
Abstract
Infarct volume and edema were assessed after transient focal ischemia in mice lacking neuronal nitric oxide synthase (NOS) gene expression. With use of an 8–0 coated monofilament, the middle cerebral artery (MCA) of mutant (n = 32) and wild-type mice [SV-129 (n = 31), C57Black/6 (n = 18)] were occluded for 3 h and reperfused for up to 24 h. Regional CBF (rCBF), neurological deficits, water content, and infarct volume were examined in all three strains. rCBF, blood pressure, and heart rate did not differ between groups when measured for 1 h after reperfusion. Neurological deficits were less severe in mutant mice after MCA occlusion. Brain water content at 3 h after reperfusion and infarct volume at 24 h after reperfusion were greater in wild-type mice. These data indicate that genetic deletion of neuronal NOS confers resistance to focal ischemic injury in a reperfusion model. The findings agree with previous studies showing that tissue injury is less extensive after both permanent MCA occlusion and global ischemia in mice lacking neuronal NOS gene expression. Hence, NO may play a pivotal role in the pathogenesis of ischemic brain damage.
Keywords
Nitric oxide (NO) is a ubiquitous molecular messenger involved in numerous biological processes (Moncada et al., 1991; Bredt and Snyder, 1992, 1994; Vincent, 1994). NO is synthesized by NO synthase (NOS), an enzyme for which at least three isoforms have been identified, including neuronal, endothelial, and inducible NOS (Nathan and Xie, 1994). NO has been implicated as a putative mediator of N-methyl-D-aspartate (NMDA) cytotoxicity in cultured cells (Dawson et al., 1991) and in the pathophysiology of ischemic brain damage (Dawson et al., 1992b; Dalkara and Moskowitz, 1994; Iadecola et al., 1994). This evidence is largely derived from in vivo experiments showing that NOS inhibitors ameliorate tissue damage after middle cerebral artery (MCA) occlusion (Nowicki et al., 1991; Buisson et al., 1992; Nagafuji et al., 1992; Ashwal et al., 1993). However, some reports found that these inhibitors increase ischemic infarction after MCA occlusion (Dawson et al., 1992a; Yamamoto et al., 1992; Kuluz et al., 1993; Morikawa et al., 1994).
Knock-out mice deficient in the neuronal isoform have helped to clarify this controversy. Our laboratories observed that infarct volume was smaller in mutant mice with selective neuronal NOS gene deletion than in wild-type controls, whereas inhibition of endothelial NOS activity increased ischemic brain damage in these mutants. Hence, neuronal but not endothelial NOS has a negative impact on ischemic damage (Huang et al., 1994). However, our previous report did not address the role of reperfusion injury in the context of focal ischemia. NO may promote tissue injury because oxygen free radicals and related species such as superoxide anion are formed in abundance during reperfusion and generate peroxynitrite anion, a putative mediator of tissue injury (Beckman, 1991; Dawson et al., 1992b).
The present study examines the consequences of transient focal ischemia on edema formation and infarct volume after transient MCA occlusion in neuronal NOS deficient mice. We now report that mutant mice exhibit less brain edema and smaller infarct volumes after transient MCA occlusion.
MATERIALS AND METHODS
NOS activity
NOS activity was measured by the conversion of [3H]arginine to [3H]citrulline according to the method described by Bredt and Snyder (1990) with a minor modification. Samples were homogenized in cold N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) (Research Organics, OH, U.S.A.) buffer (50 mM, 500 μl) containing 1 mM ethylenediaminetetraacetate (EDTA) (pH 7.4; Sigma, St. Louis, MO, U.S.A.). Homogenates were centrifuged (2,500 rpm, 5 min, 4°C) and the supernatant used for assay. The incubation mixture contained 100 μl of HEPES (50 mM, pH 7.4), EDTA (1 mM), β-nicotinamide adenine dinucleotide phosphate reduced form (β-NADPH, 1 mM; Sigma), dithiothreitol (1 mM; Sigma), calmodulin (10 μg/ml; Calbiochem, CA, U.S.A.), Ca2+ (1 mM), and 25 μl of 100 mM L-[3H]arginine (1 mCi; Dupont NEN, MA, U.S.A.). The reaction was started by adding supernatant (25 μl) and stopped by adding HEPES (20 mM)/EDTA (2 mM), pH 5.5 solution after 20 min at 37°C. The mixture was then applied to anion-exchange columns containing Dowex AG50WX-8 (Na+ form; Bio-Rad, Richmond, VA, U.S.A.) and eluted with 2 ml of distilled water. [3H]Citrulline was measured within eluates by scintillation spectrometry (Taurus, 36000 Series; Micro-medic Systems, AL, U.S.A.).
Physiology
Animals were initially anesthetized with 1.5% and maintained by 1.0% halothane in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH, U.S.A.). Anesthesia was promptly changed to α-chloralose (Sigma) when animals were artificially ventilated (SAR-830-P; CWE, Ardmore, PA, U.S.A.) 30 min prior to ischemia. α-Chloralose was administered intravenously from the left femoral vein catheterized with a PE-10 polyethylene tubing (Clay Adams, Parsippany, NJ, U.S.A.) initially at a dose of 80 mg/kg and then every 1 h at a dose of 40 mg/kg.
