
Abstract
Traumatic brain injury (TBI) causes delayed neuronal deficits that in principle could be prevented by timely intervention with therapeutic genes. However, appropriate vectors for gene transfer to the brain with TBI remain to be developed. First-generation adenoviruses (fgAd) are usually associated with inflammatory and toxic effects when inoculated into brains, despite their high efficiency of gene transfer to these tissues. In this study the authors attempted to determine whether a less immunogenic gene-transfer protocol can be established in the traumatically injured rat brain using helper-dependent adenoviruses (hdAd), a novel adenoviral construct with full deletion of viral coding sequences. Their results show that transgene expression from intrahippocampally inoculated hdAd is maintained for at least 2 months after TBI, in contrast to the much shorter duration of fgAd-mediated gene expression. There was only minimal secretion of proinflammatory IL-1β and TNF-α after inoculation of hdAd. Furthermore, the hdAd-mediated gene expression was associated with less microglial proliferation, astrocytic activation, and macrophage infiltration than observed in fgAd-inoculated brains. There was no additional tissue loss after hdAd inoculation compared with PBS injection. Although both anti-adenoviral and neutralizing antibodies were found in serum after brain inoculation of hdAd, they did not appear to affect transgene expression. The results suggest that hdAd are less immunogenic vectors than conventional adenoviral vectors, and offer improved vehicles for long-term therapeutic transgene transfer to traumatically injured brains.
Traumatic brain injury (TBI) is the leading cause of traumatic death in the United States (Sauaia et al., 1995). TBI-associated death and tissue damage is mediated through both primary and secondary injuries. The former is essentially irreversible, while the latter can potentially be attenuated or even prevented by an appropriate therapeutic intervention. The cortex and hippocampus are the most affected regions in TBI and thus represent major therapeutic target sites. Because TBI-induced secondary injury is primarily mediated by ischemia, inflammation, oxidative stress, and apoptosis, rational therapeutic strategies should include delivery of reagents with vessel-dilating, antiinflammatory, antioxidant, antiapoptotic, or neuroprotective action (Hilton, 1994;McIntosh et al., 1998;Sapolsky and Steinberg, 1999;Dirnagl et al., 1999). For example, superoxide dismutase (SOD), a specific scavenger of free radical superoxide anions, has been reported to attenuate secondary injury and to improve outcome after TBI in experimental animals (Yunoki et al., 1997). Other reagents showing promise in brain injury treatment include endothelium-derived nitric oxide synthases (eNOS) (Bredt, 1999), which synthesizes nitric oxide and dilates microvessels (Chabrier et al., 1999;Iwata et al., 2000); interleukin-1 receptor antagonist (Yang et al., 1997), which inhibits inflammation; glucose transporter (Lawrence et al., 1996;Fink et al., 2000), which facilitates glucose transportation; heat-shock protein (Yenari et al., 1998), the antiapoptotic protein Bcl-2 (Shimazaki et al., 2000); and trophic factors such as nerve growth factor (NGF) (Sinson et al., 1997). Systemic administration of these agents is hindered by their limited biological half-lives (Igarashi et al., 1994), and by the blood—brain barrier, leading to efforts to devise effective methods of in vivo gene transfer.
We have successfully used cationic liposomes to deliver the NGF gene to traumatically injured rat brains, noting attenuation of forebrain cholinergic neuronal deficits 2 weeks after injury (Zou et al., 1999). However, it was unclear whether long-term transgene expression could be achieved after TBI. This capability is desirable because substances such as SOD may require long-term delivery to produce therapeutic effect, and brain cells have an overall compromise in protein synthesis after ischemic injury (Abe et al., 1988). During the pathologic course of TBI, inflammation produced by the initial traumatic injury and secondary ischemia pose a formidable obstacle to successful gene transfer and expression. However, trauma-compromised brains are potentially vulnerable to additional inflammatory reactions caused by vectors. This situation requires an optimal gene delivery system with the ability to mediate prolonged transgene expression and to induce fewer inflammatory reactions.
Recombinant adenoviruses are powerful tools for gene delivery to the brain, where they transduce a broad range of cell types, including dividing glial cells and nondividing neurons (Le Gal et al., 1993;Navarro et al., 1999). However, first-generation adenoviruses (fgAd) have been associated with adverse side effects, including tissue damage due to vector-induced cytotoxicity and inflammation (Wood et al., 1996;Kajiwara et al., 1997;Parr et al., 1998). Recently, we have tested helper-dependent, or so-called gutless, adenoviruses (hdAd) as vehicles for gene delivery into the rat CNS (Zou et al., 2000, 2001). These novel constructs lack almost the entire viral genome, excluding noncoding packaging sequences and terminal repeat sequences (Parks et al., 1996;Morsy and Caskey, 1999). We (Zou et al., 2000, 2001) and others (Thomas et al., 2000;Cregan et al., 2000) have demonstrated that stable, safe gene transfer and expression can be achieved in the CNS with hdAd. Still to be determined is the feasibility of using these modified viruses for gene delivery into the trauma-compromised CNS and whether they cause less inflammatory reaction in brains with TBI.
Only a few studies have investigated humoral responses after CNS delivery of recombinant adenoviruses. Kajiwara et al. reported limited production of antibodies against adenovirus, but found no neutralizing antibodies in serum (Kajiwara et al., 2000). To date, there have been no reports demonstrating hdAd-induced humoral responses in the CNS. Although it is generally accepted that humoral responses do not affect viral vectors in transduced cells after initial inoculation, they could compromise a second inoculation by inhibiting viral activity. It would be of fundamental interest to know if deletion of the viral genome confers any advantage to hdAd in terms of humoral responses. In the study reported here, we monitored transgene expression in traumatically injured rat brain after gene delivery by hdAd, and compared cellular and humoral immune responses to hdAd with those induced by fgAd.
MATERIALS AND METHODS
Preparation of first-generation adenoviruses and helper-dependent adenoviruses
FgAd and hdAd harboring the β-geo gene, a fusion product of the β-galactosidase and neomycin phosphotransferase genes, were constructed and prepared as previously described (Zou et al., 2000). All vector preparations were evaluated by particle number, as determined by optical density measurements of DNA and by their ability to express β-galactosidase activity in HeLa cells. After purification, both fgAd and hdAd were concentrated to about 1.0 × 1012 particles per milliliter. The numbers of blue-forming units in HeLa cells were 1.1 × 1011 and 1.0 × 1011 for fgAd and hdAd, respectively. Helper virus contamination, determined by plaque assay according to the method described elsewhere (Graham, 1991), was less than one plaque-forming unit in 1.0 × 107 particles of hdAd.
Traumatic brain injury and vector injection
Controlled cortical impact (CCI), a TBI model that causes damage in the cortex and hippocampus, was produced with a cortical impact device. Briefly, each male Sprague-Dawley rat (250–350 g) was anesthetized with a ketamine-based drug combination (42.8 mg ketamine, 8.6 mg xylazine, and 1.4 mg acepromazine per milliliter) at 0.5 mL/kg. Its head was secured in a stereotaxic frame, a midline incision was made, and a circular section of skull 8 mm in diameter was removed from the right margin of the skull midway between the frontal and occipital sutures, 2 mm medial to the temporal ridge. The exposed site was injured with a controlled lateral cortical impact device at a velocity of 4 to 5 meters/s and a deformation depth of 2.0 mm. Immediately after impact, a Hamilton syringe with a 27-gauge needle was placed in the hippocampus (coordinates: 4.3 mm posterior to bregma, 3.0 mm lateral to the sagittal suture, and 3.6 mm below the skull, with reference to bregma). Either fgAd or hdAd (5 × 108 particles in 5 μL) were injected over 10 minutes into the hippocampus ipsilateral to the injury. The needle was left in the place for an additional 5 minutes to prevent outflow. The skin was then sutured.
