
Abstract
It is well established that tissue damage and functional outcome after experimental or clinical stroke are shaped by biologic sex. We investigated the novel hypothesis that ischemic cell death from neuronally derived nitric oxide (NO) or poly-ADP ribose polymerase (PARP-1) activation is sexually dimorphic and that interruption of these molecular death pathways benefits only the male brain. Female neuronal nitric oxide synthase (nNOS) knockout (nNOS−/−) mice exhibited exacerbated histological injury after middle cerebral artery occlusion (MCAO) relative to wild-type (WT) females, unlike the protection observed in male nNOS−/− littermates. Similarly, treatment with the nNOS inhibitor (7-nitroindozole, 25 mg/kg) increased infarction in female C57Bl6 WT mice, but protected male mice. The mechanism for this sexually specific response is not mediated through changes in protein expression of endothelial NOS or inducible NOS, or differences in intraischemic cerebral blood flow. Unlike male PARP-1 knockouts (PARP1−/−), female PARP1−/− littermates sustained grossly increased ischemic damage relative to sex-matched WT mice. Treatment with a PARP inhibitor (PJ-34, 10 mg/kg) resulted in identical results. Loss of PARP-1 resulted in reversal of the neuroprotective activity by the female sex steroid, 17β estradiol. These data suggest that the previously described cell death pathways involving NO and PARP ischemic neurotoxicity may be operant solely in male brain and that the integrity of nNO/PARP-1 signaling is paradoxically protective in the female.
Introduction
Clinical ischemic stroke is increasingly recognized as a sexually dimorphic disease. Most international databases consistently show that women enjoy lower stroke incidence relative to men until advanced age, yet early outcome from injury may be more favorable in males (Thorvaldsen et al, 1997; Sudlow and Warlow, 1997; Wyller, 1999). It is equally well established that tissue damage and functional outcome after experimental brain injury are shaped by biologic sex (for reviews, see Hurn and Macrae, 2000; McCullough and Hurn, 2003; Roof and Hall, 2000). Reproductive hormones clearly contribute to such differences in male and female pathobiology. A wealth of data emphasizes that estrogens and progestins alter neuronal survival after injury in vivo and in vitro (Wise et al, 2001; Garcia-Segura et al, 2001; Stein, 2001; Sribnick et al, 2004). However, the hormonal milieu may not fully account for sexual dimorphism in cerebral ischemia (Hurvitz et al, 1999; Vukmir, 2003). Emerging data suggests that cell death in brain may follow differing mechanistic paths depending on gender (Nunez et al, 2001; Lauterbach et al, 2001; Zhang et al, 2003; Schuessel et al, 2004), in addition to sex steroid exposure.
One well-studied mechanism of neuronal cell death in cerebral ischemia and trauma occurs by a serially linked set of events: overstimulation of neuronal nitric oxide synthase (nNOS) leading to toxic local elaboration of nitric oxide (NO), subsequent peroxynitrite formation and nitrosative DNA damage, activation of the DNA repair enzyme poly ADP ribose polymerase-1 (PARP-1), and PARP-1-induced mobilization of proapoptotic molecules such as apoptosis inducing factor (AIF) (Endres et al, 1998; Yu et al, 2002, 2003; Du et al, 2003; Hong et al, 2004; Plesnila et al, 2004). Key evidence in establishing NO toxicity/PARP-1 activation as a major cytotoxic mechanism has accumulated from exclusively male animals with targeted deletions of nNOS (nNOS−/−) or PARP (PARP−/−). In each case, these knockouts are resistant to brain injury from focal and global cerebral ischemic insults, as are primary neuronal cell cultures derived from fetal brain obtained from mixed sexes (Huang et al, 1994; Dawson et al, 1996; Eliasson et al, 1997; Endres et al, 1997; Samdani et al, 1997; Goto et al, 2002). Similar findings of neuroprotection using pharmacological inhibitors of nNOS and PARP-1 in male animals and mixed sex neuronal cell cultures have been widely reported (for reviews, see Beckman and Koppenol, 1996; Dawson and Dawson, 1996; Virag and Szabo, 2002; Virag et al, 2003; Ha and Snyder, 2000; Yu et al, 2003).
In contrast, we have previously observed that female nNOS−/− mice treated with irreversible middle cerebral artery occlusion (MCAO) do not benefit from the genetic mutation as do their male counterparts (Sampei et al, 2000). Using a reperfusion injury model, we now extensively evaluate whether the nNO-PARP1 pathway of ischemic cell death is present in the female animal and if this mechanism is skewed by the presence of the principal estrogen, 17-β estradiol (E2). We examined outcome in a reversible ischemia model, as reperfusion injury is an important contributor to ischemic damage from oxidative injury. Our findings suggest that this molecular cell death mechanism is sexually dimorphic in brain, and that the integrity of nNO/PARP-1 signaling is paradoxically protective in the female.
Methods
Experimental Groups
The present study was conducted in accordance with the NIH guidelines for the care and use of animals in research and under protocols approved by the Animal Care and Use Committee of the Johns Hopkins University. Knockout strains used in this study have been previously described (Huang et al, 1994; Eliasson et al, 1997; Goto et al, 2002). Genotype was determined by PCR via tail DNA, as described previously (Eliasson et al, 1997; Sampei et al, 2000).
Neuronal Nitric Oxide Synthase−/−
Age- and weight-matched homozygous nNOS-null mice were compared with wild-type (WT) controls (22 to 28 g). Ovary intact, randomly cycling female nNOS−/− were compared with intact WT female mice (n=15/group). Male nNOS−/− and WT mice (n=13/group) were also studied to confirm protection in male nNOS mutants, as has been previously described (Huang et al, 1994).
7-Nitroindazole Treatment
C57Bl6 WT mice (Charles River) of both sexes were injected immediately before MCAO with a selective nNOS inhibitor. The dose selected was previously determined to reduce injury from focal cerebral ischemia in males (Goyagi et al, 2001; Nanri et al, 1998). 7-Nitroindazole (7NI) (25 mg/kg) or oil vehicle (peanut oil with DMSO) was administered intraperitonealy at the onset of MCAO to male (n=10 vehicle, 12 7NI) and female mice (n=12/group). In a separate cohort, females (n=5/group) were injected with either 7-NI or oil vehicle, and cerebral blood flow (CBF) was quantified by C14-iodoantipryine (IAP) autoradiography at 90 mins of occlusion.
Estradiol Treatment
Ovariectomized (OVX) WT and nNOS−/− females, with or without E2 treatment, were studied (n=12/group). 17-β Estradiol was administered by subcutaneous Silastic capsule (0.062″ID/0.125″OD) filled with either 0.035 mL of sesame oil or E2 (180 μg/mL; Sigma, St Louis, MO, USA) 7 days before ischemia. Additional cohorts of PARP 1 null females were OVX and treated with E2 or vehicle 1 week before MCAO (n=10/group). Plasma E2 was measured by radioimmunoassay (Diagnostic Products Corp, Los Angeles, CA, USA) as previously described (Dubal et al, 2001; McCullough et al, 2003). Males of both genotypes were also implanted 1 week before MCAO, as further controls.
