
Abstract
The authors previously demonstrated that Ca 2+/calmodulin (CaM)-dependent protein kinase IIα (CaM-KIIα) can phosphorylate neuronal nitric oxide synthase (nNOS) at Ser847 and attenuate NOS activity in neuronal cells. In the present study, they established that forebrain ischemia causes an increase in the phosphorylation of nNOS at Ser847 in the hippocampus. This nNOS phosphorylation appeared to be catalyzed by CaM-KII: (1) it correlated with the autophosphorylation of CaM-KIIα; (2) it was blocked by the CaM-KII inhibitor, KN-93; and (3) nNOS and CaM-KIIα were found to coexist in the hippocampus. Examination of the spatial relation between nNOS and CaM-KIIα in the brain revealed coexistence in the hippocampus but not in the cortex during reperfusion, with a concomitant increase in autophosphorylation of CaM-KIIα. The phosphorylation of nNOS at Ser847 probably takes place in nonpyramidal hippocampal neurons, which increased after 30 minutes of reperfusion in the hippocampus, whereas no significant increase was detected in the cortex. An intraventricular injection of KN-93 significantly decreased the phosphorylation of nNOS in the hippocampus. These results point to CaM-KII as a protein kinase, which by its colocalization may attenuate the activity of nNOS through its Ser847 phosphorylation, and may thus contribute to promotion of tolerance to postischemic damage in hippocampal neurons.
Transient cerebral ischemia induces increases in extracellular glutamate and intracellular calcium (Choi, 1995; Rothman and Olney, 1995), production of free radicals (Chan, 1996), and alteration of protein kinases (Cardell et al., 1990; Hu et al., 1995; Wieloch et al., 1991). At least three major protein kinases have been identified in brain: cyclic AMP-dependent protein kinase (PKA), Ca2+/phospholipid-dependent protein kinase C, and Ca2+/(CaM-K II) (Goldenring et al., 1984; Kelly et al., 1979, 1984; Wolf et al., 1986). CaM-K II, which makes up 1% to 2% of the protein content of the hippocampus (Erondu and Kennedy, 1985), is a multifunctional kinase that plays important roles in neuronal plasticity and postischemic neuronal damage (Churn et al., 1990; Shackelford et al., 1995; Taft et al., 1988). An ischemic episode causes a marked translocation of protein kinase C and CaM-K II to PSDs (Aronowski and Grotta, 1996; Domanska-Janik et al., 1999; Hu et al., 1998; Suzuki et al., 1994), specialized cytoskeletal structures lying beneath the postsynaptic membrane (Harris and Kater, 1994; Kennedy, 1997).
In the nervous system nNOS appears to be targeted to membranes by binding to PSD95, a component of PSDs that associates with N-methyl-D-aspartate receptors at synapses. nNOS is stimulated by calcium influx through the N-methyl-
Recently, we demonstrated that nNOS is directly phosphorylated at Ser847 by CaM-K IIα, leading to a reduction of its enzyme activity in vitro and in situ (Hayashi et al., 1999; Komeima et al., 2000). To test whether the phosphorylation by CaM-K II occurs in cerebral ischemia, we used a site-specific antibody, NP847, which reacts with nNOS phosphorylated at Ser847 (Hayashi, et al., 1999). In this article, we document evidence that CaM-K IIα colocalizes with nNOS in the hippocampus but not in the cortex during reperfusion, and phosphorylates nNOS at Ser847. We have also studied the distribution of nNOS and phospho-nNOS at Ser847 with an immunohistochemical technique to provide further information on the function of NO in the ischemic hippocampal neurons. The findings point to “cross-talk” between Ca2+ and the NO-regulated signal transduction pathway, which might provide a novel tolerance mechanism by an enhancement of susceptibility to postischemic damage in hippocampal neurons.
MATERIALS AND METHODS
Materials
KN-93 was purchased from Seikagaku Kogyo (Tokyo, Japan), β-NADPH was from Oriental Yeast (Osaka, Japan), and 2'-5'-ADP-agarose and other chemicals, unless otherwise specified, were from Sigma Chemicals (St. Louis, MO, U.S.A.).
Forebrain ischemic model
All experiments were carried out in accordance with guidelines for the care and use of laboratory animals in the physiologic sciences as approved by the Physiological Society of Japan.
Anesthesia was induced in male Sprague-Dawley rats (350 to 400 g) using methohexital sodium (50 mg/kg, intraperitoneally). Animals were then intubated and ventilated with 1.0% halothane in an oxygen/nitrous oxide (30%/70%) gas mixture. Temperature was monitored with a rectal probe and maintained between 36.5 and 37.5°C with a heating pad and lamp. The right femoral artery and vein were exposed and catheterized with polyethylene tubing (PE-50) to allow blood sampling and the monitoring of arterial blood pressure during ischemia. The arterial blood pressure was measured and recorded continuously until the rats were extubated. Arterial blood gases were examined before injection of the drug, 5 minutes before the induction of ischemia, and 15 minutes after the reperfusion of blood. The inspired halothane concentration was decreased to 0.5% 15 minutes before the induction of ischemia, and 150 IU/kg heparin was administered intravenously. Blood was withdrawn from the femoral vein catheter to induce hypotension with a mean arterial blood pressure of 50 mm Hg, and both carotid arteries were clamped. Blood pressure was maintained at 50 mm Hg during the 15-minute ischemic period by withdrawing or infusing blood through the femoral vein catheter. At the end of the 15-minute period of ischemia, the clamps were removed, blood was reinfused through the femoral vein catheter, and all wounds were sutured.
Animals to be killed before the induction of forebrain ischemia, immediately at the end of ischemia, or after a 30-minute reperfusion continued to be given halothane anesthesia. Those killed at later recovery times were maintained in warmed cages and were reanesthetized using methohexital sodium (50 mg/kg, intraperitoneally). Brain samples were collected at the end of the 15-minute ischemia, as well as after 0.5, 1, 2, 4, and 24 hours of reperfusion. Brain samples without induced forebrain ischemia were used as controls. Tissue samples for Western blot analysis were obtained by decapitation under deep anesthesia. The hippocampus and cortex at the same level were immediately isolated on ice, frozen in liquid nitrogen, and kept at −70°C until use.
Intracerebroventricular administration of KN-93
Under anesthesia, rats were set on a stereotactic operation frame. A scalp skin incision was made, and one burr hole was opened on the right parietal skull, 1.5 mm lateral and 0.8 mm posterior to the bregma. A Hamilton syringe was inserted into the brain at a depth of 3.5 mm from the brain cortex. KN-93, a calmodulin kinase inhibitor, was freshly dissolved in distilled water before use. KN-93 injection (1 mmol/L, 2 μL) slowly into the right ventricle. The needle was kept in place for 10 minutes to allow the drug to diffuse into the ventricle. H2O (2 μL) was employed as the vehicle control. Forebrain ischemia was induced 30 minutes later and brains were removed 30 minutes after the 15-minute period of cerebral ischemia. The hippocampus and cortex were obtained for Western blot analyses.