A total of 213 traumatically injured and 32 sham-operated rats were used in this study. They were allocated into four sets of groups. The first set, which was composed of phosphate-buffered saline (PBS), fgAd, and hdAd groups, was used to determine transgene expression levels at various time points. For this purpose, rats were allowed to survived for 3 hours (n = 4 for PBS, n = 5 for fgAd and hdAd), 3 days (n = 4 for PBS, n = 5 for fgAd and hdAd), 6 days (n = 4 for PBS, n = 5 for fgAd and hdAd), 14 days (n = 4 for three groups), 30 days (n = 4 for PBS, n = 5 for fgAd and hdAd), and 60 days (n = 4 for three groups). The second and third sets of animals were composed of three injured groups (i.e., PBS, fgAd, and hdAd) and an additional sham-operated group. They were used for counting immune cells and determining cytokine release after intrahippocampal viral inoculation. For those purposes, all animal groups were studied at 4 time points (3 hours, and 3, 6, and 30 days postinoculation; n = 4 for sham, n = 5 for PBS, fgAd, and hdAd groups). The fourth set of TBI animals, used for antibody determination, consisted of rats in PBS groups (n = 3), fgAd, and hdAd groups (n = 5). The protocol for this study was approved by the Baylor College of Medicine Animal Research Committee; care and handling of the animals were in accordance with the National Institutes of Health guidelines.
Histologic preparation of rat brain
Rats were deeply anesthetized with sodium pentobarbital and perfused transcardially with 200 mL of PBS (0.1 mol/L, pH 7.4), followed by 150 mL of fixative (4% paraformaldehyde in PBS). The brains were removed and postfixed overnight in the same fixative at 4°C, followed by immersion for 2 or 3 days in 25% sucrose in PBS until the tissues completely sank. Coronal sections (30 μm) were cut at −20°C in a cryostat.
Detection of β-galactosidase activity
Free-floating sections were washed three times with PBS, and then incubated at 37°C for 2 hours with 0.05% X-gal in 0.5 M HEPES buffer (pH 7.4) containing 1 mol/L MgCl2, 5 mol/L NaCl, 50 mmol/L K3Fe(CN)6, and 50 mmol/L K4Fe(CN)6. The reaction was stopped with PBS. The sections were counterstained with 0.05% cresyl violet for morphologic observation. After slide mounting, the sections were examined under the microscope, and transgene expression was quantified. Briefly, every fourth section through the hippocampus (rostral to caudal orientation) of each animal was analyzed. Images were captured with a computer equipped with image-analyzing software (Optimas 6.5; Optimas Bioscan Inc., Edmonds, WA, U.S.A.).
Immunohistochemical analysis
To determine cell phenotypes and immune infiltration, we double-stained the brain sections. Free-floating sections were first stained with X-gal as described in the above section, and then incubated overnight at 4°C with the following cell type-specific antibodies: anti-NeuN (1:500; Chemicon, Temecula, CA, U.S.A.), a neuronal marker; anti-ED1 (1:500; Serotec Inc., Raleigh, NC, U.S.A.), which recognizes a cytoplasmic antigen unique to monocytes and macrophages (Damoiseaux et al., 1994); anti-GFAP (1:200; Chemicon), which selectively stains glial fibrillary proteins in astrocytes (Debus et al., 1983); and anti-CD11b (1:200, Serotec Inc.), which labels microglia in normal brain tissue (Milligan et al., 1991). After incubation with a biotin-labeled secondary antibody (Vector Labs Inc., Burlingame, CA, U.S.A.), the bound antibodies were detected with an avidin—biotin peroxidase kit (Vector Labs Inc.) with diaminobenzidine used as a substrate.
Cytokine assay
Cytokine levels were determined with an enzyme-linked immunosorbent assay (ELISA) as previously reported (Zou et al., 2001). At specific time points after intrahippocampal inoculation of adenovirus, hippocampal tissues were dissected and placed in sterile PBS containing a protease inhibitor cocktail, after which they were homogenized and centrifuged (10,500 rpm, 15 minutes, 4°C), and the supernatant was collected and stored at −70°C. All samples were assayed for immunoreactive TNF-α and interleukin (IL)-1β with rat-specific sandwich ELISA kits (Chemicon).
Assessment of tissue damage
Tissue damage was assessed in cresyl violet—stained brain sections, with the lesion area being defined by a lack of staining due to neuronal loss. Seven coronal brain sections (30 μm) were analyzed for each animal and included one section with the maximal lesion area (middle section), three sections taken 0.3, 0.6, and 0.9 mm caudal to the middle section, and three sections with the same spacing rostral to the middle section. This sampling pattern ensured analysis of the most severely damaged tissue. The digital images of these sections were produced with a scanner, and then were measured with the computer-assisted image analysis software Optimas. The lesion area in each section was quantified and expressed as a percentage of the total brain cross-sectional area calculated as follows: [contralateral brain area (without TBI and viral inoculation) − ipsilateral brain area (with TBI and viral inoculation)]/[(contralateral brain area + ipsilateral brain area)/2] × 100. The mean lesion area from the aforementioned seven sections was reported.
Antibody determinations
Antibody responses to adenovirus were determined by a previously reported method (Kajiwara et al., 2000) with minor modifications. Briefly, 96-well plates were coated with ultraviolet-inactivated fgAd or hdAd (1 × 108 viral particles in 50 μL of NaHCO3 per well) at 4°C overnight. Wells were blocked with 200 μL of blocking buffer (3% milk in 0.1% Tween-20 in PBS) for 1 hour at room temperature. After five washes with PBS, the wells were incubated for 2 hours at room temperature with diluted serum (3-fold dilution with blocking buffer, beginning at 1:10 of the original serum sample). After five rinses with PBS, the wells were incubated with 50 μL of 1:200-diluted horseradish peroxidase—conjugated goat anti-mouse immunoglobulin G Fc (Sigma, St. Louis, MO, U.S.A.) for 2 hours at room temperature. The plates were washed five times with PBS, and 50 μL of developing solution was added to each well and developed for 10 minutes at room temperature. The reaction was stopped by adding 50 μL of 2.5 mol/L H2SO4, and the plates were read immediately at 492 nm.
Neutralizing antibody assay
This assay was performed as previously described (Kajiwara et al., 2000). In brief, serum samples were collected from experimental animals and stored at −80°C until use. The samples were heat inactivated at 55°C for 30 minutes and diluted in Dulbecco's Modified Eagle Medium (DMEM) containing 2% fetal calf serum (FCS) in 2-fold steps beginning at 1:4. One hundred microliters of each sample were mixed with 10 μL of viral vectors of either hdAd or fgAd (containing 3 × 107 viral particles) and incubated for 1 hour at 37°C. The mixtures were then applied to nearly confluent 293 cells in a 96-well plate (5 × 104 cells per well) and incubated in a 5% CO2 tissue culture incubator for 60 minutes. After incubation, the mixture was aspirated and replaced with 150 μL of DMEM containing 10% FCS. Twenty-four hours later, the cells were fixed with 2% formaldehyde and 0.2% gluteraldehyde and stained with X-gal (Sigma). The titer of neutralizing antibody in each sample was determined as the highest dilution that inhibited staining of 293 cells by 75%.