Poly-ADP Ribose Polymerase−/−
Ovary intact, random cycling female PARP-1 null mice were compared with heterozygous (PARP+/−) or WT littermates (n=12, 14, and 12, respectively). Male PARP−/− and WT mice (n=10/group) were also studied to confirm protection in male mutants (Eliasson et al, 1997).
Poly-ADP Ribose Polymerase Inhibition with PJ-34
SV/129 WT mice (Taconic) of both sexes were injected intraperitoneally immediately before MCAO with the potent PARP-1 inhibitor, PJ-34 (10 mg/kg) or saline control (Garcia Soriano et al, 2001) to WT male (n=10 vehicle, 12 PJ-34) and female mice (n=12/group).
Ischemic Model
Focal cerebral ischemia was induced by 2 h of reversible right MCAO under halothane anesthesia followed by 22 h of reperfusion, as previously described (McCullough et al, 2004). Intraischemic and 22 h neurological deficit (NDS) was scored as follows: 0, no deficit; 1, forelimb weakness and torso turning to the ipsilateral side when held by tail; 2, circling to affected side; 3, unable to bear weight on affected side; and 4, no spontaneous locomotor activity or barrel rolling. Any animal without deficit was excluded from the study. At end-ischemia, the animal was briefly reanaesthetized and reperfusion was initiated by filament withdrawal. Monitoring of physiological variables and laser Doppler flowmetry (LDF) was performed in companion cohorts for all groups as previously described (n=5/group in all genotypes and pharmacological cohorts; McCullough et al, 2004; Sawada et al, 2000).
Terminal Histopathology
Slices were incubated in 2,3,5-triphenyltetrazolium chloride (McCullough et al, 2004; Sawada et al, 2000) and analyzed as previously described (Inquiry, Loats Associates, Westminster, MD, USA). Infarct size was expressed as a percentage of the contralateral hemisphere/structure after correcting for edema (McCullough et al, 2004; Sawada et al, 2000).
Intraischemic Cerebral Blood Flow
Regional CBF was measured with 14C IAP autoradiography, as previously described (McCullough et al, 2004; Alkayed et al, 1998). Autoradiographic images representing 5 coronal levels (+2, +1, 0, −1, and −2 mm from bregma, 3 images each) were digitized and regional CBF determined by image analysis software (Inquiry, Loats Associates).
Western Blot Analysis of Nitric Oxide Synthase Isoforms
WT and nNOS mice were decapitated after 2 or 22 h of reperfusion or sham surgery (n=5/group). The cerebellum was removed, and the brain was divided into ischemic (right) and nonischemic (left) hemisphere and flash-frozen in 2-methyl butane cooled on dry ice and stored at −80°C until use. The brain tissue was homogenized in 1/3 (w/v) of 10 mmol/L Tris-HCl, pH 7.6, 1 mmol/L EDTA, 0.1% SDS, 1 mmol/L phenylmethylsulphonylfluoride (PMSF) then centrifuged for 1 h (10,000g). Protein concentrations in the supernatants were determined by BCA protein assay (Pierce, Rockford, IL, USA). Equal amounts of protein were loaded per lane and separated on a 7% SDS–poly acrylamide gel (Bio Rad, Santa Cruz, CA, USA) with transfer to a nitrocellulose membrane. Blots were probed with anti-eNOS (1:2,000, Transduction Labs), anti-P-eNOS (Ser 1177) (1:200, Santa Cruz Biotechnology, Hercules, CA, USA) (Dimmeler et al, 2000), anti-nNOS (1:2,000; Santa Cruz Biotechnology), or anti-iNOS (1:1,000 Santa Cruz Biotechnology) in 5% milk overnight at 4°C, with subsequent incubation with biotinylated goat-anti-rabbit IgG and horseradish peroxidase (Amersham, Piscataway, NJ, USA). Blots were incubated with ECL detection reagents and visualized on Hyperfilm ECL (Amersham). Actin was used as a control for protein gel loading (1:5000, Sigma, St Louis, USA). Blots were analyzed by NIH image software.
Statistical Analysis
All data are expressed as means±s.d. Physiological variables and infarct volume were analyzed by one-way ANOVA with a post hoc Newman–Keuls test to correct for multiple comparisons. Estradiol levels were compared by t-test with Bonferoni correction. Regional CBF was assessed by one-way ANOVA with a post hoc Newman–Keuls test to evaluate differences between groups by treatment at end-ischemia.
Results
Neuronal Nitric Oxide Synthase Signaling Paradoxically Protects the Female Brain During Ischemia
Infarction volume was dramatically increased in ovary-intact female nNOS−/− as compared with female WT mice (Figure 1), unlike the protection observed in both the cortex (WT 59±21.2 versus 37.6±14.9 nNOS) and the striatum (WT 60.8±20.3 versus 36.5±12 nNOS) of male nNOS−/− mutants (total infarct; WT 55.6±18.9 versus nNOS 30.6±11.9). NDS at the time of killing reflected these differences in infarct size. As expected, intact WT females exhibited low scores (1.5±0.7) and small infarcts because of the protective effect of endogenous estrogen, similar to nNOS males (1.7±0.6). Higher scores were seen in WT males (3.1±0.8) and nNOS females (3.4±0.9). Baseline (not shown) and intraischemic physiologic variables were not different among the groups (Table 1). Intraocclusion CBF as assessed by LDF was similar in all groups (female nNOS−/− 16.7%±6.7% of baseline signal; female WT 16.4%±8.5%; male nNOS−/− 14.3%±8.9%; male WT 15.9%±10.5%). Plasma E2 levels varied in the randomly selected females because of the estrus cycle, but E2 did not differ between nNOS−/− (48±45 pg/mL) and WT (56±46 pg/mL) females.
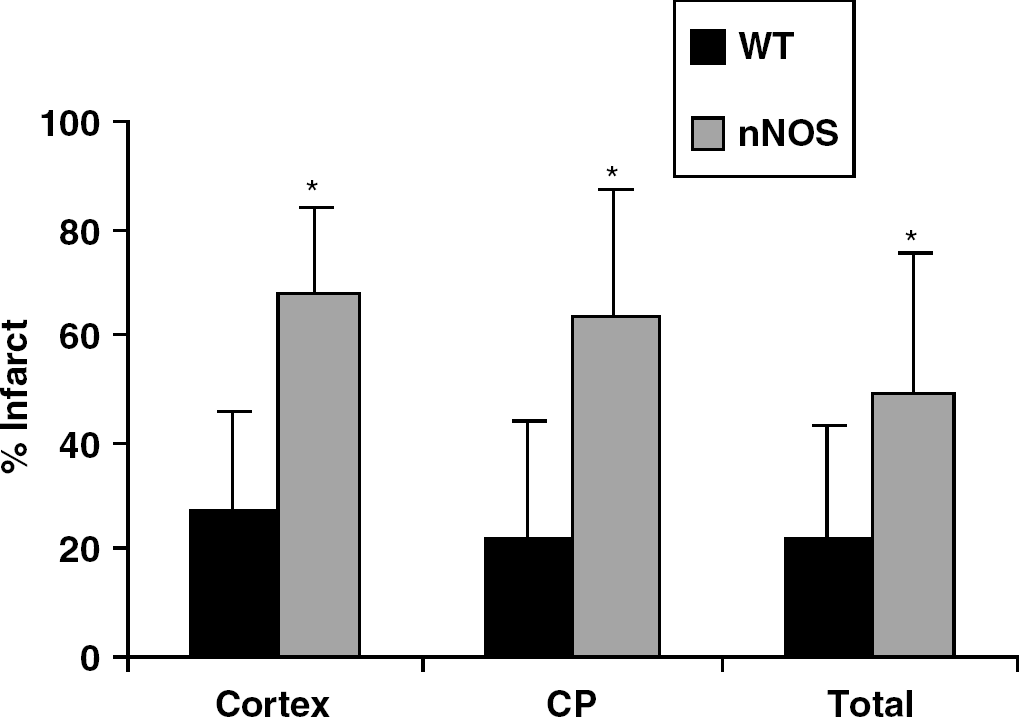
Infarct size (expressed as % of contralateral structure) in ovary-intact nNOS−/− versus wild-type (WT) female mice. There is a significant increase (*P<0.001) in total, cortical (CTX) and striatal (CP) infarct volume in nNOS−/− females compared to WT.