Sample preparation for Western blot analysis
Samples were prepared from four or five different animals in each group. Manipulations were carried out at 4°C. Brain tissues were homogenized using a homogenizer in 10 volumes of homogenization buffer containing 50 mmol/L Tris base/HCl (pH 7.5), 0.1 mmol/L dithiothreitol, 0.2 mmol/L ethylenediamine tetraacetic acid, 0.2 mmol/L ethylene glycol-bis(β-aminoethyl ether N,N,N',N'-tetraacetic acid, 0.2 mmol/L phenylmethylsulfonyl fluoride, 1.25 μg/mL pepstatin A, 0.2 μg/mL aprotinin, 0.2 μg/mL leupeptin, 5 nmol/L tetrahydrobiopterin (BH4), 1 mmol/L sodium orthovanadate, 50 mmol/L sodium fluoride, 2 mmol/L sodium pyrophosphate, and 1% Nonidet P-40 (NP-40). The homogenates were then centrifuged at 15,000 g at 4°C for 10 minutes. Protein concentrations of the supernatants were determined by the method of Bradford using bovine serum albumin as the standard. Supernatant fractions were applied as crude fractions.
For preparation of the nNOS fraction, NOS was partially purified by 2'-5'-ADP-agarose, as described previously (Richards and Marletta, 1994). Briefly, 15 μL of 2'-5'-ADP-agarose with the same concentration of crude fraction (1,000 μg/200 μL) was incubated gently for 1 hour at 4°C. The agarose was washed with 200 μL of the homogenization buffer without NP-40 and nNOS was eluted from 2'-5'-ADP-agarose with 50 μL of 10 mmol/L NADPH.
Western blotting analysis
Extract samples and 25 μg of crude samples were subjected to 7.5% SDS-PAGE, and the proteins were transferred to PVDF membranes and incubated with primary polyclonal antibodies against phospho-nNOS (NP847) at a dilution of 1:100, and CaM-K IIα (Oncogene, Cambridge, MA, U.S.A.) at a dilution of 1:1,000 for 45 minutes at room temperature. NP847 recognizes only Ser847 phosphorylated nNOS (Hayashi et al., 1999; Komeima et al., 2000). The membranes were then incubated for 30 minutes at room temperature with horseradish peroxidase-conjugated secondary antibodies, which were visualized using an enhanced chemiluminescence (ECL) or ECL plus Western blotting detection system (Amersham, Little Chalfont, U.K.). Phospho-nNOS and CaM-K IIα immunoblots were stripped from PVDF membranes and reblotted with primary monoclonal nNOS (Transduction Laboratories, Lexington, KY, U.S.A.) at a dilution of 1:3,000, and phospho-CaM-K IIα at Thr286 (Promega, Madison, WI, U.S.A.) at a dilution of 1:500 for 45 minutes at room temperature. Finally, the membranes were developed with the ECL system, and band intensities were quantitated by densitometric scanning using the NIH IMAGE program.
Immunohistochemistry
The rats subjected to a 15-minute ischemia followed by a 30-minute reperfusion were perfused with ice-cold 200 mL 4% paraformaldehyde in 0.1 mol/L sodium phosphate (pH 7.4). The brains were removed and preserved in the fixation solution for 3 hours and rinsed with 0.1 mol/L lysine hydrochloride in 0.1 mol/L phosphate-buffered saline for 3 hours. Serial coronal cryostat sections (10 μm) were collected on silane-coated slides. Staining was performed according to the ABC technique at room temperature. The staining sequence was as follows: 2% goat serum for 30 minutes; primary polyclonal antibodies against CaM-K II and phospho-Thr286/287 CaM-K II (Liu et al., 1999; Yokota et al., 2001) at a dilution of 1:2,000 each, monoclonal antibodies against nNOS (Sigma Chemicals) at a dilution of 1:100 and NP847 at a dilution of 1:2,000; biotinylated anti-rabbit or mouse IgG for 1 hour; and avidin-biotinylated peroxidase complex for 1 hour. The solution for antibody dilution was made with 20 mmol/L sodium phosphate (pH 7.4) in 150 mmol/L sodium chloride containing 2% normal goat serum. Sera for the blocking step, biotinylated antibodies, and avidin-biotinylated peroxidase complex were purchased from Vector Laboratories (Burlingame, CA, U.S.A.). Reaction products were developed by incubation in 0.05% 3, 3'-diaminobendizine tetrachloride and 0.01% H2O2 in 50 mmol/L Tris-HCl (pH 7.5) for 10 minutes. To amplify the reaction where necessary, development was carried out for 15 minutes in 0.05% 3, 3'-diaminobendizine tetrachloride and 0.01% H2O2 in 100 mmol/L sodium phosphate (pH 7.2), containing 0.025% cobalt chloride and 0.02% nickel ammonium. Rats not undergoing forebrain ischemia were used as controls.
Statistical analysis
Data are expressed as mean ± SD values. Statistical analyses were performed by the multiple comparisons test and a Mann-Whitney test. Statistical significance was concluded at the P < 0.05 level.
RESULTS
Physiologic parameters
Major physiologic parameters for the animals were unaltered. No significant changes in mean arterial blood pressure, temperature, or arterial blood gas data were detected among any of the experimental groups by repeated-measure analysis of variance (Table 1).