Statistics
The results of X-gal staining, a measure of transgene expression, were quantified by determining the densities and areas of staining in each section, and then multiplying the resultant values to obtain the staining intensity. Total staining intensity was the sum of the staining intensity values for all sections analyzed per test animal. Mean staining intensities (with standard deviations) were then calculated from pooled data from four or five rats and reported as percentages of the maximal staining intensity achieved with hdAd. An unpaired t test was used to evaluate differences in transgene expression (a two-tailed P value of less than 0.05 indicated statistical significance).
ELISA, antibody assay, and cell counting results for each experimental group were pooled, and mean values (with standard deviations) were calculated. Statistical testing was performed by repeated measurement ANOVA, followed by the Student-Newman test for multiple comparisons (a two-tailed probability of less than 0.05 denoted statistical significance).
RESULTS
Transgene expression
To investigate the duration and kinetics of fgAd- or hdAd-mediated transgene expression in traumatically injured brain, animals were immediately inoculated with either fgAd or hdAd after TBI. We measured X-gal staining intensities at six time points postinoculation (3 hours and 3, 6, 14, 30, and 60 days). The reason for choosing these time points was to have a cross-reference to our previous study that measured transgene expression in uninjured rats at a similar set of time points (Zou et al., 2000). In the hippocampus (vector inoculation sites), transgene expression was first detected on day 3 after gene transfer. There were no appreciable differences in the size and density of stained areas in fgAd-versus hdAd-infected samples at this time point (Fig. 1). By 6 days after injection, transgene expression had increased dramatically over the 3-day values, regardless of the vector used. This allowed for the examination of spatial and cellular distributions of transduced cells. The X-gal reaction product localized primarily in cells of the dentate gyrus (Fig. 2a) and pyramidal cell layer after hippocampal inoculation. Analysis with cell-type specific antibodies revealed transduction of neurons (Fig. 2b) and astrocytes (Fig. 2c) after hippocampal inoculation of hdAd. The distribution patterns of fgAd-mediated transgene expression were similar to those of hdAd, despite a significantly lower intensity for fgAd. With extended observation times, the intensity of staining in fgAd-infected samples decreased rapidly, reaching an undetectable level on day 30 after intrahippocampal inoculation (Fig. 1). By contrast, hdAd-mediated transgene expression showed less decline over the test period, regardless of the inoculation site (Fig. 1). For example, the value obtained on day 60 (end time point studied) after inoculation was 27.8% of peak value. Statistical analyses revealed significantly higher levels of transgene expression in hdAd-inoculated samples compared with fgAd-inoculated samples at each time point except day 3 (Fig. 1). The rat with TBI that received PBS instead of viral vector demonstrated no X-gal staining in the hippocampus and ependyma (data not shown), indicating that TBI did not confer any background staining in the areas studied.
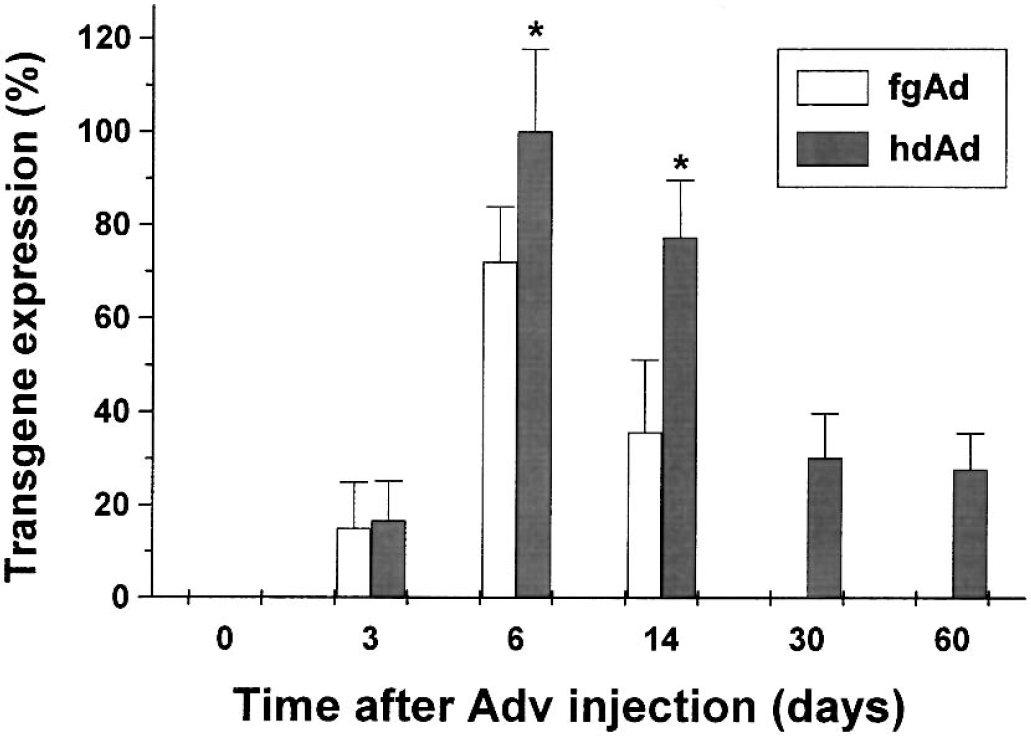
Time-dependent transgene expression after inoculation of adenoviral vectors. Sprague-Dawley rats were inoculated with first-generation adenoviruses (fgAd) or helper-dependent adenoviruses (hdAd) in the hippocampus. X-gal staining was monitored for up to 2 months and semiquantified by measuring the area and intensity of staining, reported as an index relative to the maximum value of staining in hdAd-injected samples. Four or five rats were tested in each group with results given as means and standard deviations. * P < 0.05 and ** P < 0.01 versus fgAd.
