
Abstract
The authors used cultured mouse cortical neurons to study mechanisms of DNA damage-induced apoptosis in immature and mature neurons. Neurons were maintained viably for 60 days in vitro (DIV60). The increased levels of glutamate receptors, synaptic proteins, and glycolytic enzyme were used to track maturation. Exposure of neurons to the DNA-damaging agent camptothecin induced apoptosis in immature (DIV5) and mature (DIV25–30) neurons. Internucleosomal fragmentation of DNA emerged more rapidly in mature neurons than in immature neurons. Immunoblotting revealed that cleaved caspase-3 increased in apoptotic DIV5 neurons but not in DIV30 neurons, but immunolocalization showed accumulation of cleaved caspase-3 in DIV5 and DIV30 neurons. A reversible caspase-3 inhibitor blocked apoptosis in DIV5 neurons but not in DIV30 neurons. Phosphorylation of extracellular signal-regulated kinase/mitogen-activated protein kinase (Erk/MAP kinase)-42/44 occurred preapoptotically in mature but not immature neurons, while Erk54 nuclear translocation and MAP kinase kinase kinase-1 cleavage into putative caspase-3–generated proapoptotic fragments occurred in DIV5 but not DIV30 neurons. Inhibition of Erk activation with MAP kinase kinase inhibitor blocked apoptosis at both ages. The results show that immature and mature cortical neurons engage different signaling mechanisms in MAP kinase and caspase pathways during apoptosis; thus, neuron age influences the mechanisms and progression of apoptosis.
Keywords
Understanding the molecular regulation of apoptosis is relevant to neuronal death in nervous system development and to neurodegeneration in pathological conditions affecting the CNS of individuals throughout fetal, newborn, childhood, and adult life. Defective apoptosis can cause cerebral malformations during CNS development (Hakem et al., 1998;Kuida et al., 1996). Dysregulated apoptosis contributes to the pathogenesis of CNS damage in acute neuropathological disorders, such as cerebral ischemia (MacManus et al., 1997;Martin et al., 1998;Martin et al., 2000) and spinal cord trauma (Liu et al., 1997), and in chronic neurodegenerative diseases, such as Alzheimer disease (Anderson et al., 1996;Kitamura et al., 1999) and amyotrophic lateral sclerosis (Martin, 1999, 2001). The state of CNS maturity at the time of injury appears to influence the emergence of different types of cell death and the rate of cell death (Martin et al., 1998;Martin, 2001;Martin et al., 2001;Natale et al., 2002;Portera-Cailliau et al., 1997).
DNA damage is a potent signal for apoptosis (Levine, 1997). Neurons sustain DNA damage (e.g., hydroxyl radical adducts or single-strand breaks) after cerebral ischemia (Martin et al., 2000) and axotomy/target deprivation (Al-Abdulla and Martin, 1998;Martin and Liu, 2002). Cells that have sustained DNA lesions can undergo apoptosis by engaging molecular cascades involving expression, activation by phosphorylation or proteolysis, or translocation of p53, Bax, and caspases (Polyak et al., 1997). Recently, the extracellular signal-regulated protein kinase/mitogen-activated protein kinase (Erk/MAP kinase) pathway, including the upstream MAP kinase kinase kinase-1 (MEKK1) and MAP kinase kinase (MEK), have been implicated in the signaling of apoptosis in response to DNA damage in nonneuronal cells (Widmann et al., 1998). Erk activation may also have a role in excitotoxic (Jiang et al., 2000) and ischemic (Alessandrini et al., 1999) neuronal death, but the link to DNA damage in neurons is unknown.