Physiologic measurements in nNOS−/− and WT mice
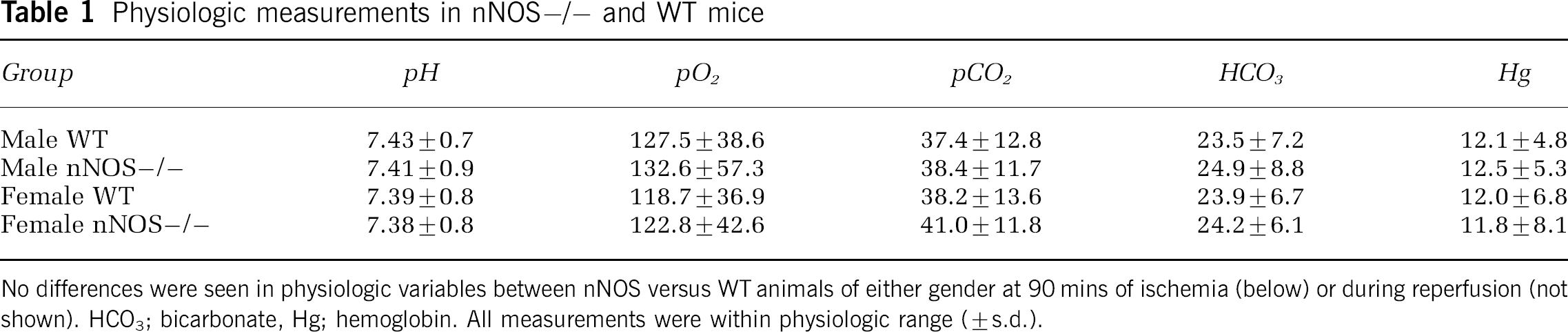
No differences were seen in physiologic variables between nNOS versus WT animals of either gender at 90 mins of ischemia (below) or during reperfusion (not shown). HCO3; bicarbonate, Hg; hemoglobin. All measurements were within physiologic range (±s.d.).
No Gender Differences were Found in Nitric Oxide Synthase Isoforms
As expected, both male and female nNOS−/− had complete absence of nNOS protein at all time points examined (Figure 2 at 22 h of reperfusion; 2 h data not shown). Similarly, endothelial nitric oxide synthase (eNOS) expression was not different in the female versus male (Figure 2), either at baseline or after stroke. P-eNOS was lower in sham WT (left hemisphere) and in sham nNOS−/− females compared with all other groups. A slight increase in P-eNOS in stroke animals compared with sham at 24 h was seen; however, no gender differences in p-eNOS expression existed at either 2 h or 24 h after stroke. Evaluation of iNOS blots showed an increase in iNOS in the contralateral nonischemic hemisphere in both male and female nNOS−/− 24 h after stroke (Figure 2) compared with WT but no differential expression between genders.
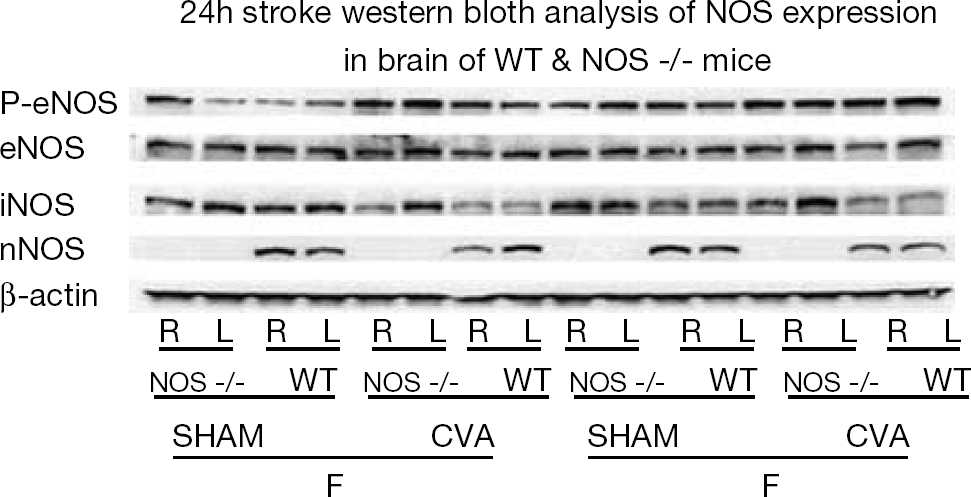
Expression of nitric oxide synthase (NOS) isoforms in nNOS−/− and wild-type (WT) mice 24 h after stroke (CVA; cerebrovascular accident) or sham surgery. No nNOS protein was seen in nNOS−/− mice (M=male; F=female) in either the ischemic (R) or the contralateral nonischemic hemisphere (L). No gender differences in eNOS (endothelial NOS), P-eNOS (phosphorylated eNOS; Ser1177) or iNOS (inducible NOS) after stroke were noted.