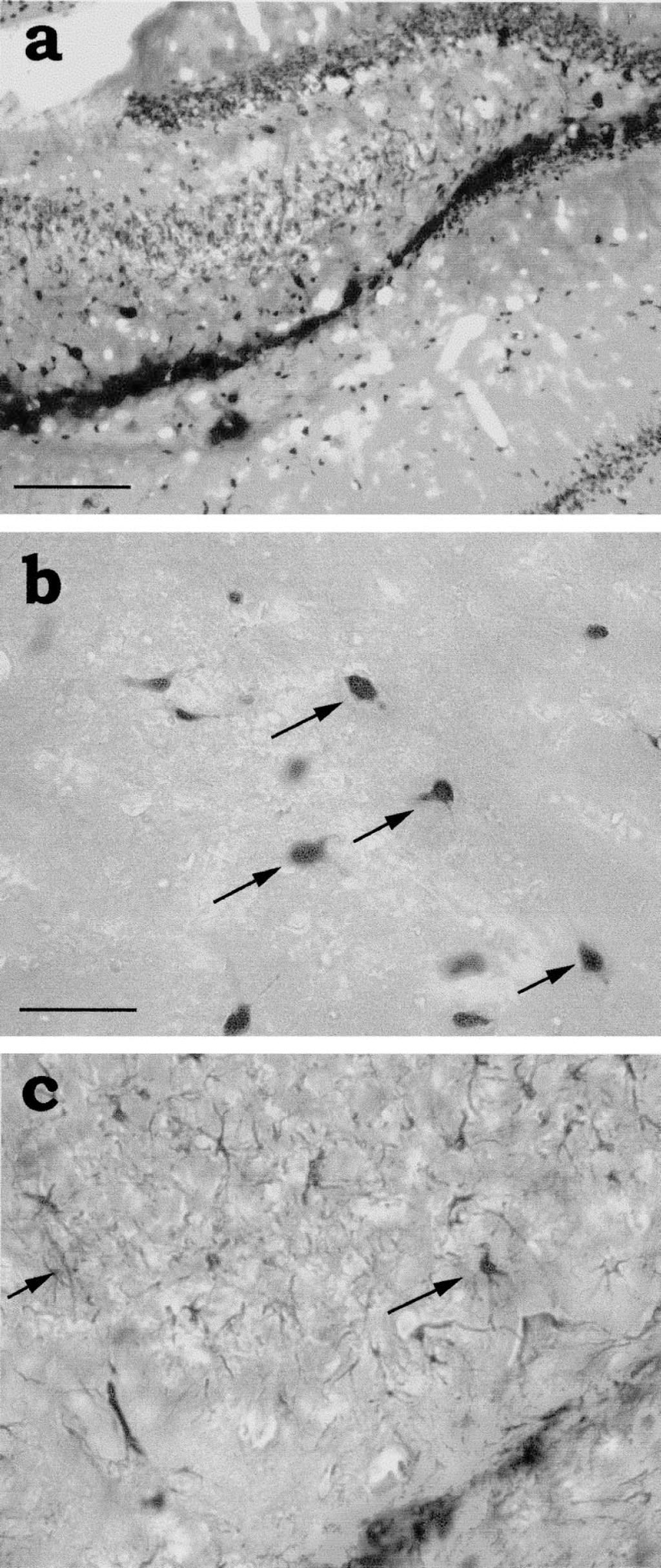
Spatial and cellular distribution of β-galactosidase activity 6 days after intrahippocampal inoculation of helper-dependent adenoviruses (hdAd).
Glial activation and macrophage infiltration
Inflammation, characterized by gliosis and phagocytic cell infiltration, poses a potential obstacle to long-term transgene expression. To investigate host inflammatory responses after TBI and adenoviral inoculation, we monitored brain tissues for the proliferation or activation of microglia and astrocytes as well as infiltration of macrophages, using CD11b, glial fibrillary acidic protein (GFAP) and ED1 immunohistochemical methods (Fig. 3). After TBI and intrahippocampal inoculation of an adenoviral vector, CD11b immunostaining detected numerous, activated microglia throughout the entire hippocampus at each postinjury time point (3 hours; 3, 6, and 30 days). Slightly activated microglia could be identified by their short and highly ramified processes, whereas highly activated microglia possessed amoeboid structural features (Cregan et al., 2000). TBI combined with PBS injection significantly enhanced the number of microglial cells present in hippocampal tissue (Fig. 4A), with further increases noted after injection of fgAd or hdAd. However, most of the microglia did not appear to be highly activated at 3 hours (image not shown), suggesting early proliferation without full activation. Highly activated microglial cells were largely seen at 6 days after injection (Fig. 3a), reaching peak numbers by day 6 in fgAd-inoculated hippocampus (Fig. 4A), where they persisted for as long as 30 days. In hdAd-inoculated hippocampus, the number of microglial cells decreased relatively rapidly, at rates approximating those for brain-injured rats injected with PBS. Significantly lower numbers of microglia were noted in hdAd-versus fgAd-inoculated animals on days 6 and 30 (for day 6 image, see Fig. 3b). The astrocyte response to TBI was less pronounced (Fig. 4B), as shown by the similarity of cell counts of GFAP-positive astrocytes at 3 hours after injury. The administration of fgAd significantly enhanced the number of GFAP-positive astrocytes counted on days 3 and 6. These cells showed morphologic signs of activation that included thicker processes and more intense GFAP