Physiologic variables (mean ± SD, n = 4) measured before cerebral ischemia (preischemia) and 15 minutes after reperfusion (postischemia)
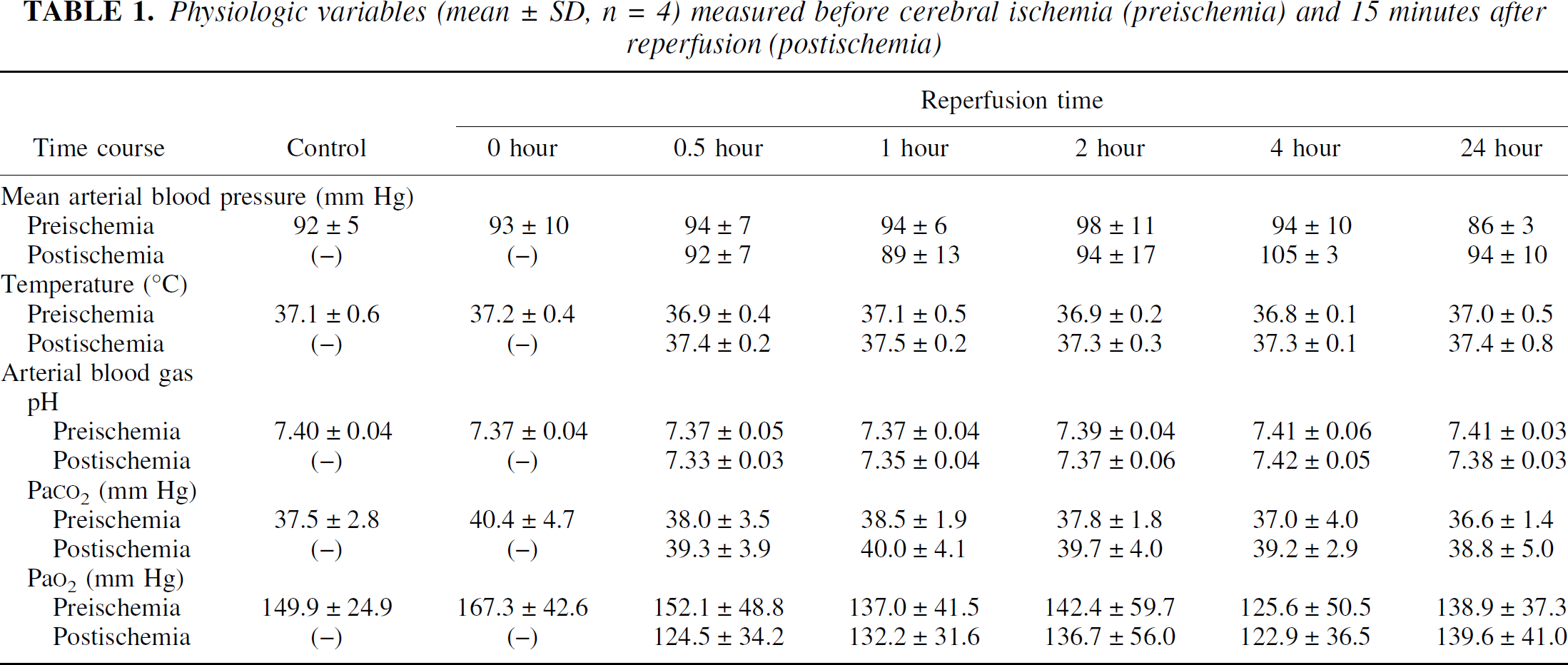
Effects of ischemia on phosphorylation of neuronal nitric oxide synthase at Ser847
We first examined the levels of phosphorylated nNOS at Ser847 in both the hippocampus and cortex after transient ischemia. For this we developed a polyclonal antibody, NP847, which reacts with nNOS phosphorylated at Ser847 (Hayashi et al., 1999). We partially purified nNOS by ADP-agarose affinity chromatography and quantified the phosphorylation state at Ser847 and the extracted protein levels. After 15 minutes of ischemia, the carotid clamp was released, and reperfusion was continued for 24 hours or less. Transient ischemia resulted in a twofold enhancement in the phosphorylation of nNOS at Ser847 after a 30-minute reperfusion in the hippocampus, relative to nonischemic samples (Fig. 1A). At this time, no increase in phosphorylation was detected in the cortex (Fig. 1B). Equal levels of nNOS were purified from lysates even after reperfusion, indicating that an ischemic episode modulates nNOS primarily through the phosphorylation at Ser847 in the hippocampus.
Phosphorylation of neuronal isoforms of nitric oxide synthase (nNOS) at Ser847 in the ischemic brain. After a 15-minute bilateral carotid artery occlusion and 0, 0.5, 1,2,4, or 24-hour reperfusions as indicated below the panel, nNOS was affinity-purified by ADP agarose affinity chromatography from a 1% NP-40 soluble fraction of hippocampus
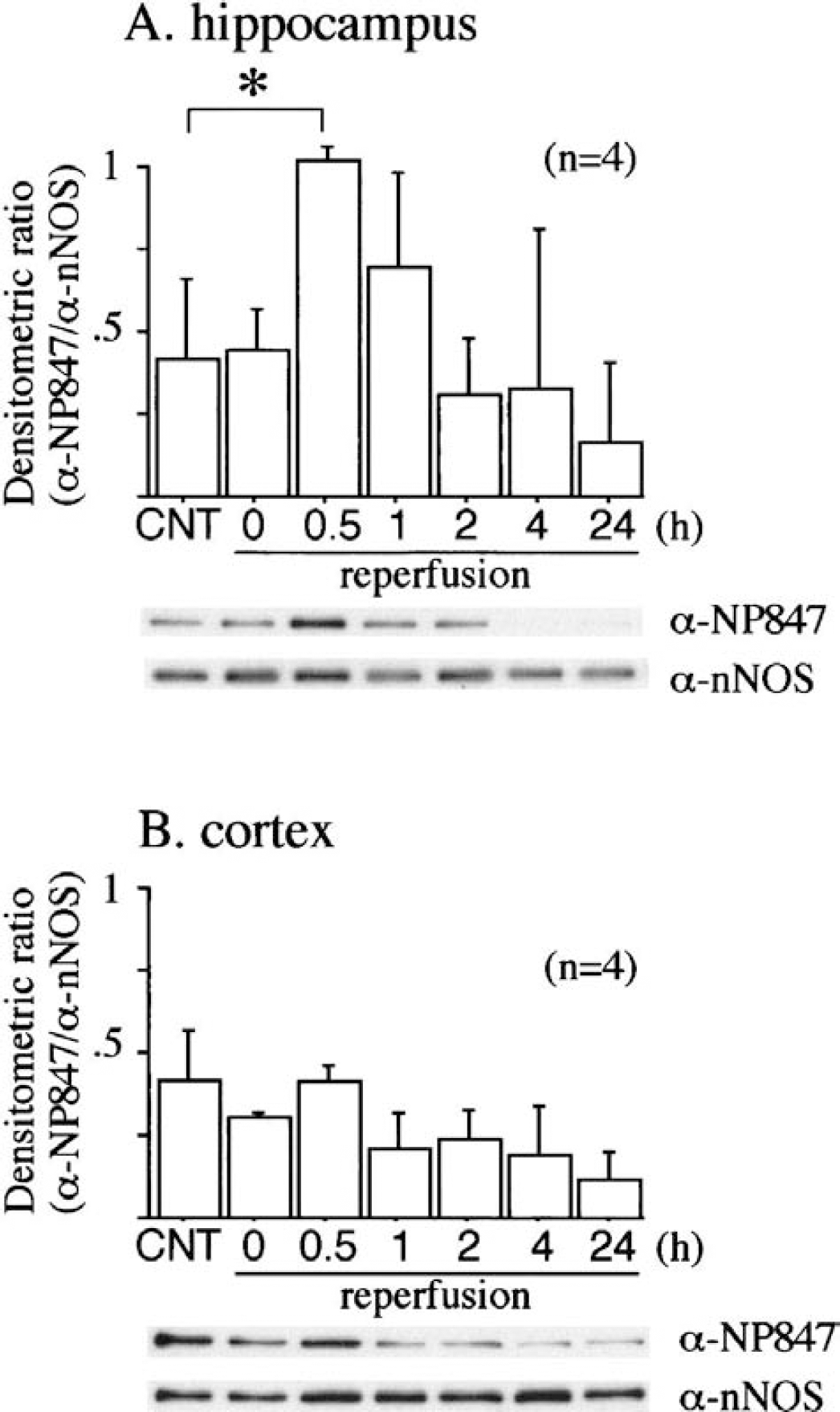
Colocalization of neuronal nitric oxide synthase and Ca2+/calmodulin-dependent protein kinase IIα in rat brain
The Ser847 residue of nNOS was recently identified as the potential phosphorylation site of CaM-K II in vitro and in situ (Hayashi et al., 1999; Komeima et al., 2000). We therefore examined the spatial relation between CaM-K II and nNOS in rat brain using a cosedimentation assay by ADP-agarose affinity chromatography. The content of CaM-K IIα was slightly decreased in supernatant fractions of both the hippocampus and cortex during reperfusion (Fig. 2A,B). CaM-K IIα cosedimented with nNOS on