Regional CBF (rCBF) was determined by laser-Doppler flowmetry (Perimed, PF2B, Stockholm, Sweden) using a flexible 0.5-mm fiberoptic extension to the masterprobe. The tip of the probe was fixed by an adhesive agent and its accelerator (Aron Alpha, Toa, Tokyo, Japan) on the intact skull over the ischemic cortex (2 mm posterior and 6 mm lateral from the bregma). Steady-state baseline values were recorded before MCA occlusion, and rCBF during and after occlusion was expressed as percentage of the baseline values. rCBF, blood pressure, and heart rate were monitored using MacLab/8 data acquisition system (AD Instruments, Milford, MA, U.S.A.) equipped with ETH 400 transducer amplifier via femoral, artery catheterized with a PE-10 polyethylene tubing. Heart rate was calculated from arterial pressure pulses or ECG and continuously displayed by the data acquisition software. Arterial blood samples were obtained from the same line. Fifty-microliter samples were withdrawn into heparinized capillary tubes and analyzed at 37°C for partial pressures of oxygen (Pao2) and carbon dioxide (Paco2) before ischemia and 1 h after reperfusion using a blood gas/pH analyzer (Corning 178; Ciba Corning Diag., Med-ford, MA, U.S.A.). An equal volume of saline was injected after every sampling. Withdrawal of more than three samples usually led to systemic hypotension. Body temperature was maintained at 36.5–37.5°C with a heating pad (T/PUMP, TP400, Gaymar, NY, U.S.A.) and heating lamp (Skytron, Daiichi Shomei, Tokyo, Japan) until 1 h after reperfusion.
Ischemic model
Adult male wild-type, SV-129, and C57Black/6 (Taconic Farms, Germantown, NY, U.S.A.) and adult male and female mutant mice (Huang et al., 1993) weighing 22–29 g were allowed free access to food (Prolab, rat, mouse, hamster 300 w formula; Agway, CG, Syracuse, NY, U.S.A.) and water (pH 7.1–7.2). All animals were kept under diurnal lighting conditions. Anesthesia was induced by 1.5% halothane and maintained with 1% halothane in 70% N2O and 30% O2 or α-chloralose. The left MCA was occluded with an 8–0 nylon monofilament (Ethicon, NJ, USA) coated with a mixture of silicone resin (Xantopren; Bayer Dental, Osaka, Japan) and a hardener (Elastomer Activator; Bayer Dental). This coated filament was introduced into the internal carotid artery through the external carotid artery, up to the origin of the anterior cerebral artery via the internal carotid artery, so as to occlude the MCA and posterior communicating artery. At the same time, the left common carotid artery was occluded by a temporary clip (10-B Zen type clip; Ohwa Tsusho, Tokyo, Japan). Three hours after ischemia, animals were briefly reanesthetized with halothane and the filament withdrawn through the external carotid artery. Thus, the MCA, posterior communicating artery and common carotid artery were reperfused, but not the external carotid artery. Three hours of ischemia and reperfusion was selected because it produced a reproducible subtotal infarction in our preliminary studies and because rCBF could be restored at that time point.
Neurological deficits
Thirty minutes after ischemia, mice were tested for behavioral changes and scored as described by Bederson et al. (1986a) with a minor modification as follows: 0—no observable neurological deficits (normal); 1—failure to extend right forepaw (mild); 2—circling to the contralateral side (moderate); 3—loss of walking or righting reflex (severe). The 30-min time point was selected because behavioral changes are marked at this time after ischemia.
Brain edema
Brains were quickly removed 3 h after reperfusion, and both the ipsilateral (ischemic) and the contralateral (nonischemic) cortex and striatum were dissected and immediately assayed by the dry weight method (Hara et al., 1990). Water content in samples was calculated as follows: 100 (wet wt—dry wt)/wet wt (%).
Brain infarction
Twenty four hours after reperfusion, the brains were removed and the forebrains sliced into five coronal 2-mm sections using a mouse brain matrix (RBM–2000C; Activational System, MI, U.S.A.). Slices were then stained with 2% 2,3,5-triphenyltetrazolium chloride (Sigma) as described previously (Bederson et al., 1986a). The infarcted areas were quantitated by an image analysis system (Bioquant IV). The volume of infarction was obtained by numeric integration of areas of marked pallor × section thickness (Huang et al., 1994).
Statistical analysis
Data are presented as means ± SD. Statistical comparisons were made by a one-way analysis of variance followed by Mann–Whitney U test in the neurological deficit study and by the one-way analysis of variance and Tukey multiple-range test in other studies using Statview 4 software (Abacus Concepts, Berkeley, CA, U.S.A.)
RESULTS
NOS activity
In forebrain, NOS activity in SV-129 and mutant mice was 8.7 ± 4.5 (n = 4) and 0.2 ± 0.2 (n = 3, p < 0.05) fmol/mg wet wt/min, respectively. In cerebellum, NOS activity in SV-129 and mutant mice was 15.7 ± 4.9 (n = 3) and 0.2 ± 0.2 (n = 5, p < 0.01) fmol/mg wet wt/min, respectively. NOS activity in cerebellum of SV-129 was 1.8-fold higher than that in forebrain.
Physiological data
rCBF decreased to ∼20% of baseline immediately after MCA occlusion and was sustained during 3 h of ischemia in both wild-type and mutant mice. After reperfusion, rCBF immediately increased to 70–100% of baseline. There were no significant group differences in blood pressure, heart rate, blood gas measurements, and arterial pH before ischemia or after reperfusion (Fig. 1; Tables 1 and 2).