reactivity (for day 6 image, see Fig. 3c). By contrast, inoculation of hdAd yielded essentially the same results as PBS injection. Markedly higher numbers of GFAP-positive astrocytes were also seen in the dentate gyrus region after inoculation of fgAd, in contrast to results for hdAd (Figs. 3c and 3d). Infiltrating macrophages were first apparent on day 3 postinoculation, increasing in number by day 6 in fgAd-but not hdAd-inoculated animals (Fig. 4C), as reflected by ED1 immunohistochemical staining (Figs. 3e and 3f). The numbers of infiltrating macrophages decreased on day 6 in the PBS- and hdAd-injected groups, remaining significantly higher in fgAd-inoculated tissue even by day 30 after injection (Fig. 4C). Overall, inoculation of hdAd did not produce any increase in the numbers of infiltrating macrophages that would be considered additive to the TBI effect.
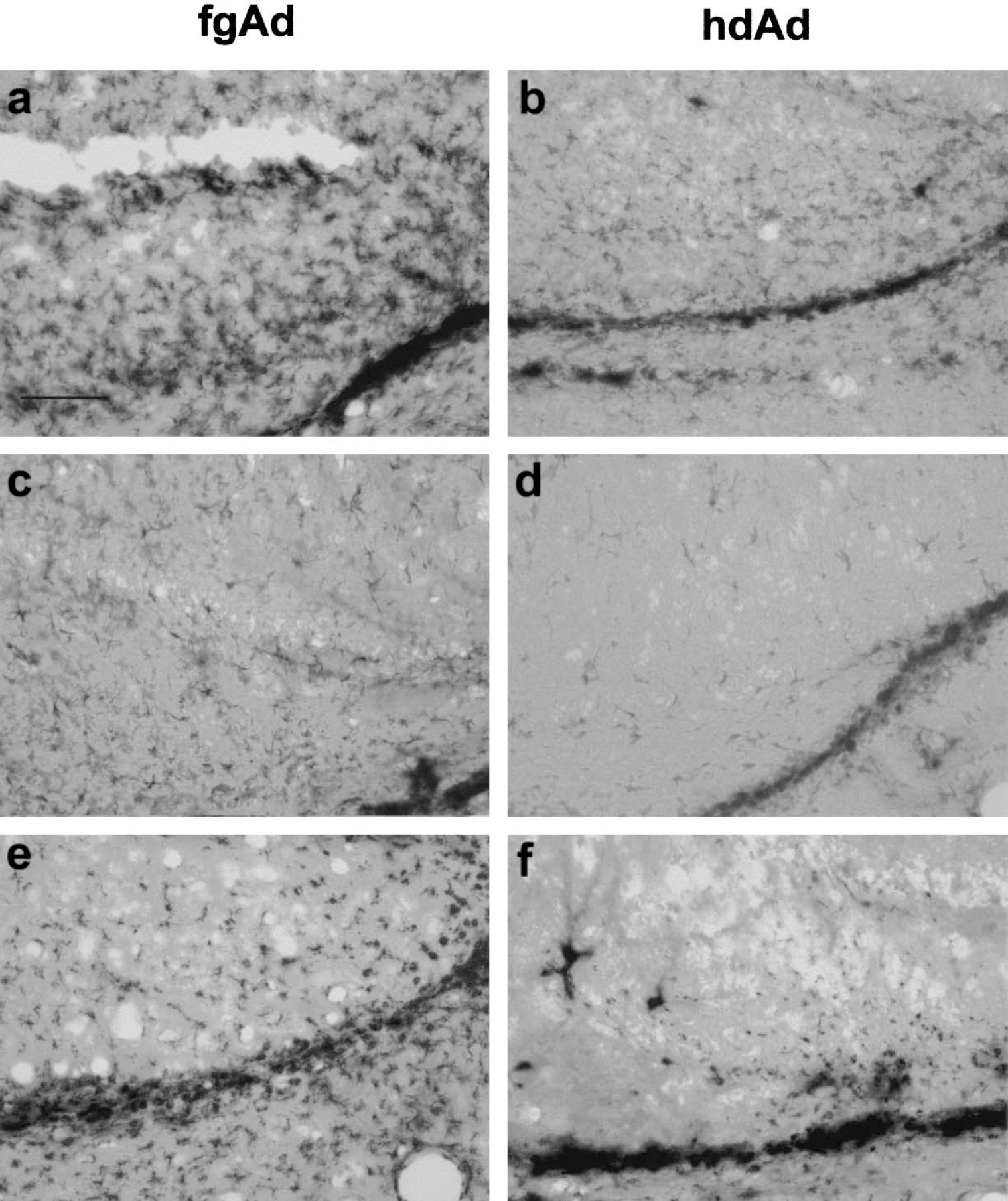
Proliferation and activation of microglial cells, activation of astrocytes, and infiltration of macrophages 6 days after inoculation with adenoviral vectors. Microglia
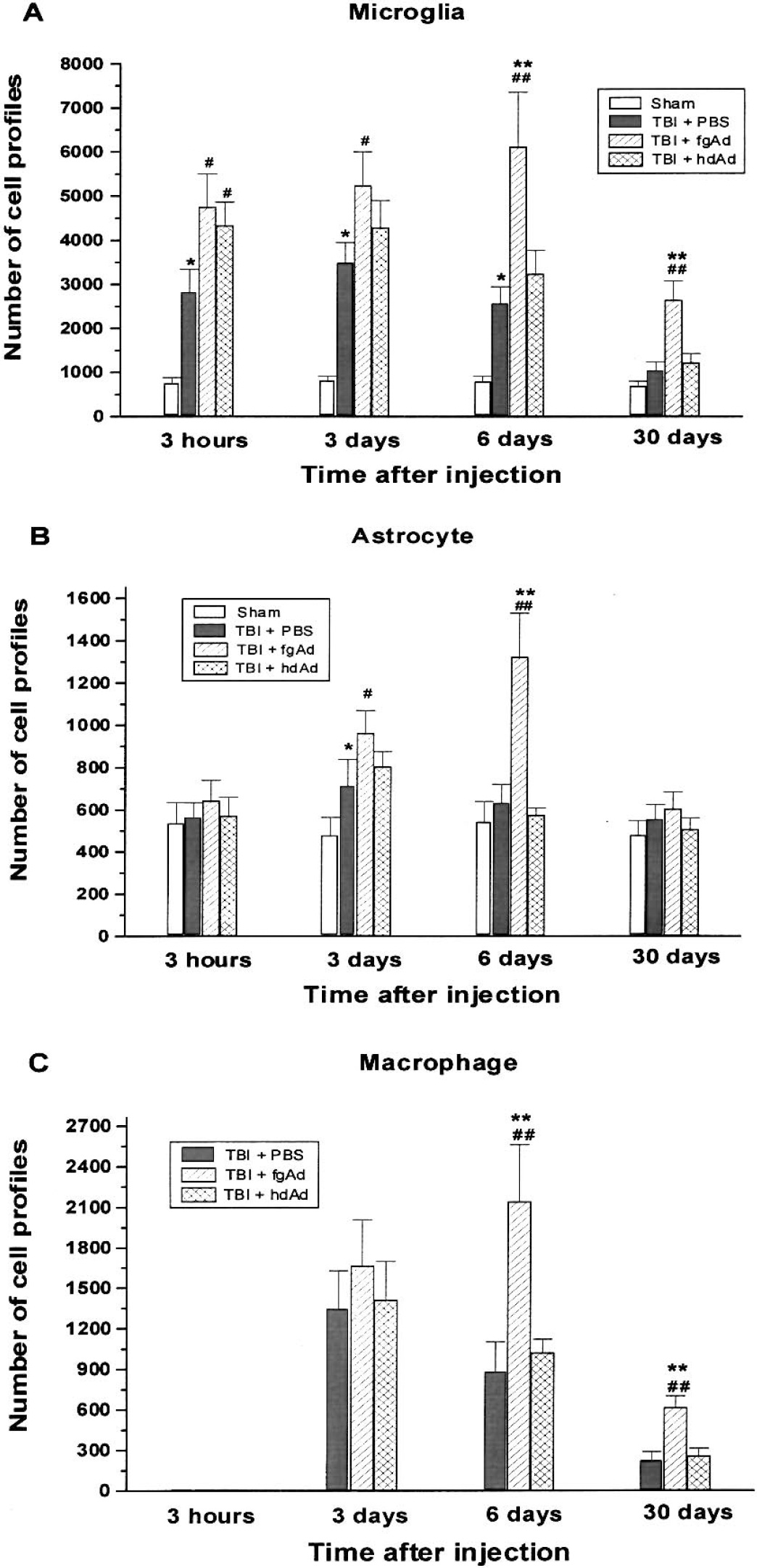
Quantification of glial cells and macrophages after inoculation of adenoviral vectors. Microglia
Proinflammatory cytokine secretion
The proinflammatory cytokines IL-1β and TNF-α are critical mediators of host immune responses (Marquette et al., 1996;Sauder and De la Torre, 1999). We therefore measured concentrations of IL-1β as well as TNF-α in hippocampal homogenate at 3 hours and 3, 6, and 30 days after intrahippocampal inoculation of adenoviral vectors in the injured brain. This assay also serves to detect the downstream immune responses related to macrophage infiltration and microglial activation.
As shown in Figure 5A, TBI and intrahippocampal PBS injection induced significant secretion of IL-1β as early as 3 hours after injury. Inoculation of either adenoviral vector caused an additional increase of IL-1β at this time point, which represented the peak level attained during the entire experiment. By day 3, cytokine levels resulting from TBI and injection with PBS or hdAd had returned to the sham control level (Fig. 5A), whereas the fgAd-inoculated group still showed significantly increased IL-1β levels (P < 0.05, Fig. 5A). All groups had low IL-1β levels on day 6 and day 30.