ADP-agarose affinity chromatography of nonischemic samples from both the hippocampus and cortex. We next examined whether CaM-K IIα was present in the nNOS-containing fraction even after cerebral ischemia. CaM-K IIα was still detected in the nNOS fraction during reperfusion in the hippocampus but not in the cortex. The densitometric ratio of immunoreactive CaM-K IIα in the nNOS fraction relative to that in the hippocampal extract did not decrease significantly (Fig. 2A), whereas that in the cortex dramatically decreased during reperfusion (Fig. 2B).
Immunoblot analysis of calcium/calmodulin-dependent protein kinases IIα (CaM-K IIα) in the ischemic brain. Ischemia was induced as for Fig. 1 without 4- and 24-hour reperfusions. One percent NP-40 soluble fraction (crude fraction) and a neuronal isoforms of nitric oxide synthase (nNOS)-containing fraction (nNOS fraction), prepared by ADP agarose affinity chromatography, from hippocampus
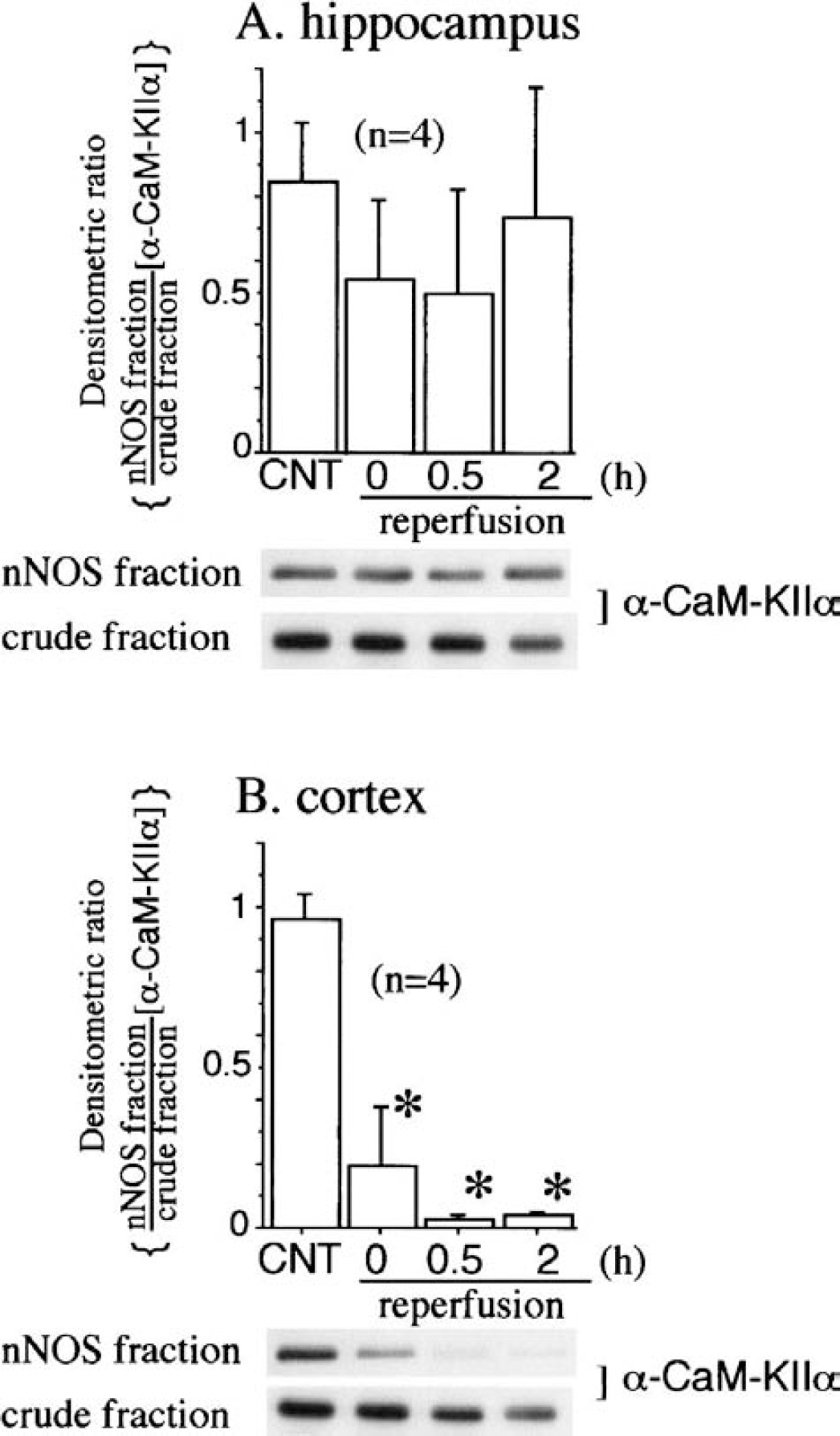
Autophosphorylation of Ca2+/calmodulin-dependent protein kinase IIα at Thr286 in rat brain
It is well established that CaM-K IIα can undergo rapid autophosphorylation of its Thr286, which appears to precede the phosphorylation of substrates (Kwiatkowski et al., 1988). We therefore examined the levels of autophosphorylated CaM-K IIα at Thr286 in an ischemic brain using a phosphospecific antibody, Abp-Thr286, specific for Thr286-phosphorylated CaM-KIIα. Protein immunoblot analyses with Abp-Thr286 of ischemic hippocampal extract detected an immunoreactive band corresponding to the 50-kd, which gradually decreased after cerebral ischemia (Fig. 3B). In addition, the densitometric ratio of the immunoreactivity of Abp-Thr286 relative to that of a general CaM-K IIα antibody significantly increased in the NOS-contained fraction at the end of ischemia compared to the nonischemic control in the hippocampus (Fig. 3A). In the cortex as in the hippocampus, a gradual decrease in autophosphorylation of CaM-K IIα at Thr286 was detected in a crude fraction. No Abp-Thr286-immunoreactive band was observed due to an insufficient amount of CaM-K IIα (Fig. 2B) copurifying with nNOS during reperfusion in the nNOS fraction (data not shown).