Pharmacological Neuronal Nitric Oxide Synthase Inhibition Increases Damage in the Female
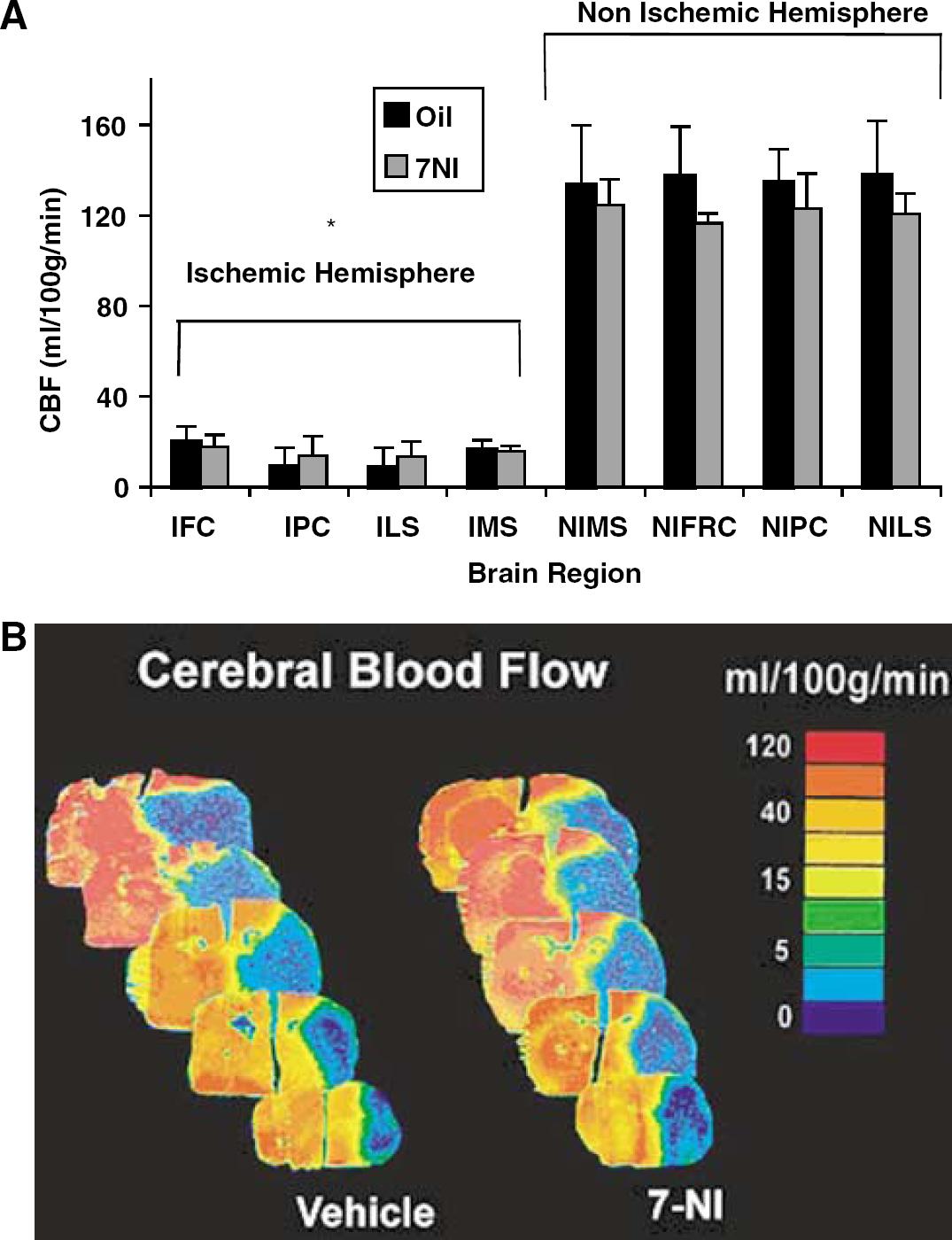
We further tested the effect of blocking nNOS signaling with a well-studied pharmacological inhibitor and obtained results consistent with those from female knockouts. 7NI exacerbated hemispheric tissue damage in WT females (Figure 3) in both cortex (7NI: 58%±26.3%; oil: 21%±17.3%) and striatum (7NI: 64%±25.9%; oil: 22%±18.1%; % of contralateral structure). Similarly, oil-treated females had lower NDS relative to 7NI-treated females (female WT 1.4±0.07 versus female 7NI 3.5±0.9, P<0.01). As expected, males treated with 7NI sustained significant reduction in infarct volume as compared with oil-treated males (Figure 3). Physiologic variables were not different among groups (data not shown). Intraischemic LDF fell during occlusion in all animals (female 7NI: 15.7%±5.9% of baseline; female oil: 13.9%±7.8%; male 7NI: 16.8%±4.7%; male oil: 14.6%±7.6%) and returned to baseline on reperfusion. To further confirm that intraischemic CBF was equivalently reduced in both female treatment groups, we mapped hemispheric blood flow with C14-IAP autoradiography. End-ischemic regional blood flow was not different in 7NI relative to oil-treated females in either the ischemic or contralateral nonischemic hemisphere (Figures 4A and 4B).
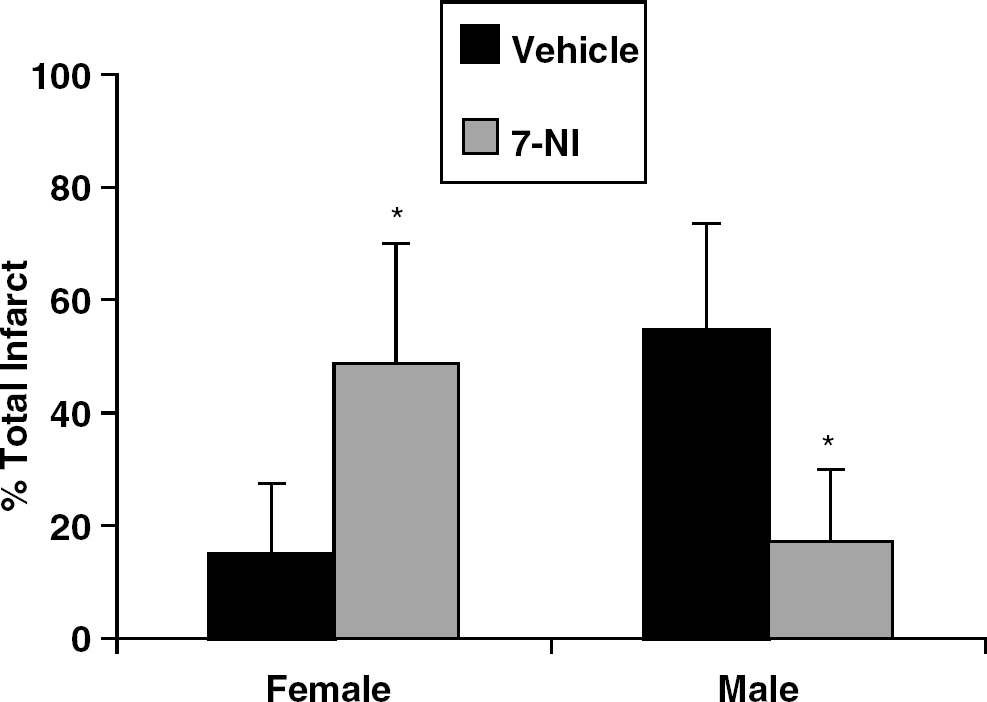
The effects of 7-nitroindazole (7-NI) in male and female WT mice. 7-Nitroindazole-treated male mice had significant reductions (*P<0.001) in total infarct volume (expressed as % of contralateral hemisphere) compared with oil treated male controls. Ovary intact females had a significant exacerbation in tissue damage after treatment with 7-NI compared with treatment with oil vehicle (*P<0.001).
(
Loss of Neuronal Nitric Oxide Synthase Blocks Protective Estradiol Signaling in Female
As only female nNOS−/−mice show increased ischemic damage, we hypothesized that neuroprotective estradiol signaling might be impaired. As expected, ovariectomy increased, and physiologic E2 replacement decreased, infarction in WT females (Figure 5). NDS were significantly improved by E2 treatment.