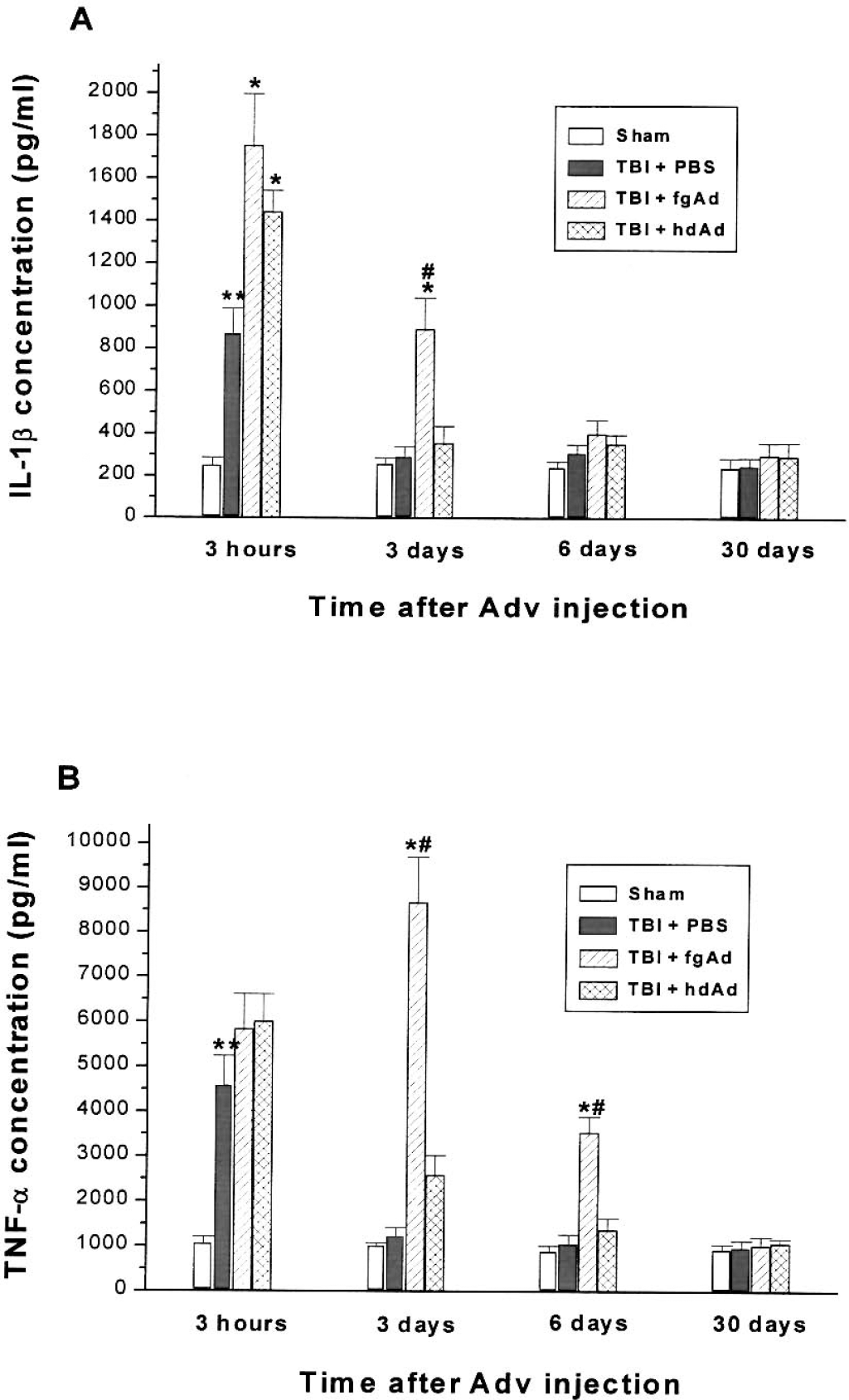
Secretion of proinflammatory cytokines after intrahippocampal inoculation of adenoviral vectors. Interleukin 1β
There was also rapid and transient secretion of TNF-α attributable to TBI and PBS injection at 3 hours after injury (Fig. 5B). Inoculation of the adenoviral vectors was not associated with significant increases of the cytokine at this time point compared with PBS injection. However, by day 3, the inoculation of fgAd had elicited a striking augmentation of the TNF-α concentration, which was significantly higher than the result in the hdAd-inoculated group. TNF-α levels were uniformly low on days 6 and 30 after inoculation, with the exception of a significantly increased value associated with inoculation of fgAd on day 6 (Fig. 5B).
Tissue damage
Goodman et al. documented a significant tissue loss at 14 days after injury (Goodman et al., 1994). To investigate tissue loss throughout an extended period of time, we measured the lesion area on both day 14 and day 60 after injury. As shown in Figure 6A, TBI induced significant tissue loss in the cortical and upper hippocampal regions, leading to formation of a cavity. Inoculation of fgAd into TBI rat brain exacerbated tissue loss induced by TBI, as reflected by significant additional tissue loss (P < 0.01) on day 60 after injection. By comparison, hdAd inoculation was not associated with additional tissue loss (Fig. 6B).
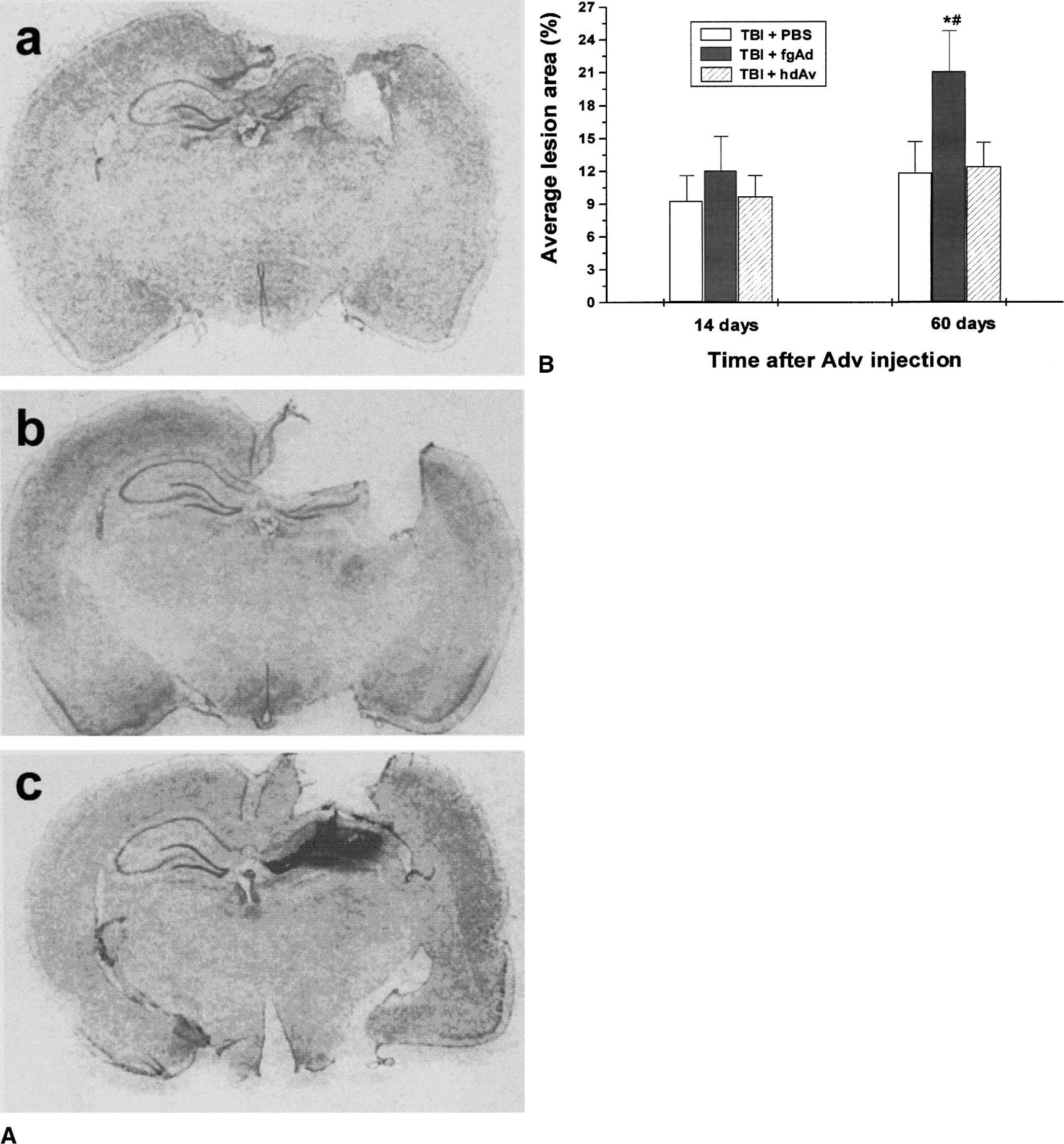
Tissue lesion follows traumatic brain injury (TBI) and adenoviral vector inoculation.