Autophosphorylation of calcium/calmodulin-dependent protein kinases IIα (CaM-K IIα) at Thr286 in the ischemic hippocampus. Ischemia was induced as for Fig. 1 without 4- and 24-hour reperfusions. At the indicated times, neuronal isoforms of nitric oxide synthase (nNOS)-containing
Effects of KN-93 on the phosphorylation of neuronal nitric oxide synthase at Ser847
The above results demonstrated that CaM-K IIα can colocalize with nNOS, undergoing autophosphorylation of its Thr286, and regulate the enzyme through its Ser847 phosphorylation in ischemic hippocampal neurons. To further investigate the role of CaM-K IIα in the phosphorylation of nNOS at Ser847, we used KN-93, a well-known specific inhibitor of CaM-K II. An intraventricular injection of KN-93 caused no significant changes in physiologic parameters compared to H2O by repeated-measure analysis of variance (Table 2). Immunoblot analyses showed that pretreatment with KN-93 decreased the phosphorylation of nNOS at Ser847 after a 30-minute reperfusion in the hippocampus but not the cortex. The densitometric ratio of immunoreactivity with NP847 relative to that with a general nNOS antibody was significantly decreased in the hippocampus but not the cortex (Fig. 4).
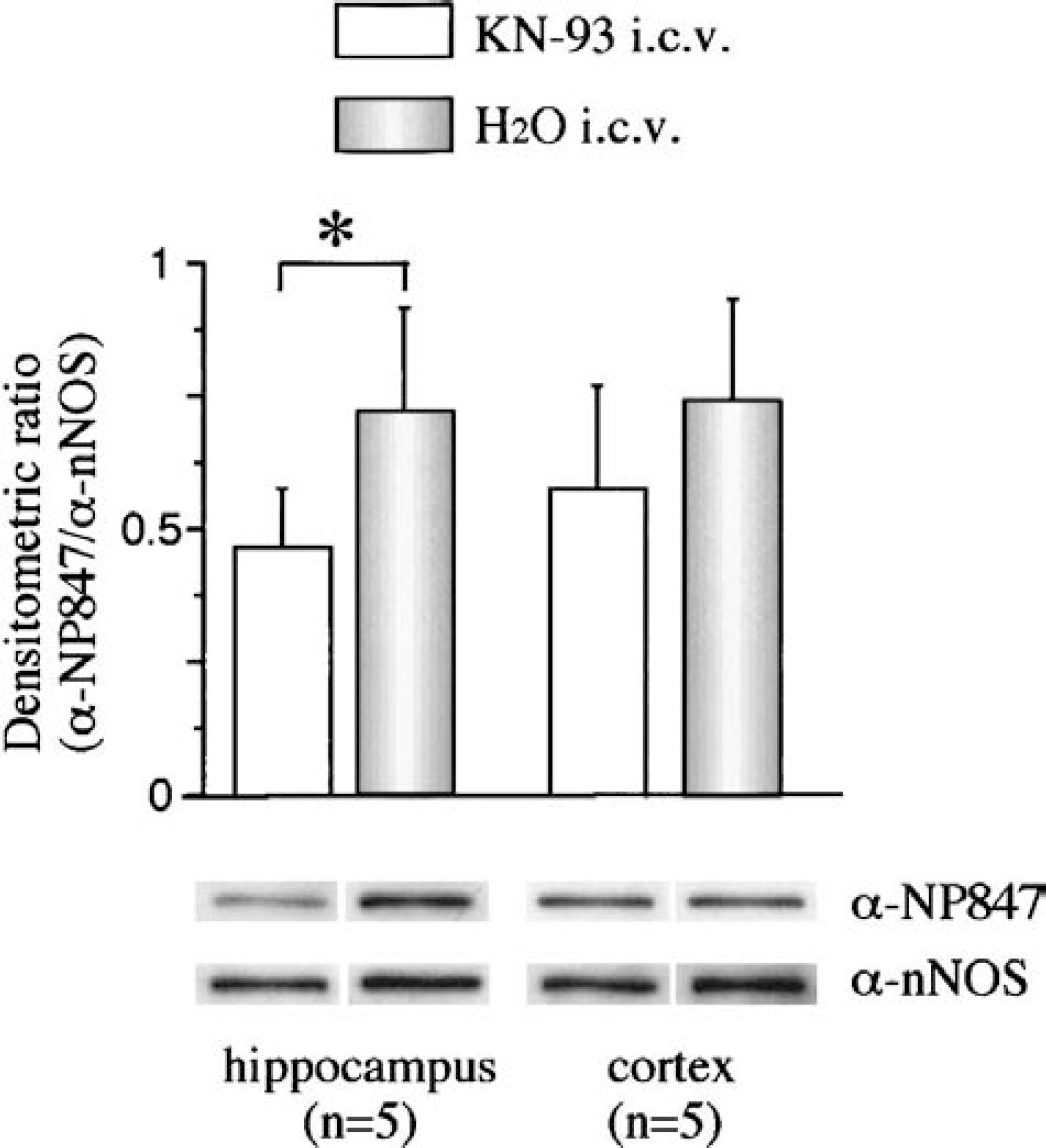
Effects of intraventricular injection of KN-93 on the phosphorylation of neuronal isoforms of nitric oxide synthase (nNOS) at Ser847. After ischemia and a 30-minute reperfusion either with (KN-93 intracerebroventricularly) or without (H2O intracerebroventricularly) KN-93 pretreatment, nNOS was affinity-purified from the hippocampus or cortex and subjected to Western blotting. The histogram shows the amount of α-NP-847 relative to that of α-nNOS. Mean ± SD values from five animals are shown; the asterisk indicates a significant difference in hippocampus by the Mann-Whitney U test (P < 0.05).