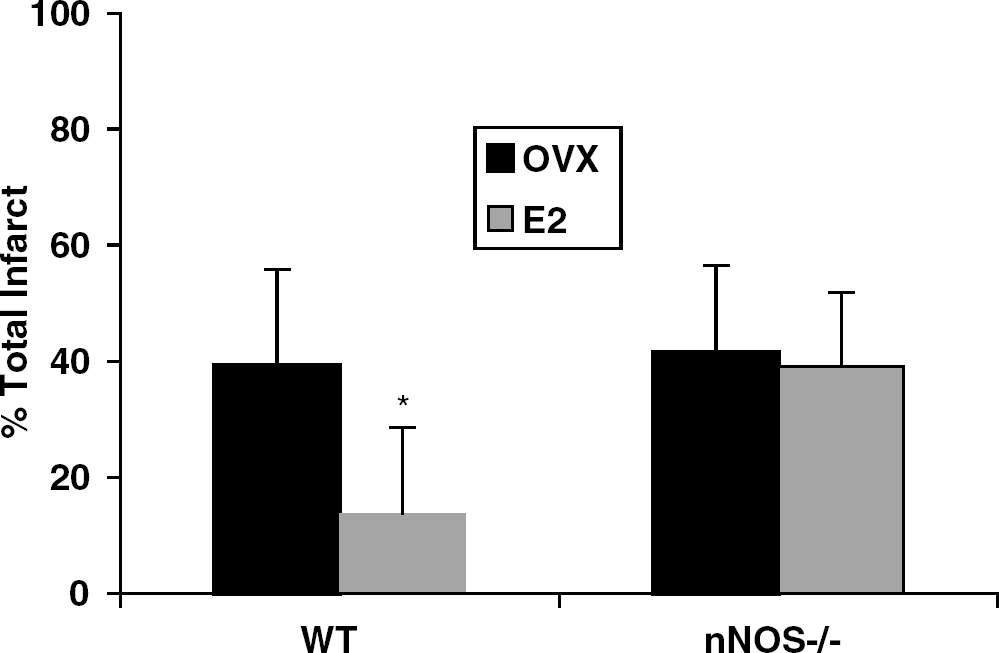
Total infarct volume in ovariectomized (OVX) nNOS−/− and wild-type (WT) females exposed to physiologic doses of exogenous estrogen (E2 replaced). Wild-type OVX females treated with oil had significantly larger infarct volumes (expressed as a % of the contralateral hemisphere) than WT OVX treated with E2 (*P<0.01). Ovariectomized nNOS−/− females were insensitive to the neuroprotective effects of E2.
In contrast, ovariectomy with or without E2 replacement had no effect on ischemic damage in nNOS−/− females, suggesting that estradiol's known ability to provide ischemic protection is blocked (Figure 5). Cortical and striatal infarction volumes as well as NDS were unchanged by E2 treatment in nNOS−/− females. Plasma E2 was restored to physiologic range in all implanted females (OVX WT+oil 9±7.3 pg/mL; OVX nNOS−/−+oil 8±10.6; OVX WT+E2 115.2±92.8; OVX nNOS−/− 86.7±52.4; P<0.01 for oil versus E2 in both genotypes). Identical E2 treatment reduced total, striatal and cortical infarction in WT and nNOS−/− males (Figure 6). An additive effect of E2 treatment and gene deletion was seen in nNOS−/− males.
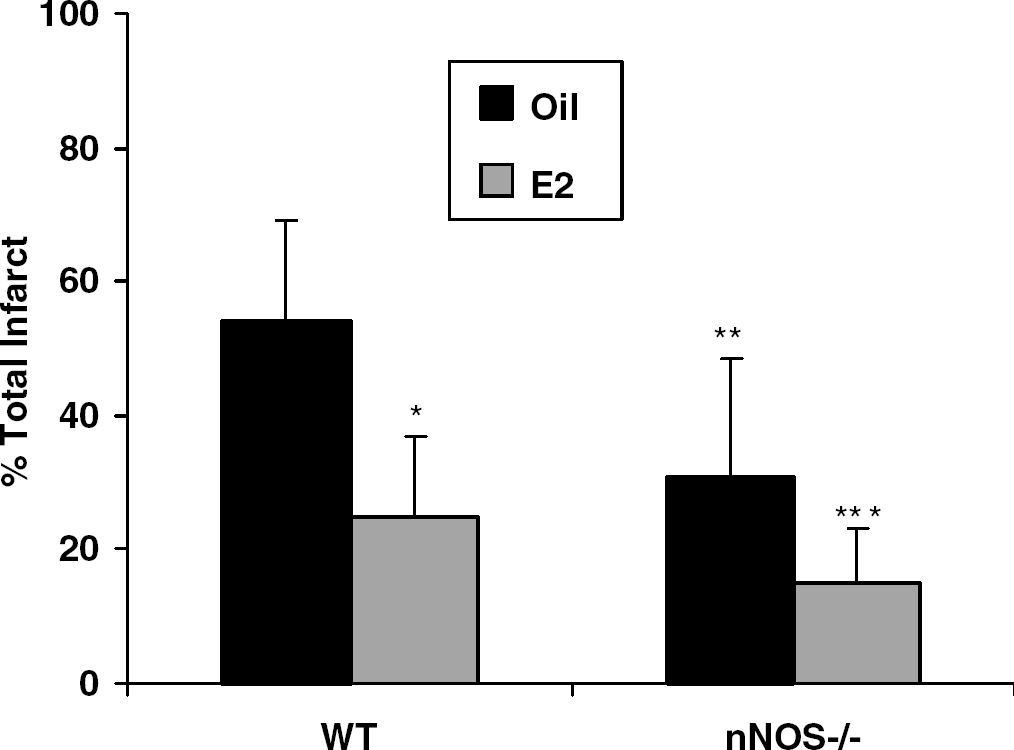
Additive neuroprotective effect of estrogen (E2) and nNOS−/− in nNOS-deficient males. There was a significant reduction in infarct damage in E2-treated versus oil-treated wild type (WT) males (*P<0.01) showing the neuroprotective effect of physiologic levels of E2. Oil-treated nNOS−/− males had significantly smaller infarcts (**P<0.05) than oil-treated WT males. The protective effect of E2 was also seen in the nNOS−/− males, reducing infarct beyond that seen with the gene deletion or E2 alone (***P<0.05).
Poly-ADP Ribose Polymerase Deletion Exacerbates Damage in Intact Female Mice
To further evaluate the nNO-PARP-1 signaling pathway in female ischemia, we evaluated outcome from MCAO in WT versus PARP−/− females. Female WT mice sustained smaller infarctions than either heterozygote PARP (+/−) or homozygote PARP (−/−) females (Figure 7). There were no differences in LDF, estrogen levels, or physiologic parameters among the groups (data not shown). Of the 19 PARP−/− females subjected to stroke, only 12 survived. NDS were higher in the PARP−/− females (3.7±1.3) and PARP+/− (2.5±0.6) compared with WT (1.3±0.06). As previously shown, male mice benefit from genetic deletion of PARP-1 in our model of focal ischemia (Eliasson et al, 1997; Goto et al, 2002) compared with WT males (total infarct: male WT, 58.9%±36.1%; PARP−/− 26.4%±26.9%, P<0.01).