Humoral immune responses
To assess humoral immune responses to adenoviral vectors, sera were collected from TBI rats at 3, 6, 20, and 26 days after hippocampal inoculation with either fgAd or hdAd, or at 6 days after a second inoculation made 20 days after the first. Traumatically injured rats injected with PBS instead of viral vector served as the PBS control group. All serum samples were assayed for the presence of anti-adenovirus antibody and neutralizing antibody. As shown in Figure 7A, animals in the PBS control group had little or no antibody to either virus. However, there were significant amounts of anti-adenovirus antibody in the sera of animals inoculated with adenovirus. The antibody was first detected 3 days after the inoculation of an adenoviral vector into brain. The amount of anti-adenovirus antibody increased gradually until day 26 (last observation day after single inoculation), and was further enhanced in the repeated inoculation group (Fig. 7A). The sera for rats that received a second inoculation contained much higher levels of anti-adenovirus antibody. There were no statistically significant differences in the extent or temporal pattern of serum antibody responses induced by fgAd and hdAd.
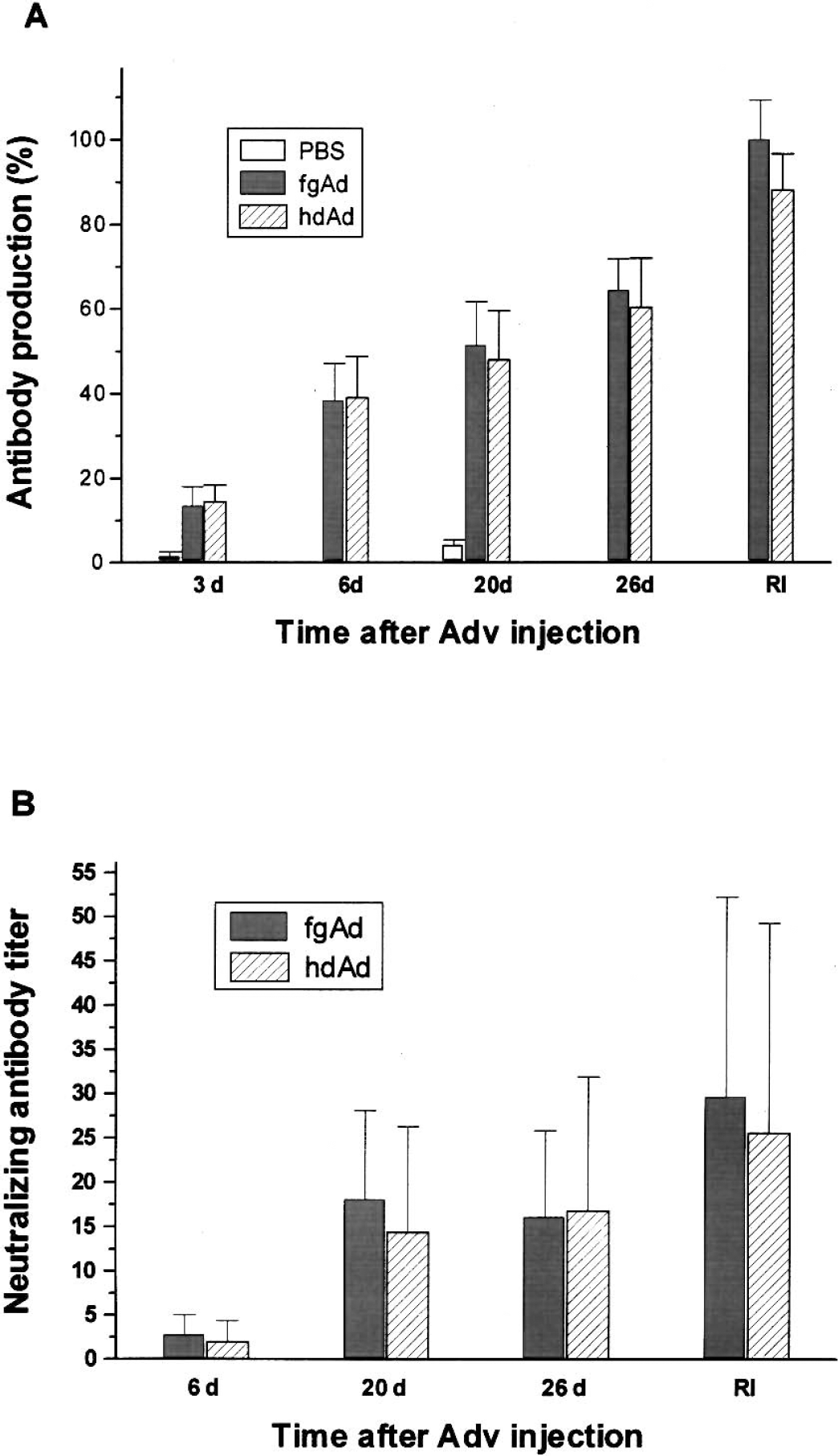
Antibody responses after single or repeated inoculation of first-generation adenoviruses (fgAd) or helper-dependent adenoviruses (hdAD) in rat hippocampus. Serum samples were collected at 3, 6, 20, and 26 days after initial inoculation or 6 days after second inoculation. The data are presented as means and standard deviations for three to five rats per time point.
Neutralizing antibody to either vector was not detected in any of the animals until day 6 after inoculation (Fig. 7B). Antibody titers increased on days 20 to 26 and after repeated inoculation. Neutralizing antibodies were not detected in PBS control animals (data not shown). Consistent with results of the anti-adenovirus antibody assay, neither the titer nor temporal pattern of neutralizing antibody development differed between fgAd and hdAd.
DISCUSSION
Several important findings were made in this study. First of all, we demonstrated that hdAd can mediate gene transfer and long-term transgene expression in traumatically injured brain, compared with relatively short-term expression mediated by fgAd. Second, we found that hdAd induced only a slight inflammatory response in traumatically injured brain as compared with fgAd. Third, there was no additional tissue loss after hdAd inoculation compared to PBS injection in traumatically injured brain. Fourth, fgAd and hdAd elicited similar systemic humoral responses when inoculated into brain, which did not adversely affect the transduced cells.
Robust transgene expression is observed in this study, especially after inoculation of hdAd. Our study largely confirms Kochanek's finding that transgene expression can be mediated from adenovirus in rat brain with TBI (Kochanek et al., 2001). Collectively, these studies demonstrate that although compromised in some aspects by TBI, hippocampus is capable of being inoculated by adenoviral vectors and expressing exogenous protein. These observations are in contrast to reports of impairment of protein synthesis and compromised transgene expression in the brains of experimental animals with focal or global cerebral ischemia (Abe et al., 1997, 1998). The discrepancy may reflect distinct pathophysiology of TBI versus ischemia. For example, there is total blockage of cerebral blood flow (CBF) in the hippocampus of the patients and animal models of global and focal ischemia, whereas the same region may not have ischemia at all after CCI brain injury (Morioka et al. 1999). Bryan et al. reported 39% and 65% reductions of CBF at 30 minutes and 4 hours after CCI, respectively (Bryan et al., 1995), whereas Hendrich et al. documented a 58% reduction at 2.5 to 3.5 hours after CCI (Hendrich et al., 1999). Thus, the nonischemic nature of the hippocampus after CCI may account for cells expressing exogenous protein.
