a significant role of endogenous BDNF for the survival of the majority of striatal neurons, i.e., the GABAergic projection neurons, after ischemic insults. For the cholinergic interneurons, on the other hand, BDNF is not only an important survival factor during development (Ward and Hagg, 1999), but also seems to have a pronounced neuroprotective action in the adult rat. These neurons express TrkB as well as the high affinity receptors for NGF and NT-3, i.e., TrkA and TrkC, respectively (Merlio et al., 1992; Sobreviela et al., 1994). In addition, the striatal cholinergic interneurons transiently upregulate the p75 neurotrophin receptor (p75NTR) after cerebral ischemia (Kokaia et al., 1998). The striatal levels of BDNF are moderately high (Nawa et al., 1995), but this factor is not synthesized locally in the striatum (Yan et al., 1997). Instead, BDNF is transported anterogradely in afferents originating in the substantia nigra and cerebral cortex (Altar and DiStefano, 1998). At least the nigral projection has direct synaptic contacts with cholinergic interneurons (Kubota et al., 1987), and could provide the trophic support exerted by BDNF. The cholinergic interneurons have previously been shown to be highly resistant to ischemic damage (Chesselet et al., 1990; Kokaia et al., 1998). Neuronal apoptosis inhibitory protein, which is expressed by the cholinergic interneurons and upregulated after ischemia, is probably an important factor underlying this low vulnerability (Xu et al., 1997). The present data indicate that BDNF, supplied by afferents, also plays a key role for the survival of these neurons after ischemic insults.
The TrkB-Fc infusion also caused significant aggravation of damage to CA4 pyramidal neurons after the ischemic insult, leading to 21% lower numbers of these cells as compared to Hu-Fc-treated controls. The CA4 pyramidal neurons express both BDNF and TrkB mRNAs (Ernfors et al., 1990; Merlio et al., 1992), and BDNF could, therefore, act in an autocrine and/or paracrine manner on these cells. In addition, the CA4 neurons are richly supplied with mossy fibers (Freund and Buzaki, 1996), transporting BDNF protein anterogradely from dentate granule cells (Smith et al., 1997). Our data indicate that endogenous BDNF protein, produced by the CA4 neurons themselves or received via mossy fiber afferents, plays a moderate role for the survival of these neurons after global forebrain ischemia. In agreement, Beck et al. (1994) found that CA4 pyramidal neurons are protected against global ischemic damage by administration of exogenous BDNF.
In the TrkB-Fc-treated rats subjected to 30 minutes of ischemia, the dentate gyrus hilus contained lower numbers of NPY - and possibly parvalbumin-immunopositive interneurons as compared to Hu-Fc-exposed animals. Parvalbumin- and probably also NPY -positive neurons express the TrkB receptor (Croll et al., 1994; Marty et al., 1996) and our findings suggest that endogenous BDNF increases their resistance to ischemic neuronal death. However, data from BDNF knockout mice and after intrahippocampal infusion of BDNF have indicated that this factor can regulate the expression of NPY and parvalbumin (Croll et al., 1994; Jones et al., 1994) in hippocampal interneurons. Therefore, it cannot be excluded that blockade of BDNF activity by TrkB-Fc merely leads to downregulation of the expression of NPY and parvalbumin and that the survival of these neurons in the present experiment was unaffected.
The extensive loss of CA1 pyramidal neurons after 30 minutes of global ischemia was not influenced by the TrkB-Fc infusion. Two main explanations for the lack of effect can be envisaged. First, it may be hypothesized that this insult was too severe to reveal a neuroprotective action of endogenous BDNF. However, this seems unlikely because when the ischemic insult was mild (5 minutes) and the CA1 pyramidal cell loss was only partial, TrkB-Fc infusion did not aggravate the neuronal death. Second, the endogenous BDNF level in the CA1 region is low and decreases after forebrain ischemia (Kokaia et al., 1996). Our data could, therefore, suggest that this low level of BDNF does not significantly contribute to the survival of CA1 pyramidal neurons. On the other hand, the findings by Beck et al. (1994) and Wu and Pardridge (1999) indicate that increasing the levels of BDNF by exogenous administration can give rise to major neuroprotective effects also in the CA1 region.
The levels of BDNF protein in the CA3 region are considerably higher compared to those in CA1, and 10 minutes of global ischemia leads to increased synthesis of BDNF in the CA3 area (Kokaia et al., 1996). The Fluoro-Jade-stained sections indicated that there was a modest damage to CA3 pyramidal neurons in both groups, and cell counting did not reveal any difference in neuronal survival after 30 minutes of ischemia. The lack of effect by the TrkB-Fc infusion suggests that BDNF has no significant neuroprotective action on CA3 neurons after ischemic insults. However, it seems possible that the concentration of TrkB-Fc reaching the CA3 region was insufficient to effectively scavenge the relatively high levels of BDNF in this area.
In conclusion, the results of the present study indicate that endogenous BDNF can exert neuroprotective effects after transient forebrain ischemia; this is similar to what has been reported previously after administration of recombinant BDNF. These effects may be mediated, e.g., through stabilization of calcium homeostasis (Cheng and Mattson, 1994) or triggering of systems that can buffer free radicals (Spina et al., 1992), and could involve also glial cells (Nakajima et al., 1998). However, our data also suggest that endogenous BDNF per se only plays a modest to moderate role in counteracting neuronal death after ischemic insults. In addition to BDNF, ischemia leads to increased synthesis of a large number of neurotrophic molecules and their receptors, such as NGF, glial cell line-derived neurotrophic factor and platelet-derived growth factor, and these responses could all be neuroprotective (Lindvall et al., 1992; Iihara et al., 1997; Kokaia et al., 1999). Therefore, blockade of the protective effect of one neurotrophic factor, as attempted in the present study, may conceivably be compensated for by the other factors. Another possibility raised by our findings is that neuroprotection is not the major role of BDNF after cerebral ischemia. It seems highly warranted to pursue studies intended to clarify the involvement of BDNF, e.g., in cell proliferation, sprouting processes and synaptic plasticity in the postischemic phase.
Footnotes
Acknowledgment:
The authors thank Dr. Susan Croll, Regeneron Pharmaceuticals, Tarrytown, NY, U.S.A for providing TrkB-Fc and Hu-Fc and for helpful discussions.