Physiologic variables (mean ± SD, n = 5) measured before intraventricular injection of drug (predrug), after drug injection 5 minutes before cerebral ischemia (preischemia), and 15 minutes after reperfusion (postischemia)
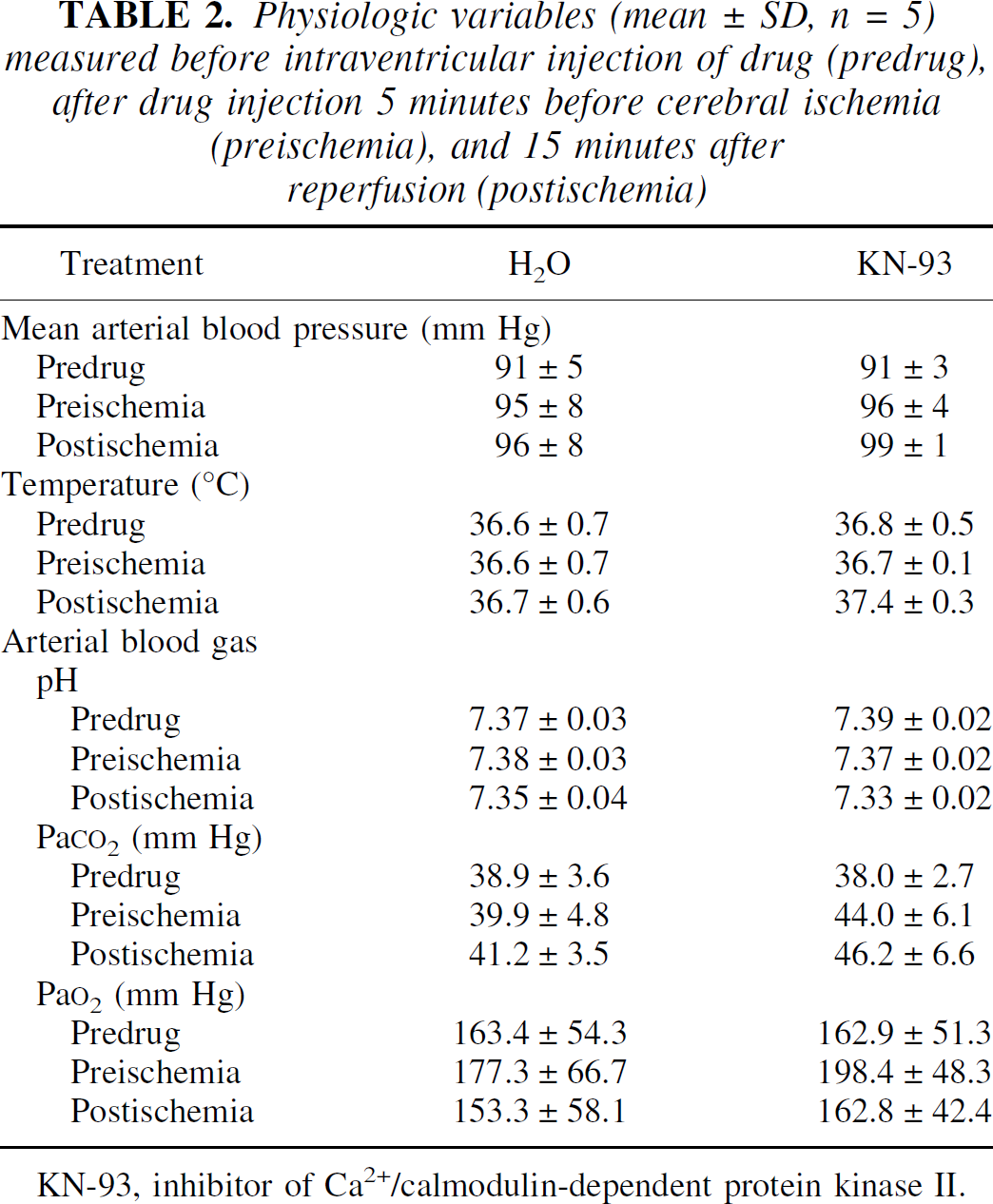
KN-93, inhibitor of Ca2+/calmodulin-dependent protein kinase II.
Neurons in rat hippocampus contain Ser847-phosphorylated neuronal nitric oxide synthase after transient ischemia
To further investigate where the phosphorylation of nNOS at Ser847 by CaM-K IIα occurs in the hippocampus, we tested the expression of phopho-Ser-847 nNOS after a 30-minute reperfusion. We initially analyzed immunohistochemical alteration of CaM-K II and Thr286/287-phosphorylated CaM-K II. Immunoreactivity against CaM-K II and phospho-Thr-286/287 CaM-K II were observed mainly in the cytoplasm and apical dendrites of CA1 pyramidal neurons (Fig. 5A,B). After a 30-minute reperfusion, immunoreactivity of phospho-Thr-286/287 CaM-K II was markedly attenuated in pyramidal neurons (Fig. 5D) without a remarkable decrease in their nonpyramidal counterparts (Fig. 5D, arrow). The immunoreactivity of CaM-K II in pyramidal neurons was relatively preserved (Fig. 5C). Immunoreactivity against nNOS was apparent in the cytoplasm and processes of CA1 nonpyramidal neurons without a remarkable change (Fig. 6A,C, arrows). Intense phopho-Ser-847 nNOS immunoreactivity was observed in the cytoplasm and processes of CA1 nonpyramidal neurons after a 30-minute reperfusion (Fig. 6D, arrow).