Mean arterial blood pressure and heart rate during and after ischemia for 3 h in wild-type and mutant mice under α-chloralose anesthesia

Blood pressure and heart rate were monitered using MacLab/8. data acquisition equipped with a transducer amplifier via femoral artery. Heart rate was calculated from arterial pressure pulses or ECG and continuously displayed by the data acquisition software. There are no significant differences between groups.
Physiological variables (pH, P a co2, and P a o2) before and after ischemia in wild-type and mutant mice under α-chloralose anesthesia
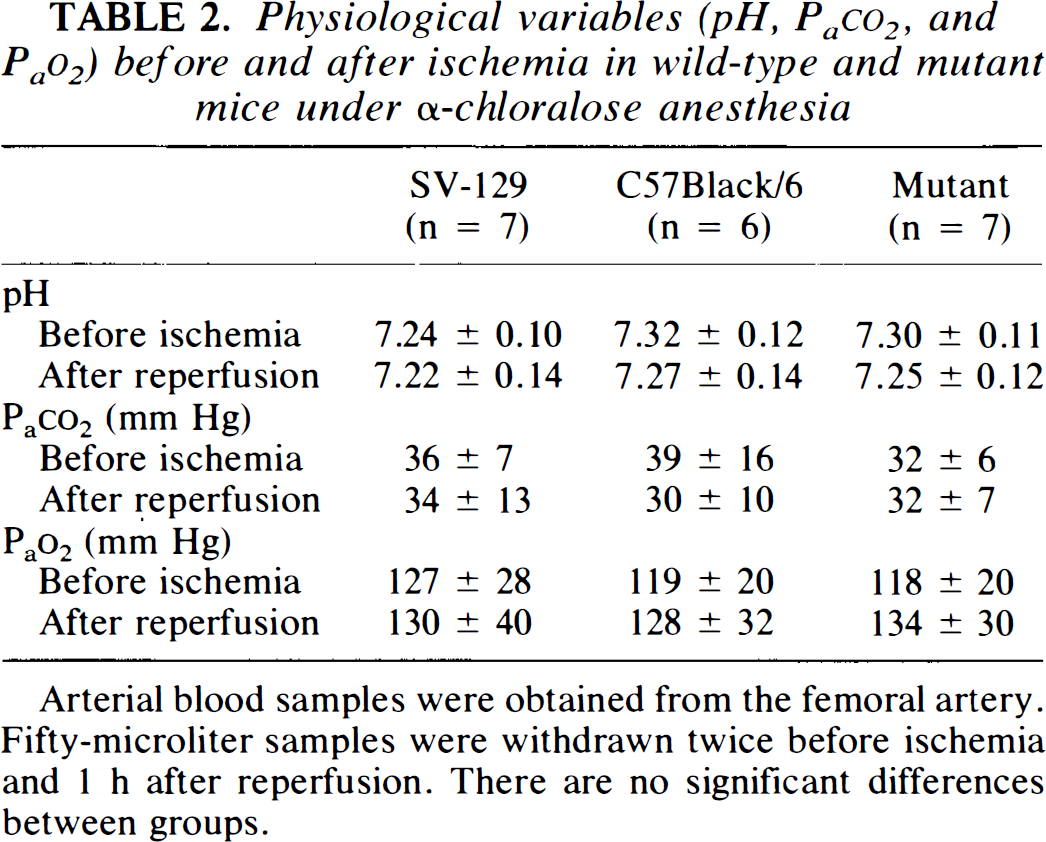
Arterial blood samples were obtained from the femoral artery. Fifty-microliter samples were withdrawn twice before ischemia and 1 h after reperfusion. There are no significant differences between groups.
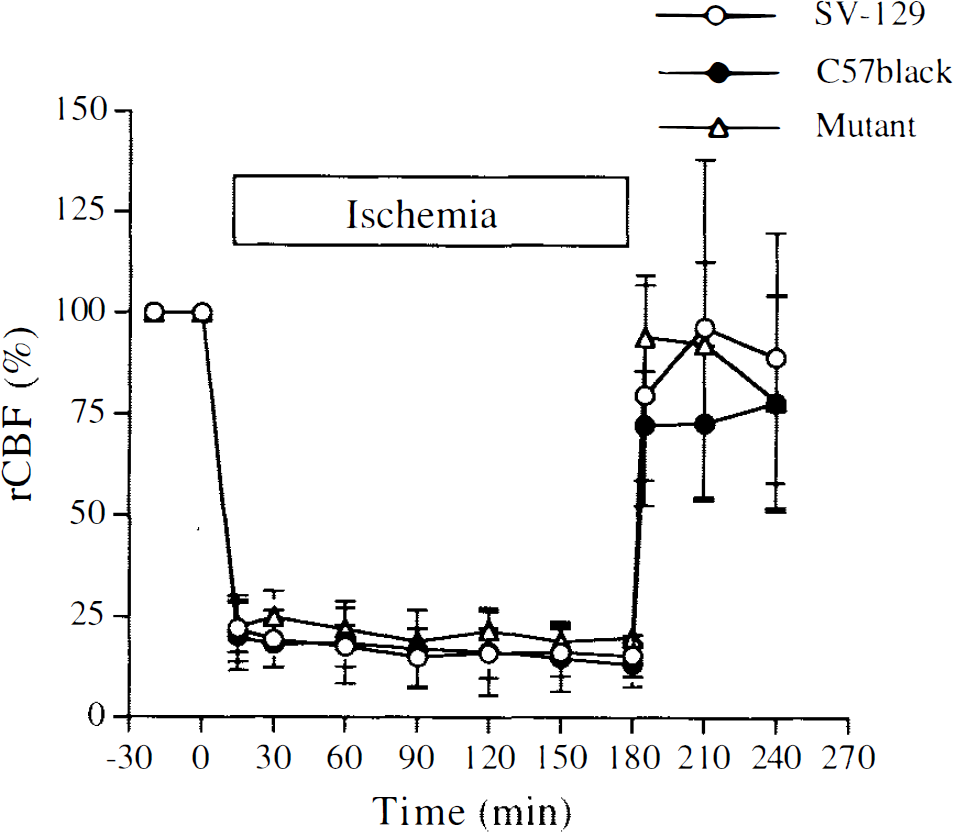
Regional CBF (rCBF) during 3 h of middle cerebral artery occlusion and 1 h of reperfusion in wild-type and mutant mice. Animals were operated initially under halothane anesthesia. rCBF was determined by laser-Doppler flowmetry under α-chloralose anesthesia. The tip of the probe was placed on the intact skull over the ischemic cortex (2 mm posterior and 6 mm lateral from bregma). SV-129 (n = 7), C57Black/6 (n = 6), mutant mice (n = 7). There are no significant differences between groups.