The mechanisms of DNA damage-induced apoptosis in postmitotic cells such as neurons are much less understood compared with cells of nonnervous tissue origin. Relatively few in vitro model systems have been developed to evaluate mechanisms of DNA damage-induced neuronal apoptosis. Generally, these models are exposure to γ-irradiation, ultraviolet light, cytosine arabinoside, or topoisomerase inhibitors such as etoposide or camptothecin (CPT) (Morris and Gellar, 1996;Park et al., 1997;Park et al., 1998). This study focused on CPT-induced neuronal apoptosis. The few available studies on CPT-induced apoptosis of cortical neurons or sympathetic neurons have suggested that the death process requires p53 (Johnson et al., 1999;Xiang et al., 1998), supporting the idea that CPT neurotoxicity involves DNA damage. However, the roles of caspases in CPT-induced neuronal apoptosis are unclear because some studies have shown that caspase inhibitors block (Johnson et al., 1999;Stefanis et al., 1999) or fail to block (Park et al., 1997) apoptosis, and caspase-3 deletion may only delay neuronal death (Keramaris et al., 2000). The role of the MAP kinase pathway in DNA damage-induced apoptosis of neurons is virtually unexplored. In this study, we used a mouse cortical neuron culture model to further delineate the molecular mechanisms of DNA damage-induced neuronal apoptosis. We tested the hypothesis that apoptosis mechanisms are different in neurons at different maturational ages. Specifically, we examined whether MAP kinase signaling participates in the mechanisms of DNA damage-induced neuronal apoptosis in vitro, and whether MAP kinase and caspase pathways and Bcl-2 family members are activated or modulated similarly or differently in immature and mature neurons. We found prominent molecular and biochemical differences during the progression of apoptosis in immature neurons compared with mature neurons. These differences were reflected in the time course or magnitude of internucleosomal DNA fragmentation, caspase-3 and MEKK1 cleavage, and Erk activation.
MATERIALS AND METHODS
Murine cortical neuron culture
Mouse cortical neuron cultures were prepared as described previously (Lesuisse and Martin, 2002). The Animal Care and Use Committee of the Johns Hopkins University School of Medicine approved the animal protocol. Cerebral cortices were dissected from embryonic day 16 mice (C57 strain, Charles River) and dissociated by treatment with 0.25% trypsin (Life Technologies, Rockville, MD, U.S.A.) followed by trituration with a fire-polished Pasteur pipette. Cells (106) were plated onto 35-mm tissue culture dishes coated with 33 μg/mL poly-D-Lysine. The cells were plated in Neurobasal medium (Life Technologies) supplemented with B27 (Life Technologies), and 25 μmol/L β-mercaptoethanol and streptomycin/amphotericin B (Life Technologies). Fifty percent of the medium was changed 3 days after plating and subsequently every 6 days. Neuronal cultures were maintained for up to 60 days in vitro (DIV60). The astroglial contamination of the cell culture was low. We have shown by immunoblotting that glial fibrillary acidic protein is undetectable at DIV5 and low at DIV25 (Lesuisse and Martin, 2002). By immunoblotting, the detection of markers for microglia and oligodendroglia is similarly low (Martin and Lesuisse, unpublished data, 2002). Another group has reported a nearly pure neuronal culture using similar conditions with B27 supplementation and low glutamine concentration to limit growth of glia (Brewer et al., 1993). For every experiment that was performed on cortical neurons at different in vitro ages, the cells in the different age groups were prepared from the same original dissociated cells, so that appropriate comparisons could be made (Lesuisse and Martin, 2002).
Camptothecin-induced apoptosis
Neurons were stimulated to undergo apoptosis in vitro by treatment with CPT, an inhibitor of topoisomerase-I (Bendixen et al., 1990;Hsiang et al., 1985). Camptothecin (Sigma, St. Louis, MO, U.S.A.) was dissolved in double-distilled water supplemented with sodium hydroxide and heated at 55°C for 30 minutes to solubilize completely the drug at a final concentration of 50 mmol/L, and was further diluted in Neurobasal medium. At DIV5 or DIV25–30, mouse cortical neurons were treated with 1, 10, or 100 μmol/L CPT for 4, 8, 24, 48, or 72 hours. A cell-permeable reversible caspase-3 inhibitor Ac-AAVALLPAVLLALLAPDEVD-CHO (Alexis Biochemicals, San Diego, CA, U.S.A.) and an inhibitor of MEK1/2 (U0126) (Cell Signaling Technology) were evaluated for effects on blocking neuronal apoptosis. Caspase-3 inhibitor was dissolved in DMSO (20-mmol/L stock). U0126 was dissolved in 50% ethanol/50% DMSO or 70% methanol/30% DMSO (50-mmol/L stock). The drugs were used at final concentrations of 10 and 100 μmol/L in cultures. Caspase-3 inhibitors are often used at this concentration range (Park et al., 1997;Stefanis et al., 1999). Cells were pretreated for 2 hours before exposure to CPT. Neuronal cultures were used for quantification of apoptosis, protein/DNA extractions, or electron microscopy (EM). For quantification of neuronal apoptosis, cultures were stained with a nuclear dye (Hoechst 33258), digital images were captured from six nonoverlapping microscopic fields (20× objective), and apoptotic nuclei were counted. The analysis of the cell counts was performed using ANOVA, and subsequent statistical post hoc evaluation of significance was performed using the Student's t-test.