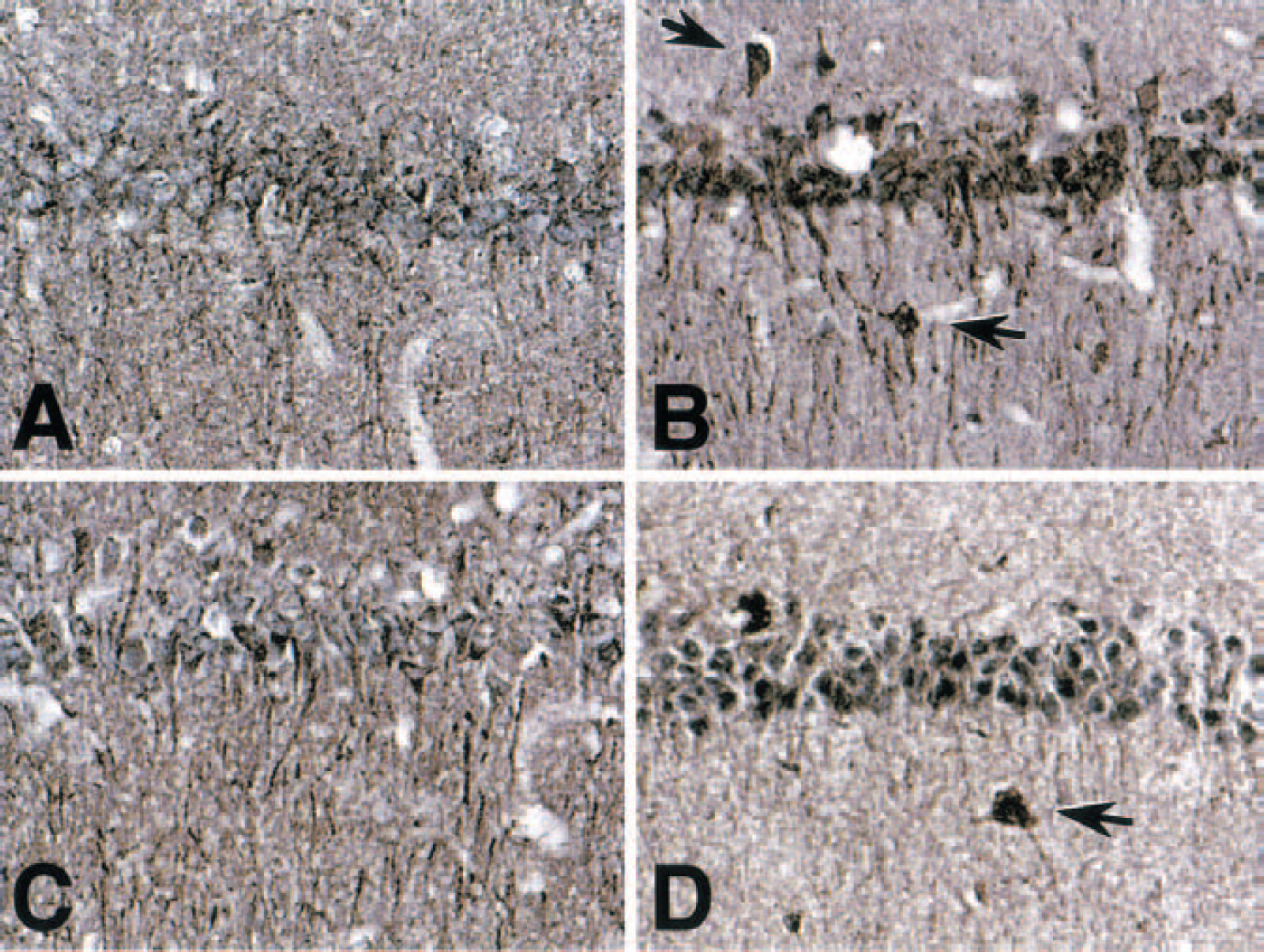
Immunohistochemical alteration of calcium/calmodulin-dependent protein kinases II (CaM-K II) after transient ischemia. Rats subjected to sham operation (
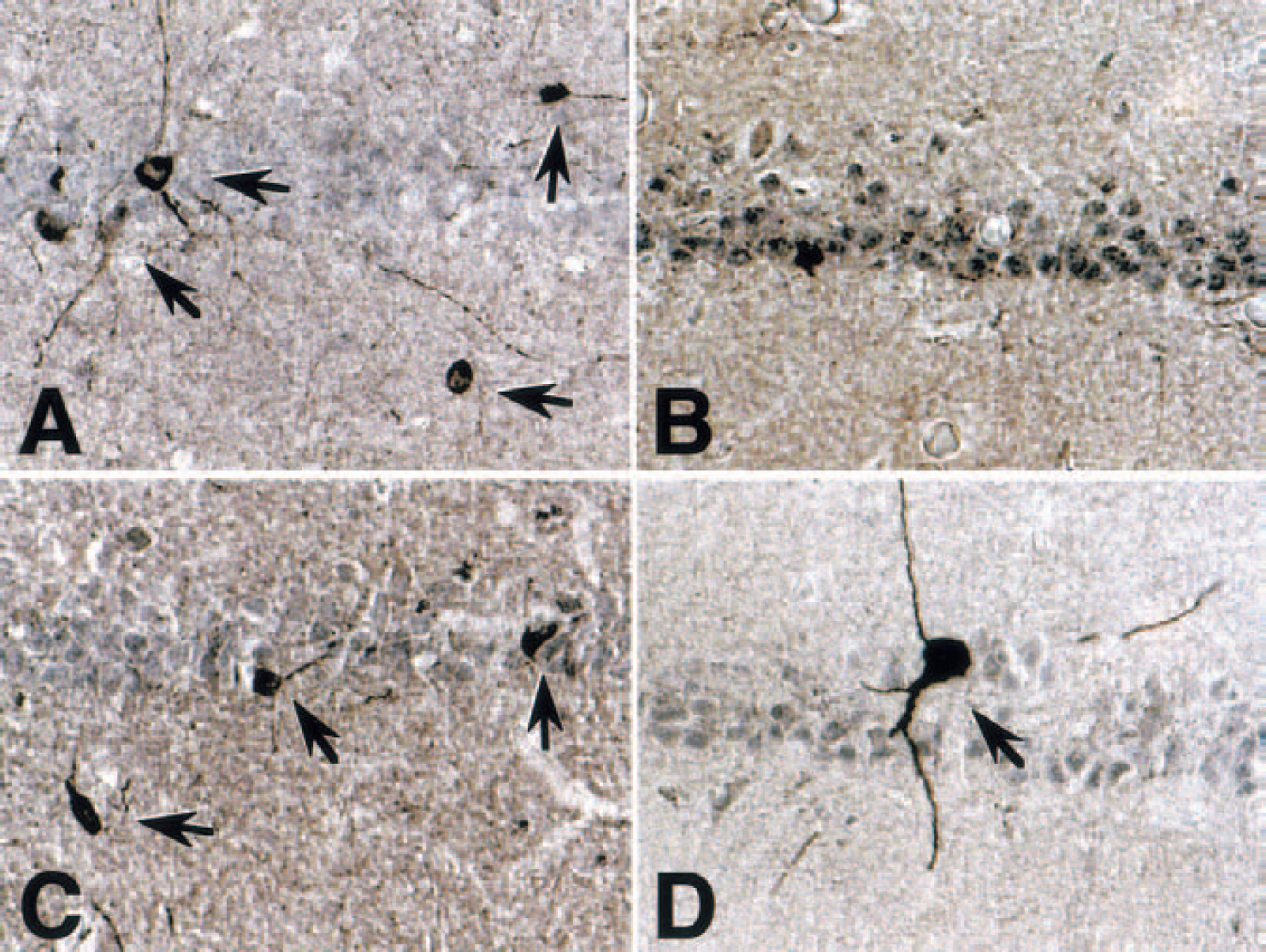
Immunohistochemical analysis of neuronal isoforms of nitric oxide synthase (nNOS) and phospho-Ser847 nNOS expression after transient ischemia in the hippocampal CA1 area. Rats subjected to sham operation (
DISCUSSION
We previously showed that CaM-K IIα can directly phosphorylate nNOS at Ser847, leading to a reduction of NOS activity in vitro and in situ (Hayashi et al., 1999; Komeima et al., 2000). In the present study, we showed that CaM-K IIα plays an important role in the phosphorylation of nNOS in the postischemic hippocampus. In our conditions, amounts of CaM-K IIα in supernatant fractions from both the hippocampus and the cortex were similar during reperfusion (Fig. 2). It has been shown that CaM-K II translocates from cytosol to the particulate fractions in postischemic brain tissues (Aronowski et al., 1992; Cardell et al., 1990; Wieloch et al., 1991). The supernatant fractions used in this study are not equivalent to cytosolic fractions used in other studies. This study used detergent and a low-speed spin to recover the supernatant. Meanwhile, cytosolic fractions in other studies used no detergent and a high-speed spin to separate cytosol from particulate fractions including PSDs. In the cells, CaM-K II and nNOS may be colocalized at PSDs but differentially solubilized by their procedures.