Neurological deficit
Mutant mice did not display gross behavioral abnormalities before ischemia. After 30 min of ischemia, both SV-129 and C57Black/6 exhibited moderate to severe neurological deficits such as circling, decreased resistance to lateral push and locomotor activity, flexion of contralateral torso and forelimb upon lifting the animal by its tail, abnormal posture, and loss of righting reflex. The neurological score was significantly reduced in mutant mice (Table 3).
Neurological scores in wild-type (SV-129 and C57Black/6) and mutant mice 30 min after middle cerebral artery occlusion
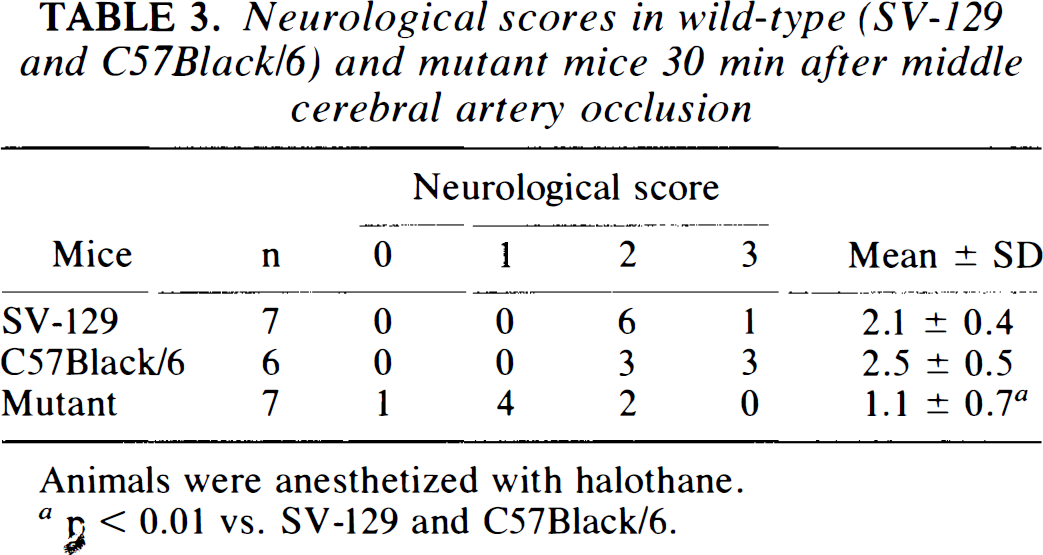
Animals were anesthetized with halothane.
n < 0.01 vs. SV-129 and C57Black/6.
Brain edema
Ischemia increased water content in the ipsilateral but not contralateral striatum and cortex (Fig. 2). Water content in mutant mice was significantly smaller than in wild-type mice.
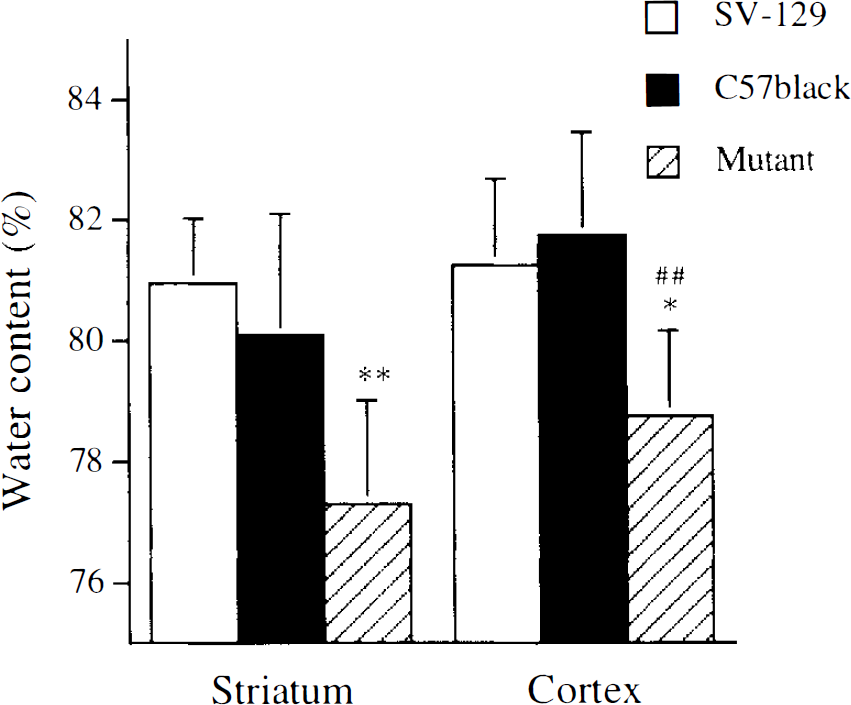
Brain edema 3 h after reperfusion following 3 h of middle cerebral artery occlusion in wild-type and mutant mice. Animals were anesthetized with halothane. The water content was calculated as follows: 100 (wet wt — dry wt)/wet wt(%) by a dry weight method (Hara et al., 1990). Water content in the contralateral striatum was 77.3 ± 0.99 (n = 7), 77.4 ± 0.45 (n = 6), and 76.3 ± 0.72% (n = 7) in SV-129, C57Black/6, and neuronal nitric oxide synthase mutant mice, respectively, and 78.0 ± 0.41 (n = 7), 78.6 ± 0.23 (n = 6), and 77.9 ± 0.81% (n = 7) in the contralateral cortex, respectively. *p < 0.05 vs. SV-129, **p < 0.01 vs. SV-129 and C57Black/6; ##p < 0.01 vs. C57Black/6.