Genomic DNA fragmentation analysis
Cortical neurons (from three different platings) cultured in 35-mm plates were washed twice in ice-cold phosphate-buffered saline (PBS), resuspended in 300 μL DNA extraction buffer (10 mmol/L Tris, pH 7.4; 10 mmol/L NaCl; 25 mmol/L EDTA) containing 100 μg proteinase K (Boehringer, Mannheim, Germany) and incubated at 37°C overnight. DNA was purified by phenol/chloroform extraction and resuspended in 200 μL TE. Approximately 20 microliters of each sample was fractionated in 1.5% TBE agarose gels containing ethidium bromide. DNA was visualized with a Fluor-S Multilmager (Bio-Rad, Hercules, CA).
Cellular and subcellular fractionation and immunoblotting
Total cell extracts were used to characterize normal developing cultures at DIV5 to DIV60. For total protein extracts, cortical neuron cultures (three wells for each maturational time point, two different platings) were lysed in TNE buffer (10 mmol/L Tris-HCL, pH 7.4; 150 mmol/L NaCl; 5 mmol/L EDTA) containing protease inhibitors (1 mmol/L PMSF, 10 μg/mL leupeptin, 10 μg/mL pepstatin A) and detergents (2% SDS, 1% deoxycholate, 1% NP-40), and sonicated for 15 seconds.
Subcellular fractionations were performed on CPT-treated neurons. DIV5 and DIV25–30 cortical neuron cultures (three different platings) were treated with 100 μmol/L CPT, and cells were harvested 4, 8, and 24 hours later. Soluble, insoluble, and nuclear proteins were analyzed by Western blot analysis to identify changes in the levels or activation of death and survival proteins and changes in organelle and synapse markers during the progression of neuronal apoptosis. For subcellular fractionations, cortical neurons (18 wells for each treatment) were resuspended in 200 mmol/L mannitol, 70 mmol/L sucrose, 1 mmol/L EGTA, and 10 mmol/L HEPES (pH 7.5) containing protease inhibitors (1 mmol/L PMSF, 10 μg/mL leupeptin, 10 μg/mL pepstatin A), homogenized in a glass-Teflon grinder with 10 up-and-down strokes, and centrifuged at 1,000 g to collect the nuclear fraction. The nuclear fraction was resuspended in the same extraction buffer and homogenized for the second time in a glass-Teflon grinder. The soluble and insoluble (pellet) fractions were isolated by centrifugation of the supernatant at 100,000 g. Protein concentrations of each homogenate were determined by protein assay (Pierce, Rockford, IL, U.S.A.). Homogenates were diluted further in Laemmli sample buffer.
Protein (20 μg) from neuronal cultures was resolved on gradient SDS polyacrylamide gels (4–20%) and transferred to nitrocellulose filter membranes by electroblotting. Nitrocellulose membranes were blocked in 5% (w/v) nonfat dry milk in PBS. After overnight incubation with the primary antibodies in PBS supplemented with 5% nonfat dry milk and 0.05% (v/v) Tween-20, membranes were rinsed and then incubated with peroxidase-conjugated secondary antibody. Immunoreactive proteins were visualized with enhanced chemiluminescence (Amersham).
The developmental maturation of cortical neuron cultures was evaluated by expression of subtypes of glutamate receptors, synaptic proteins, a glycolytic enzyme, and death/survival proteins. Protein expression profiles were also evaluated during the progression of neuronal apoptosis after exposure to CPT. The following antibodies were used for immunoblotting: monoclonal antibodies against α-synuclein (Transduction Laboratories, Lexington, KY, U.S.A.), cytochrome c oxidase subunit 1 (Molecular Probes, Eugene, OR, U.S.A.), glyceraldehyde phosphate dehydrogenase (Research Diagnostics, Flanders, NJ, U.S.A.), p53 (Pharmingen, San Diego, CA, U.S.A.), NMDA receptor R1 subunit (Pharmingen), synaptophysin (Boehringer), pan-Erk (Transduction Laboratories), active Erk42–44 (Promega), and poly-ADP ribose polymerase (PARP; Calbiochem, San Diego, CA, U.S.A.); and rabbit polyclonal antibodies against β-synuclein (Oncogene Science, Cambridge, MA, U.S.A.), AMPA receptor subunits GluR2/3 (Martin et al., 1993), Bcl-2 (N-19; Santa Cruz Biochemicals, Santa Cruz, CA, U.S.A.), caspase-3 (H-277; Santa Cruz), cleaved caspase-3 (D175; Cell Signaling Technology), Bcl-xL (Ab-3; Calbiochem), Bak (N-terminal; Upstate Biotechnology, Lake Placid, NY, U.S.A.), Bax (N-terminal, Santa Cruz), and MEKK1 (Santa Cruz). A synthetic peptide (Santa Cruz) was used to perform peptide competition experiments for MEKK1.