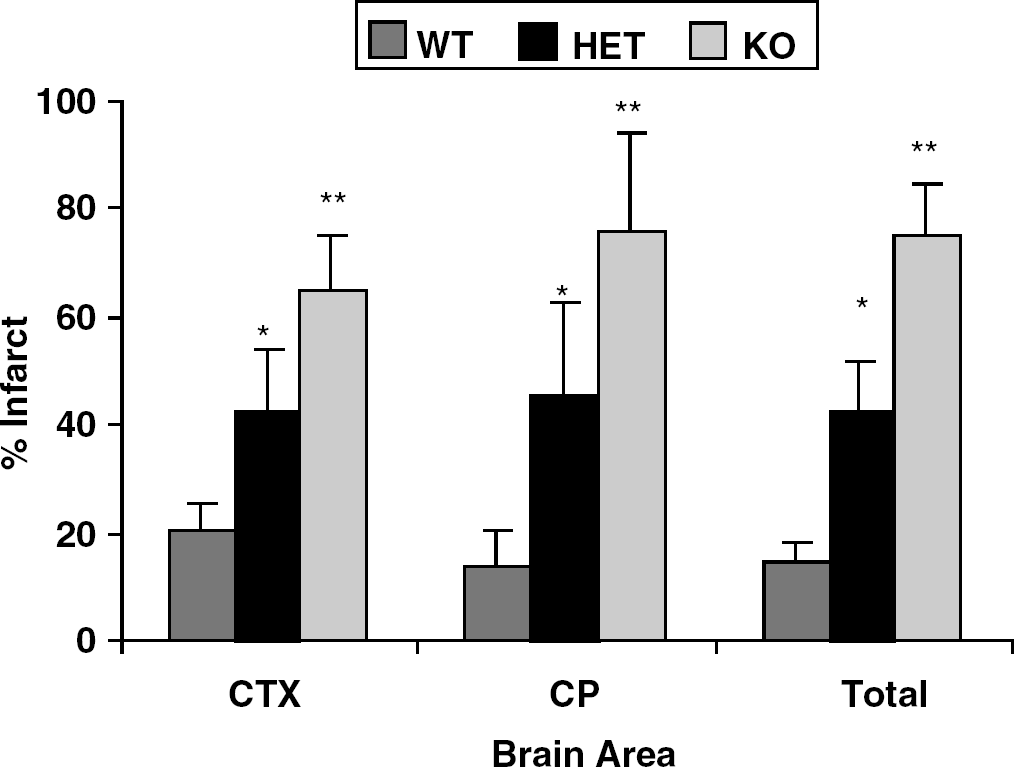
Infarct volume in with WT, PARP heterozygote (HET) and PARP knockout (KO) ovary-intact female mice. A significant increase in infarction was seen in both HET and KO compared with WT (P<0.01). There was an additional increase in total, cortical (CTX) and striatal (CP) infarction volume in the KO compared with HET (**P<0.01) mice.
Poly-ADP Ribose Polymerase Loss Results in Full Reversal of Estradiol's Neuroprotection
While nNOS−/− females were simply refractory to estradiol's neuroprotection, female PARP−/− showed a fully reversed response to the steroid during ischemia. In the absence of PARP-1, ovariectomy paradoxically decreased ischemic damage, while E2 replacement restores infarction volume to that of a PARP−/− female producing ovarian estrogens (Figure 8). E2 increased infarction volume in both cortex and striatum (n=10/group) of PARP−/− mice. As expected, reductions in infarct were seen in estrogen-replaced OVX WT littermates. NDS were increased by E2 treatment in PARP−/− mice (OVX PARP−/− +E2; 3.8±1.6, OVX PARP−/−+oil; 3.2±1.2), and reduced in WT OVX littermates treated with E2 (OVX WT+E2; 1.3±0.8, OVX WT+oil; 2.6±1.2). Estrogen levels were increased in E2-treated females of both genotypes (OVX PARP+oil; 11.6±25.1, OVX PARP+ E2; 95.7±88.6, OVX WT+oil 14.1±23.6, OVX WT+E2; 87.6±74.8, P>0.05).
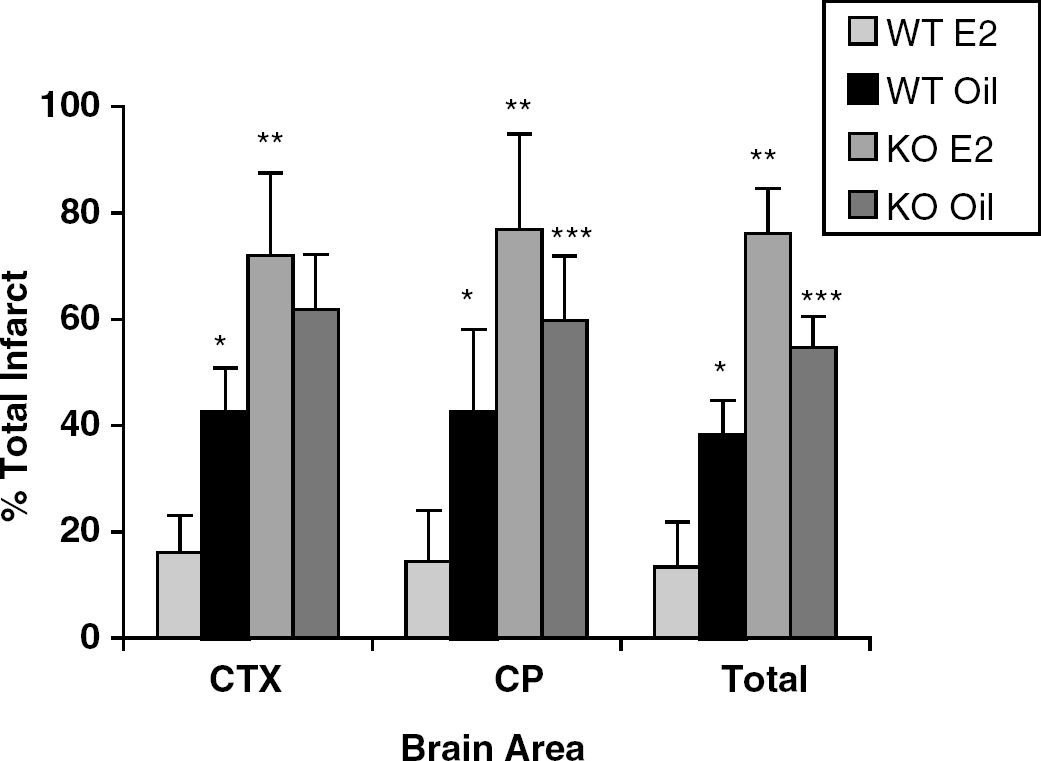
Effect of estrogen on infarction volume in PARP−/− females. Physiological levels of E2 were restored to ovariectomized (OVX) PARP−/− and wild-type (WT) female mice. Wild-type females had significant reductions in total, cortical (CTX) and striatal (CP) infarct volumes after E2 replacement compared with oil-treated WT females (*P<0.01). PARP−/− females showed increased damage compared with WT (**P<0.01). Interestingly, the neuroprotective effect of E2 was completely absent in PARP−/− females. E2 treatment exacerbated stroke damage; both striatal and total infarct volumes were significantly higher in E2-treated versus oil-treated PARP−/− mice (***P<0.05).