Brain infarction
Twenty-four hours after reperfusion, wild-type mice developed infarcts affecting both cortex and striatum (Fig. 3). Under either α-chloralose or halothane anesthesia, infarct volume was significantly smaller than that in SV-129 mice (Table 4). Infarction areas were smaller in mutants than C57Black/6 or SV-129 mice. Infarct areas were similar in SV-129 and C57Black/6 except at 8 mm (Fig. 4). In cortex, infarct area was smaller than in wild-type mice (Fig. 5). In striatum, infarct area tended to decrease in mutant mice, but this did not reach statistical significance.
Infarct volume in wild-type (SV-129 and C57Black/6) and mutant mice 24 h after reperfusion following 3-h middle cerebral artery occlusion
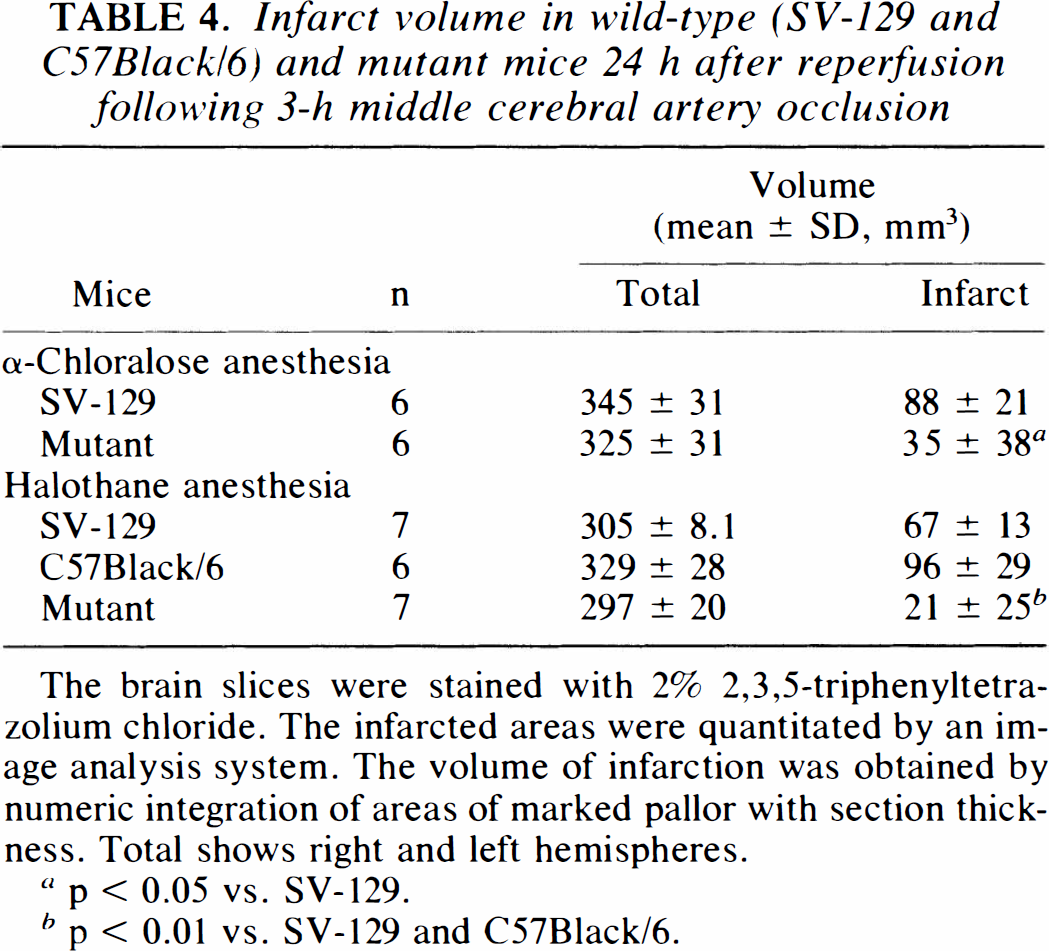
The brain slices were stained with 2% 2,3,5-triphenyltetrazolium chloride. The infarcted areas were quantitated by an image analysis system. The volume of infarction was obtained by numeric integration of areas of marked pallor with section thickness. Total shows right and left hemispheres.
p < 0.05 vs. SV-129.
p < 0.01 vs. SV-129 and C57Black/6.