Protein crosslinking and immunoprecipitation
Interactions between cleaved caspase-3 and other proteins were studied. Cortical neurons were treated for 24 hours with CPT and then incubated for 20 minutes with 1 mmol/L dithiobis-succinimidyl-propionate (DSP; Pierce). The cells were rinsed, lysed, and proteins were extracted as described previously. Proteins (200 μg) were immunoprecipitated with antibody to cleaved caspase-3 (D175; Cell Signaling Technology) and resolved on gradient SDS polyacrylamide gels with or without β-mercaptoethanol in the loading buffer (DSP-crosslinked proteins are thiol cleavable). Proteins were transferred to nitrocellulose and blots were probed with antibody to cleaved caspase-3 (D175).
Immunocytochemistry
Immunofluorescence was used to visualize cleaved caspase-3 in nontreated and CPT-treated cortical neurons. Cells were fixed in 4% paraformaldehyde/4% sucrose in PBS (4°C, 20 minutes) and then with methanol (4°C, 10 minutes), and then permeabilized in 0.2% Triton X-100, 10% NGS in PBS (4°C, 10 minutes), and incubated overnight at 4°C in 10% NGS in PBS with primary antibody to cleaved caspase-3 (D175, 1:1000). Antibody binding was visualized with Alexa Fluor-conjugated goat antirabbit (Molecular Probes) that was diluted at 1:600 in PBS. Digital images were captured with a Zeiss Axiovrt/Quantix CCD camera.
Electron microscopy
Neuronal cultures treated with CPT at DIV5 and DIV25 were used for EM. Media were removed and the neuronal cultures were washed briefly with PBS, and the cells were fixed with 1% glutaraldehyde in PBS for 1 hour. Cells were processed for conventional EM directly in culture plates. Plastic-embedded neurons were removed from the culture plates as large blocks that were cut into pieces. Plastic blocks containing neurons were trimmed and sectioned for EM.
RESULTS
Mouse cortical neuron cultures mature and remain viable for the long-term
To study the effects of DNA damage on neurons at different stages of maturity, we used a long-term culture system of mouse cortical neurons that were maintained in an extremely viable state (Fig. 1). At DIV5 cortical neurons are bipolar with prominent neurites (Figs. 1A and 1D). At DIV25, cortical neurons are large and are embedded within a dense plexus of axons and dendrites that form numerous synaptic junctions (Figs. 1B, 1E, and 1F). The neurons have abundant polyribosomes distributed throughout the cytoplasmic matrix, intact arrays of rough endoplasmic reticulum and Golgi stacks, and uniformly shaped mitochondria with intact cristae. The nucleus has a predominantly pale matrix and is surrounded by a continuous, bilaminar nuclear membrane. The structural viability was well preserved in old DIV60 neurons (Fig. 1C).