Pharmacological Inhibition of Poly-ADP Ribose Polymerase also Exacerbates Stroke Damage in Female Mice
We further tested the effect of blocking PARP-1 signaling with the pharmacological inhibitor PJ-34 (10 mg/kg intraperitonealy at MCAO onset, Jagtap et al, 2002; Abdelkarim et al, 2001) in ovary-intact WT female mice and obtained results consistent with those from female PARP-1- knockouts (Figure 9). Increased damage was observed in both cortex and striatum of PJ-34-treated mice with corresponding NDS (female sal; 1.2±0.6 versus female PJ-34; 3.6±1.2, P<0.001). Males treated with PJ-34 sustained significant reduction in both cortical and striatal infarct volume compared to saline-treated males.
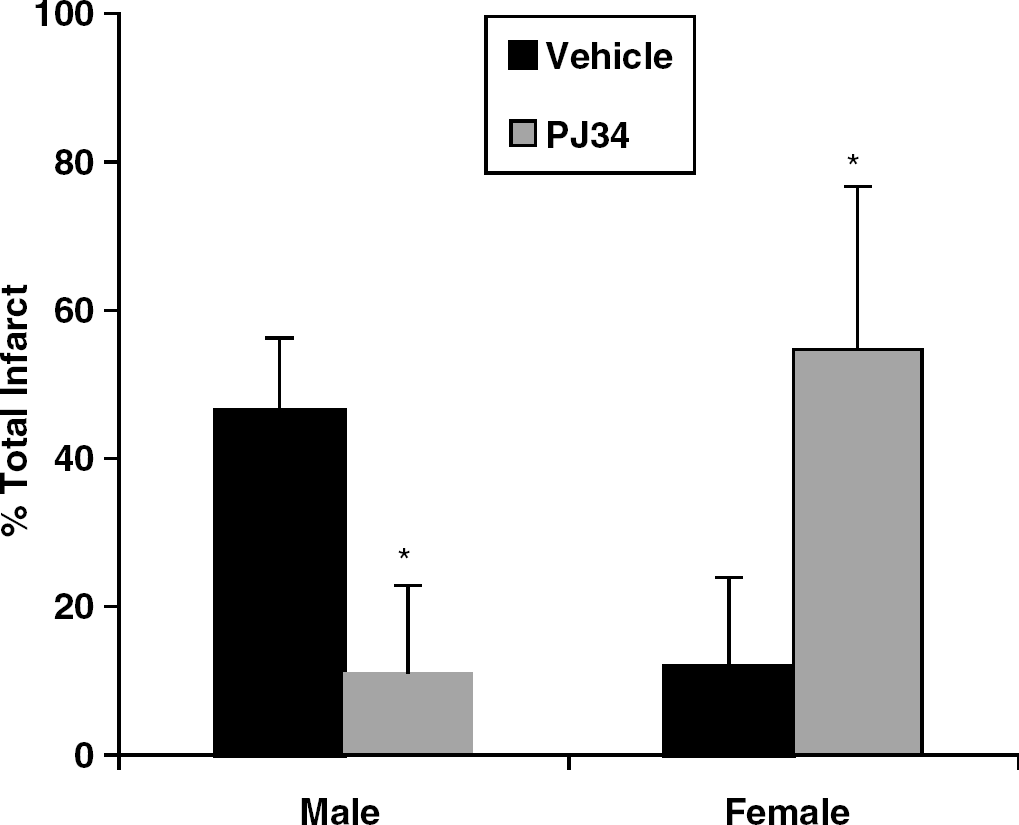
The effects of the selective poly-ADP ribose polymerase (PARP-1) inhibitor PJ-34 in wild-type (WT) mice of both genders. Treatment with PJ-34 at ischemic onset reduced total infarction in male mice compared with saline-treated controls (*P<0.001). A significant increase in ischemic damage was seen in PJ-34-treated females compared with control (*P<0.001).
Discussion
There are three novel and important findings in this study. First, ischemic NO toxicity in brain is not a universal mechanism but is highly sex-specific. In female, but not male brain, loss of NO through targeted gene deletion or pharmacological inhibition increases ischemic damage and renders neurons insensitive to the neuroprotection ordinarily provided by an important female sex steroid, 17β estradiol. These observations are not explained by compensatory upregulation of other NOS isoforms as can occur with hormone exposure, (Singh et al, 2000; Hayashi et al, 1997; Garcia-Duran et al, 1999; Dimmeler et al, 2000; Grohe et al, 2004) preservation of ischemic CBF.
Second, PARP-1's importance in ischemic neuronal death is also sexually dimorphic. Poly-ADP ribose polymerase activation has been widely studied as a potentially deleterious response to nitrosative or oxidative DNA damage subsequent to ischemic insult (Hong et al, 2004; Virag and Szabo, 2002; Virag et al, 2003; Ha and Snyder, 2000; Eliasson et al, 1997; Mandir et al, 2000; Pieper et al, 1999; Endres et al, 1997, 1998). In contrast to the male where stroke-induced PARP-1 activation promotes bioenergetic failure, proapoptotic signaling, mitochondrial dysfunction, and cell death, PARP-1 is utilized to restrict tissue damage in the female. Furthermore, we demonstrate that loss of PARP-1 abrogates estradiol's neuroprotective potential and produces a new state completely at paradox to WT females. In this new state, ovariectomy improves ischemic outcome rather than enhancing damage, and estradiol replacement increases, rather than decreases, infarction. These observations are complex in their whole animal origin, and precise events leading to nNO and PARP-1's sexual dimorphism in ischemic pathology are only initially dissected in these experiments. However, the findings exemplify a novel example of one molecular pathway of ischemic pathology that clearly diverges in male and female brain. The data also emphasize that preclinical research in stroke and neuroprotection must stratify by biological sex if we are to understand important sexually dimorphic mechanisms of cell death and recovery.
The role of NO/peroxynitrite (ONOO) toxicity and PARP-1 activation in mediating cell death is well understood in the male (Ha and Snyder, 2000; Mandir et al, 2000; Narasimhan et al, 2003). Others have reported similar findings in iNOS mutants where females, unlike males, sustain no reduction in ischemic damage relative to WT controls (Loihl et al, 1999). However, our observation that removal or inhibition of nNOS or PARP-1 signaling exacerbates ischemic damage in females was unexpected.
One obvious implication is that nNOS and PARP-1 are not molecular targets for therapeutic inhibition in the female. We and others have characterized the male response to neuronal injury with the knockout strains used in the present experiments (Huang et al, 1994; Eliasson et al, 1997; Endres et al, 1997; Sampei et al, 2000). In addition, contemporaneous control experiments in males were incorporated into the present study design to assure consistency with previous observations. Because these mice have targeted gene deletion for life, we confirmed each finding with preischemic administration of a pharmacological inhibitor. 7NI and PJ-34 are among the most specific inhibitors available for nNOS and PARP, respectively, and both greatly exacerbate ischemic damage selectively in females. Doses administered in the study were based on efficacy in previous experiments in males, and we did not examine a full range of doses in these experiments (Goyagi et al, 2001; Abdelkarim et al, 2001; Coert et al, 1999). While it is possible that pharmacokinetics for these agents differ by sex, it seems unlikely that such a factor could explain the combined and perfectly consistent observations in knockouts and drug-treated cohorts. The gender of the experimental animal, which is often not reported, could also explain some of the published discrepancies on the effectiveness of nNOS inhibitors in vivo (see Coert et al, 2003).