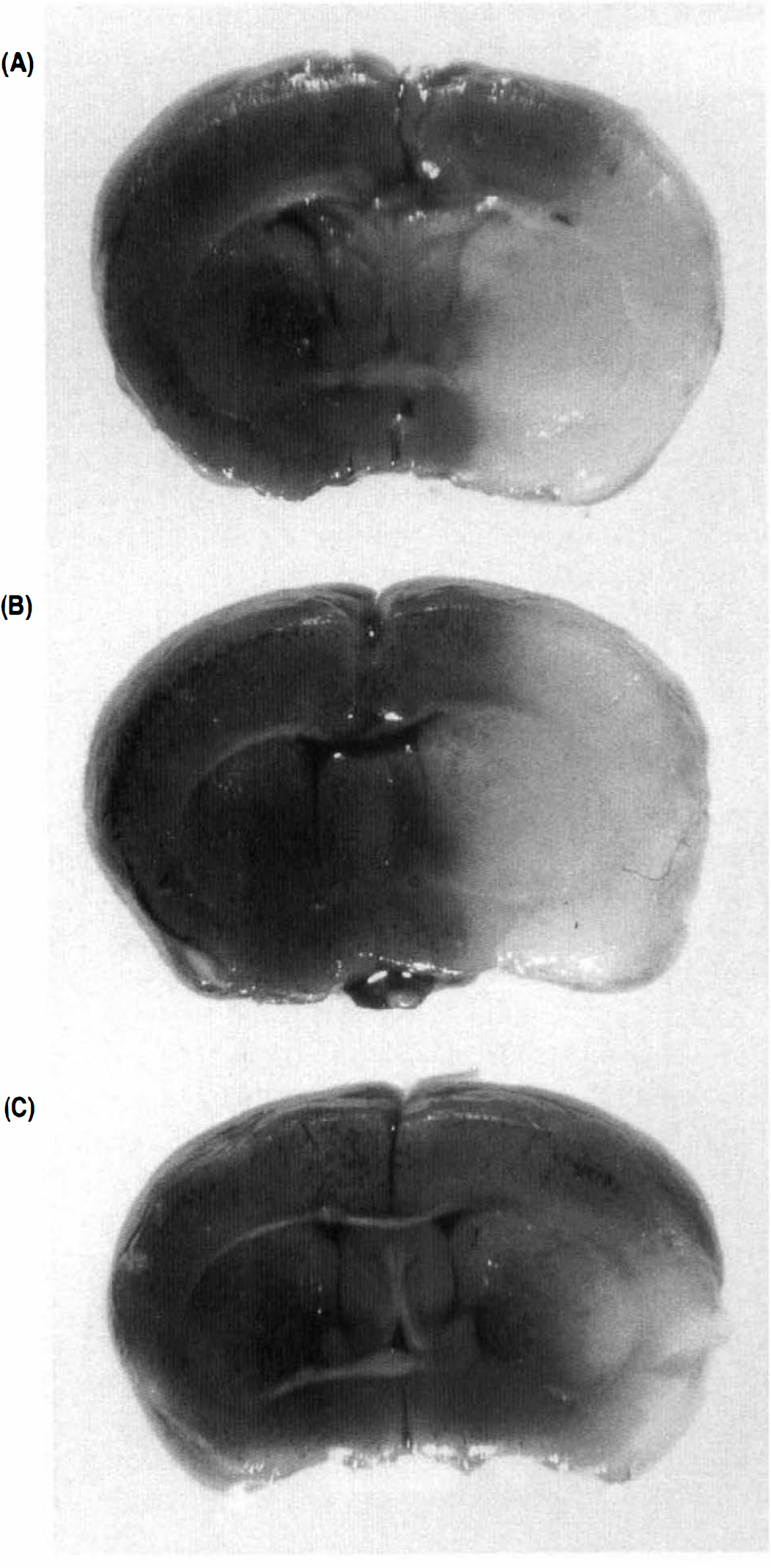
2,3,5-Triphenyltetrazolium chloride staining of coronal brain sections (2 mm) 24 h after reperfusion following 3-h middle cerebral artery occlusion in representative wild-type and mutant mice. Animals were anesthetized with halothane. Unstained areas show infarct.