We observed that CaM-K IIα can exist in an nNOS-containing fraction in which NOS could be partially purified from the 1% NP-40 soluble fraction of nonischemic samples using ADP agarose affinity chromatography (Fig. 2). This may not be due to nonspecific binding of CaM-K II to ADP agarose, since purified CaM-K IIα does not interact with ADP agarose in vitro (data not shown). Previous studies have indicated a decrease in the total CaM-K II activity and loss of enzyme immunoreactivity in the hippocampus following ischemia (Morioka et al., 1992; Yamamoto et al., 1992; Churn et al., 1992; Babcock et al., 1995). It is further known that CaM-K II is activated by autophosphorylation. We observed a decrease in immunoreactivity of phospho-Thr-286 CaM-K IIα on immunoblot analyses of ischemic hippocampal extracts (Fig. 3B) and by immunohistochemical analyses of pyramidal neurons (Fig. 5D). However, phospho-Thr-286 CaM-K IIα in the NOS-containing fraction significantly increased at the end of ischemia (Fig. 3A) and immunoreactivity of phospho-Thr-286/287 CaM-K II in nonpyramidal neurons was relatively preserved (Fig. 5B,D, arrows). It has been reported that CaM-K II is activated by 1 minute of incubation of 10- to 500-μmol/L glutamate in hippocampal-cultured eurons (Morioka et al., 1995). Thus, CaM-K II in the NOS-containing fraction might be activated immediately following ischemia under the conditions employed.
The distribution of nNOS in nonpyramidal hippocampal neurons reported in this study is in general agreement with previous findings using immunohistochemical methods (Valtschanoff et al., 1993; Dinerman et al., 1994a). However, there is controversy concerning the presence of NOS in neurons in the pyramidal layer (see Wendland et al., 1994; Vaid et al., 1996 for discussion of this topic). Based on our immunohistochemical analyses, we conclude that the phosphorylation of nNOS at Ser847 probably takes place in nonpyramidal hippocampal neurons. CaM-K IIα is not detected in CA1 GABAergic interneurons with immunocytochemistry (Sik et al., 1998), but small amounts of enzyme below the detection threshold might be functional (McBain et al., 1999; Wang and Kelly, 2001). The autophosphorylated CaM-K IIα in the NOS-containing fraction could induce phosphorylation of nNOS at Ser847 after a 30-minute reperfusion in the hippocampus, consistent with the effect of KN-93 on the nNOS phosphorylation (Fig. 4). However, there was a significant increase in phospho-Thr-286 CaM-K IIα in the NOS-containing fraction at the end of ischemia, but not after a 30-minute reperfusion (Fig. 3A). We therefore cannot eliminate the possibility that other KN-93-sensitive kinases might also phosphorylate nNOS at Ser847 in the ischemic hippocampus.
CaM-K II translocates to PSDs following synaptic stimulation (Shen and Meyer, 1999; Strack et al., 1997) and ischemia (Aronowski and Grotta, 1996; Domanska-Janik et al., 1999; Hu et al., 1998; Suzuki et al., 1994). However, previous studies have indicated that, in ischemia, the translocated kinase may not function properly because CaM-K II activity is inhibited in postischemic tissues (Hu et al., 1995; Hu and Wieloch, 1995). The increased phospho-Thr-286 CaM-K IIα in the NOS-containing fraction at the end of ischemia might not be a translocated enzyme. It is known that most nNOS is membrane-associated (Watanabe et al., 1998), but some also may exist as a free cytosolic pool (Mizukawa et al., 1988; Hecker et al., 1994) or in mitochondria (Wolf et al., 1992). Further studies using biochemical and immunocytochemical approaches are needed to pinpoint where this phosphorylation of nNOS at Ser847 by CaM-K IIα occurs in hippocampal neurons.
Waxham et al. (1996) showed that CaM-K IIα knockout mice had an infarct volume almost twice that of wild-type mice after transient focal cerebral ischemia. Thus, CaM-K II activity plays some neuroprotective role in the ischemic brain, but the mechanisms remain to be elucidated. In view of our in vivo and in vitro data, we propose that CaM-K IIα phosphorylates nNOS at Ser847 and thereby decreases its activity, so that excessive production of harmful NO is controlled. Excess NO reacts with superoxides to form the potent oxidant peroxynitrite, which can cause cell death (Eliasson et al., 1999). Brain damage is substantially reduced in nNOS knockout mice after cerebral ischemia (Huang et al., 1994). The phosphorylation of nNOS by CaM-K IIα is thus a possible neuroprotective mechanism, and could become a therapeutic strategy for protection against harmful peroxynitrite after cerebral ischemia.
In conclusion, our results show that nNOS is differently regulated by CaM-K IIα between the hippocampus and cortex. Its phosphorylation in the hippocampus could decrease enzyme activity and reduce overproduction of NO after cerebral ischemia. Further studies are required to clarify the other roles of this phosphorylated nNOS and its possible relation with apoptosis in the hippocampus.
Footnotes
Acknowledgements
We thank Dr. D. Okada (Mitsubishi Kasei Institute of Life Sciences, Japan) for valuable suggestions and Dr. Malcolm Moore for critical reading of the article.