A variety of data suggest in vivo, and more recently in vitro, that the female brain (Zhang et al, 1998; Alkayed et al, 1998; Vannucci et al, 2001; Carswell et al, 2000) and female neurons (Zhang et al, 2003; Du et al, 2004) are less sensitive to ischemic and toxic stressors than male equivalents. Without doubt, some proportion of this resilience is explained by endogenous neuroprotection via estradiol. However, the present data emphasize that female ‘tolerance’ is also strongly mediated through nNOS and PARP-1, regardless of the presence or absence of estradiol. Loss of either of these enzymes greatly enhances infarction in females. What remains to be assessed is whether ischemic activation of these enzymes directly protects against cell death or if protection is indirect, that is, blocking nNOS or PARP-1 unveils a collateral death mechanism more lethal to female than male brain. We hypothesized that ischemic NO production could contribute to preservation of cerebral perfusion in an unanticipated manner for females. A vast literature has dissected the unique blood flow regulation that occurs in female brain, as a consequence of eNOS activation via female sex steroids (for a review, see Chambliss and Shaul, 2002). However, this is not a likely mechanism, given that intraischemic LDF and C14 IAP studies failed to show differences among treatment cohorts. It is important to note that these findings do not exclude an important role of improved cerebral perfusion during reperfusion in E2-treated brains. We have previously documented a transient effect of estrogen on postischemic hypoperfusion (McCullough et al, 2001) and estrogens have been shown to improve vasodilation via an enhancement of nNOS expression and activity (Pelligrino et al, 1998). This could be a possible explanation for nNOS-mediated protection against cell death in intact and estrogen-treated females, yet the fact that OVX females showed equivalent infarcts to intact and E2-treated nNOS−/− females and the demonstration of an additive neuroprotective effect of E2 and nNOS−/− in males make this explanation less likely.
nNOS is part of a central dogma that relates glutamate and prooxidant injury mechanisms, and NO's neuronal toxicity is largely dependent on its avid reaction with the oxidant species, superoxide, to form peroxynitrite (Beckman and Koppenol, 1996; Endres et al, 1998; Beckman et al, 1994; Virag et al, 2003). One explanation for the surprising observation that nNOS function protects, rather than kills, is through a simple but unique combination of factors: a female insensitivity to ONOO and a high sensitivity to superoxide. Recent data in cortical neurons cultured separately from male and female fetuses suggest that female neurons are strikingly less sensitive to glutamate or ONOO toxicity than male neurons, applied across a range of concentrations and exposure times (Du et al, 2004). This sexually dimorphic tolerance is not mediated through estradiol, and estradiol treatment in conjunction with ONOO enhances female, but not male, cell death. Furthermore, sex-specific activation of antioxidant systems in vivo and in vitro has been reported because of basal gender differences and the activity of estrogens (Du et al, 2004; Kume-Kick et al, 1996; Kume-Kick and Rice, 1998; Gridley et al, 1998; Chen et al, 1999; Strehlow et al, 2003). In traumatic brain injury, female transgenic mice overexpressing human SOD1 benefit in selected brain regions more greatly than male hSOD overexpressors (Igarashi et al, 2001). We have also observed that female mice benefit more robustly from SOD-1 overexpression in ischemic brain injury as compared with males, although both sexes have improved outcomes (Sampei et al, 2000). SOD plays a critical role in the male response to stroke by reducing available NO/ONOO. Treatment with 7-NI gave no additional benefit to SOD overexpression after transient ischemia in males (Kamii et al, 1996). However, our findings suggest that sensitivity to superoxide can be selectively high in females and superoxide-mediated cell death could be a dominant and underappreciated feature. If so, then NO would act as an important sink for superoxide, as has been previously reported to occur in estradiol-treated cultured endothelium (Arnal et al, 1999). Rapid complexing of NO and superoxide could be greatly beneficial to superoxide-sensitive female neurons that are also able to tolerate subsequent ONOO formation.
Ischemic PARP-1 activation has been largely shown to promote neuronal cell death both in vivo and in vitro (Dawson et al, 1996; Eliasson et al, 1997; Endres et al, 1997; Goto et al, 2002). Poly-ADP ribose polymerase is a nuclear enzyme and the most heavily studied of the PARP family. The enzyme is activated specifically by DNA strand breaks and participates in DNA repair and replication, in part by an NAD-dependent ribosylation reaction (de Murcia and Menissier de Murcia, 1994; de Murcia et al, 1997; for a review, see Szabo and Dawson, 1998). Excessive DNA damage, such as occurs in ischemia, nitrosative, and oxidant injury, results in enzyme hyperactivation, energy depletion, and early cell death via necrosis. Poly-ADP ribose polymerase has also been shown to be a key factor in AIF translocation and AIF-induced, caspase-independent cell death in apoptosis-like processes (Yu et al, 2002; Komjati et al, 2004; Plesnila et al, 2004). Studies of ischemic PARP-1 activation to date have been carried out nearly exclusively in male animals and mixed sex cell culture. A single report in PARP-1−/− newborn mice of both genders indicates that outcome from hypoxia–ischemia is improved by targeted gene deletion in males, but not in females (Hagberg et al, 2004). Therefore, our present observation of protective PARP-1 signaling in female ischemia requires further understanding of PARP-1's mechanism. Although histological damage in intact WT females is ordinarily small in the reversible MCAO model (approximately 20%), loss of PARP-1 enormously increases infarction. This observation is consistent with a previous report that under conditions of mild ischemic injury without significant NAD+ depletion, PARP-1 inhibition is not beneficial and blocks DNA base excision and repair (Nagayama et al, 2000).
In the absence of nNOS, estradiol-conferred neuroprotection is not observed. This may have been because of the large amount of injury in the nNOS−/− female that obscures any of the well-characterized beneficial actions of estradiol in ischemic brain. Alternatively, 7NI treatment in human brain-derived neuroblastoma cells specifically blocks estradiol's cytoprotection through a nNOS, cGMP-PKG-dependent expression of the redox protein, thioredoxin (Lee et al, 2003). This effect may be mediated by a rapid estrogen-receptor-dependent induction of NO, which may be protective when produced at low levels. Inhibition of estrogen-induced NO production increased cell death in vitro (Wen et al, 2004). Therefore, there may be interactions between nNOS and estradiol in neuroprotection. Furthermore, in the absence of PARP-1, estradiol's protection is completely reversed to exacerbation of infarction, suggesting a more specific interaction between estradiol and PARP-1. For example, estradiol and its catechol metabolites can be carcinogenic under specific conditions (Li, 1994; Lavigne et al, 2001). Recent evidence suggests that in some cell types, even physiological levels of estradiol can cause oxidative DNA damage (Chen et al, 2003). In addition, interactions between estrogen receptor alpha and nuclear base excision repair genes have been reported (Bianco et al, 2003). If DNA repair is compromised in the PARP−/− female, then deleterious actions of estrogens may manifest.
In conclusion, striking differences exist between male and female brain after exposure to ischemic injury. Differences in the NO/PARP pathways likely play a key role in this dichotomy. These are important observations, which challenge the fundamental assumption that most important cellular mechanisms of injury or neuroprotection are not influenced by gender. It is likely that these results also provide a mechanistic clue as to how estrogen protects the brain during injury.
Footnotes
Acknowledgements
The authors thank Dr Nabil Alkayed for his thoughtful comments and assistance.