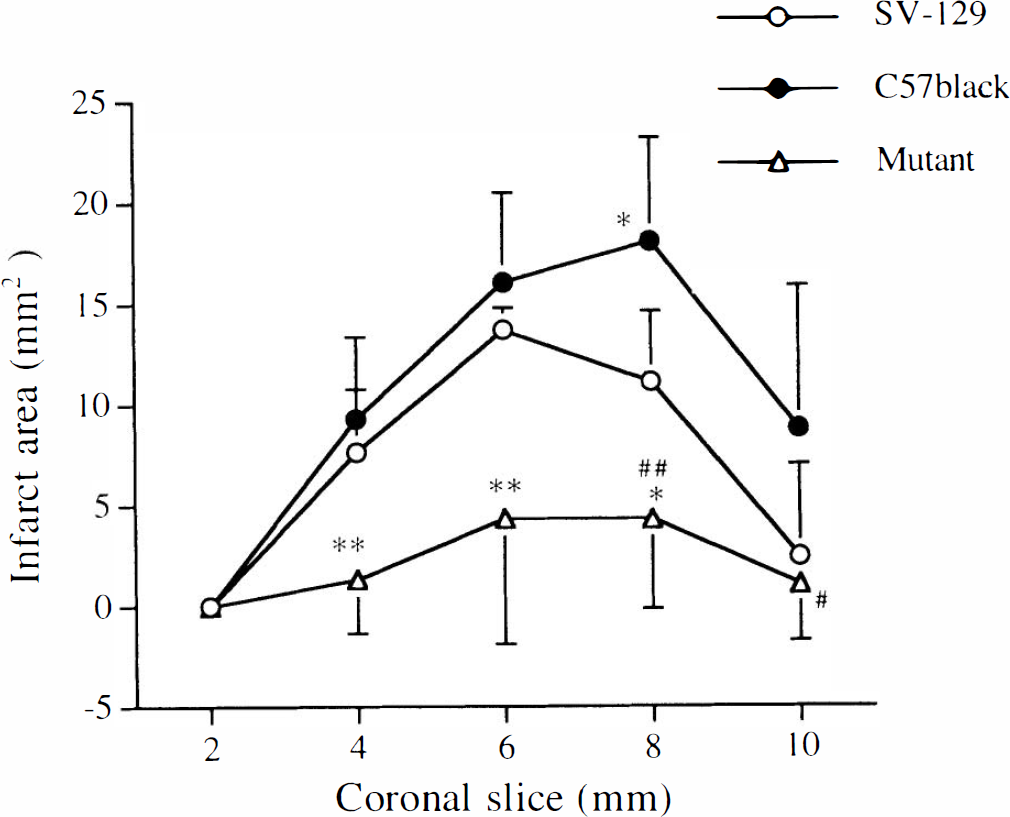
Brain infarct areas 24 h after reperfusion following 3-h middle cerebral artery occlusion in wild-type and mutant mice. Animals were anesthetized with halothane. Twenty-four hours after reperfusion, the brains were removed and the forebrains sliced into five coronal 2-mm sections. Infarct areas in brain slices were stained with 2% 2,3,5-triphenyltetrazolium chloride. *p < 0.05 vs. SV-129, **p < 0.01 vs. SV-129 and C57Black/6; #p < 0.05; ##p < 0.01 vs. C57Black/6. SV-129 and mutant: n = 7: C57Black/6: n = 6.
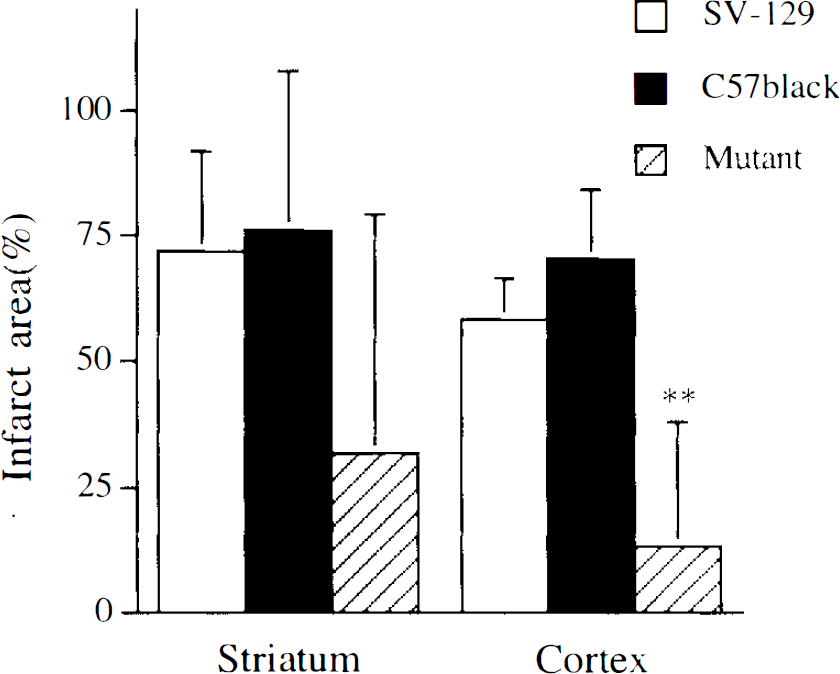
Brain infarct areas in striatum and cortex 24 h after reperfusion following 3-h middle cerebral artery occlusion in wild-type and mutant mice. Animals were anesthetized with halothane. Infarct areas in striatum and cortex were measured at 6 mm from the rostral pole after 2,3,5-triphenyltetrazolium chloride 2% staining, **p < 0.01 vs. SV-129 and C57Black/6. SV129 and mutant: n = 7; C57Black/6: n = 6.
Infarcts were also significantly smaller in the mutant group (p < 0.05) when the volume of tissue injury in the wild-type group was calculated using the method of Swanson et al. (1990).
DISCUSSION
We examined the effects of transient focal ischemia in mutant mice lacking neuronal NOS gene expression and found significantly lower brain water content and infarct volume during the reperfusion period. Differences in infarct volume were independent of the anesthetic (halothane or α-chloralose) and were not due to group differences in arterial blood pressure, heart rate, blood gases (Pao2 and Paco2), or blood pH.
C57Black/6 mice were more susceptible to ischemic injury than SV-129 or neuronal NOS mutants. After transient MCA occlusion, infarct size in SV-129 mice was small compared to permanent occlusion in the same strain. However, infarct volume was quite large in C57Black/6 mice, despite the apparent restoration of rCBF during reperfusion in both strains. Barone et al. (1993) reported that mouse strains differed in their susceptibility to focal cerebral ischemia, in part due to vascular anatomical differences at the level of posterior communicating arteries. However, we did not observe vascular abnormalities that could account for such group differences (Huang et al., 1994).