We have observed that fgAd can mediate relatively short-term transgene expression, which in principle may be useful in delivering therapeutic genes whose expression is only required for a brief time (Sanderson et al., 1999). However, in contrast to hdAd, fgAd induced marked host inflammatory reactions characterized by extensive proliferation and activation of microglia, and activation of astrocytes, as well as infiltration of macrophages. This finding agrees with the significantly higher numbers of microglia and macrophages seen in aged rat brain after treatment with fgAd versus hdAd in our previous study (Zou et al., 2001). Furthermore, their administration in this study was associated with additional tissue damage. Microglia, a sensor of threats to the CNS (Gehrmann, 1996), have been implicated in the pathologic effects of TBI (Koshinaga et al., 2000), whereas astrocytes participate in a variety of important physiologic and pathologic processes in the brain. Reactive astrocytes have been shown to upregulate a number of different molecules including other glial markers (S100β) and cytokines such as IL-1, IL-6, interferon, and TNF-α (Eddleston and Mucke, 1993). Thus, whereas fgAd vectors may offer some advantages for short-term transgene expression in TBI-affected brain, significantly increased glial activity induced by fgAd inoculation could exacerbate the pathologic processes of TBI, offsetting any beneficial effects produced by transgene expression from such vectors.
Cytokines appear to play an important role in CNS pathology developing after TBI (Ghirnikar et al., 1998). Our study demonstrates increased secretion of proinflammatory IL-1β and particularly TNF-α after TBI, with enhanced effects noted after inoculation of the adenoviral vectors, especially fgAd (Fig. 5). These observations are consistent with results of our previous study (Zou et al., 2001) and those of others (Balasingam and Yong, 1996;DeKosky et al., 1996). Activated macrophages, microglia, and astrocytes are the major cellular sources of TNF-α. It has been proposed that TNF-α induces astrogliosis and increases the number of activated microglia in the CNS (Kahn et al., 1995). Although the role of TNF-α in TBI is not fully understood, several in vitro studies have shown that both IL-1β and TNF-α exert a strong inducing signal for IL-6 production in cultured astrocytes (Benveniste et al., 1990;Murphy et al., 1993). The latter cytokine can produce several different responses in the CNS, including the activation of glia, neurons, and infiltrating cells after injury and disease (Norris et al., 1994). TNF-α has been implicated in injury-induced inflammatory events, including the activation of glia, endothelial cells, and leukocytes (macrophages, neutrophils) (Arvin et al., 1996), and enhanced expression of multiple downstream inflammatory factors (Boehme et al., 1996). It has also been shown to damage neurons (Talley et al., 1995), oligodendrocytes (Hisahara et al., 1997) and endothelial cells (de Vries et al., 1996). In the present study, lower production of TNF-α in hdAd-versus fgAd-inoculated brain might be the factor that accounts for long-term transgene expression.
The tissue loss associated with fgAd administration further supports the safety of hdAd and our hypothesis that fgAd inoculation induces greater inflammatory reaction and exacerbates tissue damage. These results have particular relevance to the selection of an adenoviral vector for gene therapy applications in TBI. Regarding TBI-induced baseline tissue loss, we detected marked tissue loss at day 14. However, there was no significant change in tissue loss at day 60. Although we recorded a higher value of lost area at day 60 than that at day 14 (11.3% versus 9.2%), it lacked statistical significance. Sutton et al. (1993) documented a peak value of TBI-induced necrosis at day 21. The observed discrepancy could be attributed to the moderate injury used in our study (4 to 5 m/s, 2.0-mm deformation depth), and the smaller number of animals included in this study.
We detected circulating antibodies against both types of adenoviral vectors after their inoculation into brain. There were also neutralizing antibodies produced in serum at certain postinoculation intervals, consistent with previous reports (Yang et al., 1995;Kay et al., 1995), and in contrast to data published by Kajiwara et al. (2000). The dosage of inoculated virus in the latter study was much lower than our own (6 × 106 versus 5 × 108), and probably accounts for the discrepancy in findings (Mastrangeli et al., 1996). Importantly, humoral responses did not differ significantly between fgAd- and hdAd-inoculated brains. These results, apparently the first pertaining to humoral responses after hdAd inoculation into brain, provide strong support for the general notion that humoral immune responses are induced by the virion protein of an adenoviral particle, rather than by the viral genome. HdAd vectors are characterized by complete deletion of the viral genome except for the packaging and replicating sequences, and the deletion eliminates the potential expression of viral genes. In accord with our previous studies (Zou et al., 2000, 2001), deletion of the viral genome reduced the immunogenicity of hdAd, but this effect may not extend to humoral immune responses, as suggested by data reported here. Hypothetically, cellular immunity is the primary factor limiting the duration of transgene expression from an inoculated adenovirus, whereas humoral immunity limits transduction with a second adenovirus (Yang et al., 1994;Dai et al., 1995). However, we have noted transduction of the contralateral hippocampus after a second inoculation of either fgAd or hdAd (unpublished observation), despite induction of neutralizing antibodies after initial brain inoculation. Thus, a second inoculation of adenoviral vector may be effective even when neutralizing antibodies are present in serum.
This study demonstrates that hdAd are capable of mediating relatively stable transgene expression even under unfavorable conditions such as TBI. Similar findings have previously been reported by Thomas et al., who compared transgene expression from inoculated high-capacity adenoviral vector (equivalent to our hdAd) with that from fgAd in the presence of subsequent challenge (Thomas et al., 2000) or under preimmunization condition (Thomas et al., 2001). However, in those studies TBI was not used; instead animals were either given a second peripheral injection of fgAd or were preimmunized with intradermal injection of fgAd. The technique described in our study used TBI, a procedure that will compromise the blood—brain barrier, and an approximately 5- to 10-times greater amount of Ad per injection. A compromised blood—brain barrier combined with a very high injection titer are likely to result in the equivalent of preimmunization, which is characterized by the presence of higher numbers of immune cells such as macrophages and cytotoxic T lymphocytes (Thomas et al., 2001). Indeed, although we did not measure T-cell infiltration in this study, we did find higher numbers of infiltrated macrophages and activated microglial cells in traumatically injured brains compared to sham-operated brains (Fig. 4). Taken together, this study largely confirms our previous findings (Zou et al., 2000, 2001) and Thomas's studies (Thomas et al., 2000, 2001) in terms of stability and safety of hdAd-mediated transgene expression.
We conclude that hdAd represents an improved vector over fgAd for TBI gene therapy applications. Not only is it less immunogenic than fgAd, but when inoculated into either hippocampus or cerebroventricles, it can secure relatively long-term transgene expression after TBI. This capability should be exploitable in the design of treatment strategies requiring long-term transgene expression in the TBI setting. By combining hdAd with emerging gene regulation technology (Burcin et al., 1999) whereby transgene expression can be controlled within desirable time frames, one could select the most appropriate scenario for post-TBI treatment, including situations requiring short-term transgene expression.
Footnotes
Acknowledgments
The authors thank Malcolm Brenner for critical reviewing, and John Gilbert for English editing of the manuscript.