We previously reported that smaller infarcts developed in neuronal NOS mutants after permanent MCA occlusion. Furthermore, we established recently that CA1 hippocampal neurons survive better in neuronal NOS mutant mice after transient global ischemia (Panahian et al., 1995). Cortical cultures from neuronal NOS mutant mice appear relatively resistant to NMDA neurotoxicity as well (Dawson et al., 1996). On the other hand, inhibition of endothelial NOS activity in neuronal NOS mutants (by treatment with L-nitroarginine) increased ischemic brain damage (Huang et al., 1994), whereas neuronal NOS inhibition by L-nitroarginine decreased ischemic brain damage in endothelial NOS deficient mice (Huang et al., 1996). Administration of 7-nitroindazole, an inhibitor with somewhat greater selectivity for the neuronal isoform in vivo, decreased infarct volume after permanent MCA occlusion in rats (Yoshida et al., 1994). Results were more ambiguous after administering nonselective inhibitors of NOS (Dalkara and Moskowitz, 1994). Taken together, our results confirm that inhibition of neuronal NOS may well be neuroprotective. By contrast, endothelial NOS inhibition may promote injury, perhaps by hemodynamic mechanisms (Dalkara and Moskowitz, 1994; Kerwin and Heller, 1994).
Oxygen free radicals may enhance ischemic neuronal damage after transient ischemia (Kontos, 1989; Hallenbeck and Dutka, 1990). For example, inhibitors of lipid peroxidation such as tirilazad afforded protection against neuronal damage after transient MCA occlusion in spontaneously hypertensive rats, but not after permanent MCA occlusion (Xue et al., 1992). Lipo some-entrapped superoxide dismutase was also effective (Imaizumi et al., 1990), as was polyethylene glycol superoxide dismutase (Liu et al., 1989). Furthermore, smaller infarcts developed in transgenic mice overexpressing human CuZn superoxide dismutase after transient MCA occlusion, but not after permanent MCA occlusion (Chan et al., 1993; Yang et al., 1994). In the neuronal NOS mutants, infarct volume after transient occlusion was considerably smaller (i.e., 69% smaller than in wild-type mice) than after permanent MCA occlusion (39% smaller) in wild-type mice (Huang et al., 1994). Hence, NO production may be more deleterious in the context of reversible ischemia possibly due to the augmentation of both superoxide and peroxynitrite anion formation. Of course, the observed differences may also reflect a slower rate of infarct maturation after transient occlusion; measurements of infarct size at 72 h could clarify this point. Several other cytotoxic mechanisms deserve consideration, including NO augmentation of ADP-ribosylating mechanisms (Zhang et al., 1994), and formation of NO-iron-sulfur complexes (Dawson et al., 1992a/Stamler et al., 1992).
NO formation may also contribute to the production of vasogenic brain edema. For example, Nω-nitro-L-arginine administration decreased brain edema after permanent MCA occlusion in rats (Nagafuji et al., 1992) and cold-induced edema in mice (Oury et al., 1993). By contrast, brain edema developed following parenchymal injections of FeCl2, which initiates oxygen free radical formation as part of the Haber-Weiss reaction (Willmore and Rubin, 1982; Willmore et al., 1983). The decreased brain water content measured in neuronal NOS mutants may indeed indicate less peroxynitrite formation and oxygen free radicals or perhaps reflects the smaller volume of ischemic tissue in this group (Slivka et al., 1995). Edema measurements in groups matched for infarct size will be required to clarify this point.
Although the formation of NO or a reaction product may promote ischemic brain damage, we cannot rule out the possible impact of unidentified compensatory mechanisms evolving during development as a consequence of neuronal NOS gene deletion. For example, infarct sparing could develop as a consequence of up-regulation of superoxide dismutase activity or a change in the sensitivity of lipid peroxidation or oxygen free radical production, or a change in the number or affinity of NMDA receptors or calcium channel binding sites in striatum or cerebellum. Whether lipid peroxidation and/or free radical formation are decreased after transient focal or global ischemia in neuronal NOS mutants would be important to determine. We recently found that spontaneous lipid peroxidation (thiobarbituric acid reaction) (Stocks et al., 1974) was not significantly different in spin homogenates of normal mutant and wild-type cortex and cerebellum (H. Hara and M. A. Moskowitz, unpublished data). Moreover, we observed a similar distribution of L-type calcium channel binding sites between wild-type and mutant mice, using in vitro [3H]PN200–110 receptor autoradiography (H. Hara and M. A. Moskowitz, unpublished data). Despite the importance of continuing such studies, the pharmacological evidence to date (Yoshida et al., 1994; Zhang et al., 1995) points to the importance of neuronal NOS deletion as the predominant neuroprotective mechanism.
In conclusion, mutant mice exhibit fewer neurological deficits and a decrease in brain water content after transient focal ischemia and decreased ischemic infarct volume. Hence, administering a specific inhibitor of neuronal NOS may become an important neuroprotective strategy in cerebral ischemia.
Footnotes
Acknowledgment:
This research was supported by the National Institute of Neurological Disorders Interdepartmental Stroke Program Project (NS10828, NS2683 to M.A.M., and NS33335 to P.L.H.) and by a sponsored research agreement with Bristol-Meyers Squibb (M.C.F.). P.L.H. is a Harcourt General Researcher. We thank Drs. Turgay Dalkara and Zhihong Huang for advice.