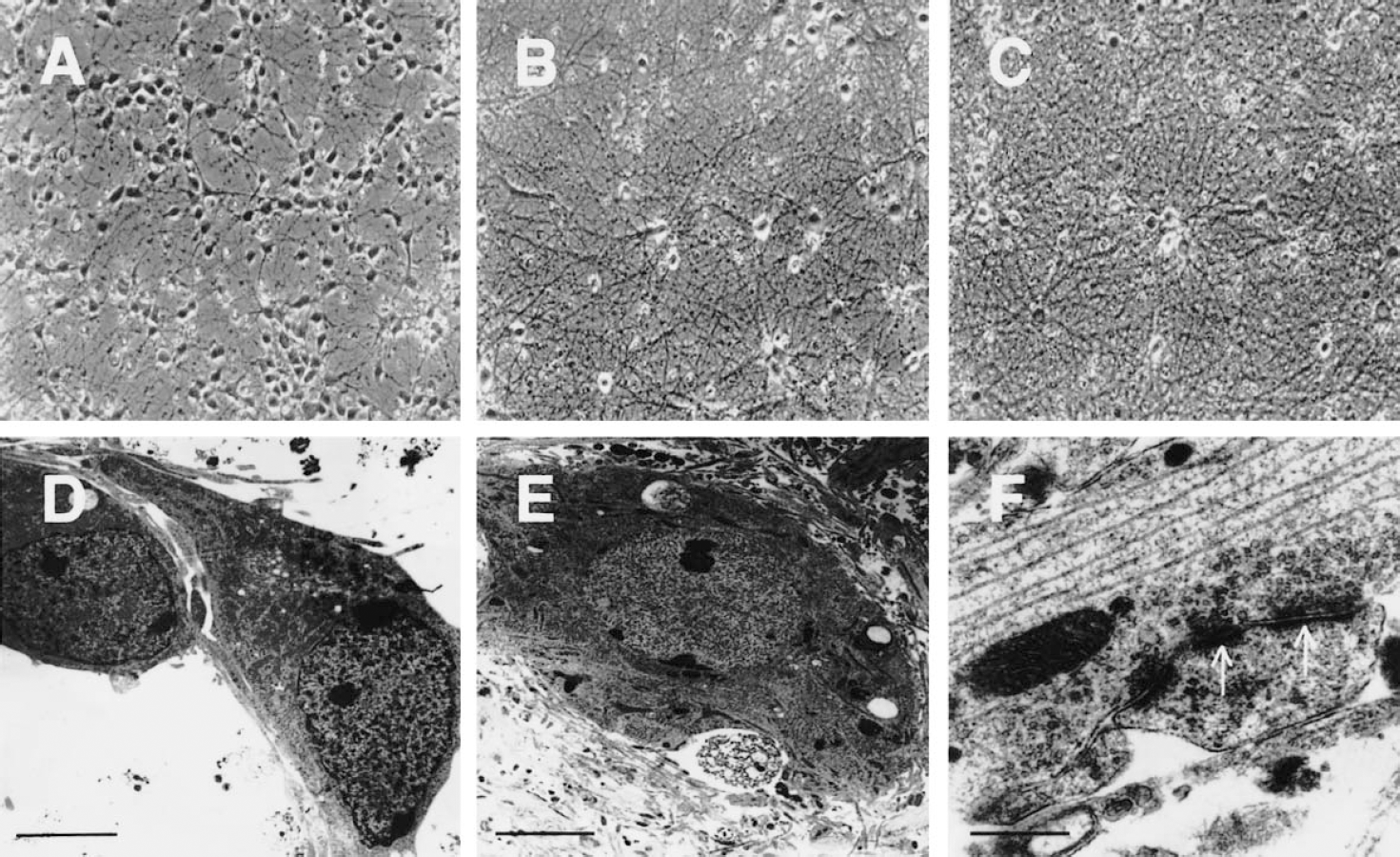
Mouse cortical neurons mature and can remain viable over the long term in culture.
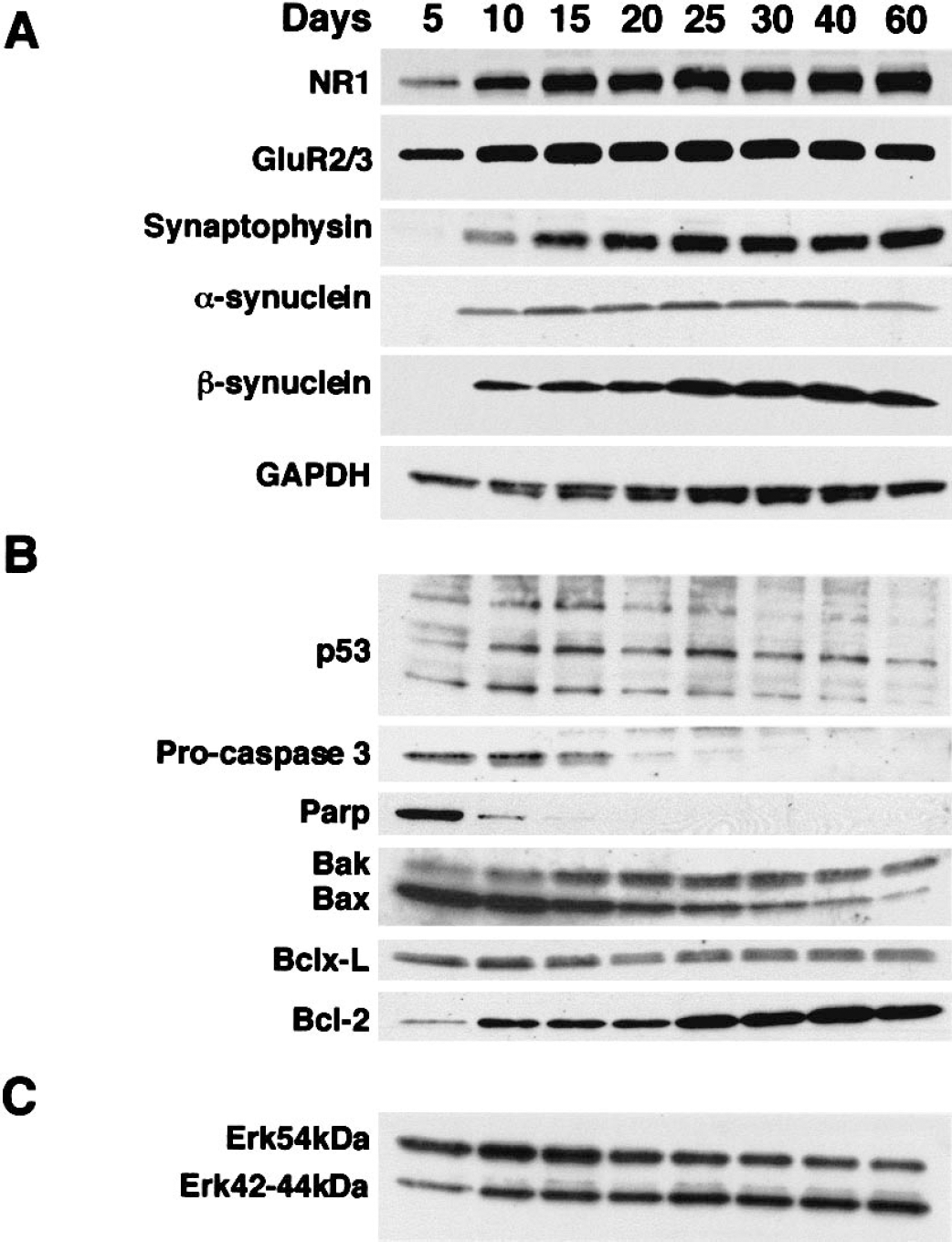
Molecular maturation of mouse cortical neurons in culture. The maturation and sustained viability of this long-term neuronal culture system were evaluated by the expression of glutamate receptors, synaptic proteins, and apoptosis-regulating proteins. For dendritic, synaptic, and metabolic maturation, NR1, GluR2/GluR3, synaptophysin, α/β-synucleins, and GAPDH (a glycolytic protein) were evaluated
To characterize the molecular viability of this long-term neuronal culture system, we evaluated the expression of glutamate receptors, synaptic proteins, and apoptosis-regulating proteins (Fig. 2). Total protein extracts from mouse cortical cultures at DIV5, 10, 15, 20, 25, 30, 40, and 60 were analyzed by immunoblotting. Expression of the NMDA receptor subunit NR1 and the AMPA receptor subunits GluR2/GluR3 was already detectable by DIV5 (Fig. 2A). Glutamate receptor levels increased to a maximum by DIV10–15 and then remained unchanged through DIV60. The levels of synaptic proteins (synaptophysin, α-synuclein, and β-synuclein) were very low at DIV5 (detectable only after prolonged exposure to film, data not shown). By DIV10, the expression of these synaptic proteins was detectable with short exposure (Fig. 2A). Synaptophysin, α-synuclein, and β-synuclein reached highest levels at about DIV15–20 and DIV25, respectively. As a metabolic marker, GAPDH (a glycolytic protein) was analyzed. The level of GAPDH peaked at approximately DIV25 and remained unchanged until after DIV40, and by DIV60 had decreased less than 30% (Fig. 2A). The high level of expression of NMDA and non-NMDA glutamate receptors, synaptic proteins, and metabolic enzyme suggests that our mouse cortical neuron culture remains viable over the long-term (60 days in culture).
Several proteins that function in cell death or survival were evaluated during the maturation of cortical neuron cultures from DIV5 to DIV60. These experiments provided important normative data because these proteins were also evaluated during CPT-induced apoptosis. The p53 in mouse cortical cultures was resolved as multiple immunoreactive bands at approximately 53 kd (Fig. 2B), consistent with phosphorylated and dephosphorylated forms of p53 (Levine, 1997). These immunoreactive proteins had mobilities similar to purified recombinant p53 (data not shown). The levels of apparent phosphorylated and dephosphorylated forms of p53 reached a maximum in expression at approximately DIV10–15, after which cortical neurons downregulated p53. Procaspase-3 was highly expressed by DIV5–10, decreased by more than 50% by DIV15, and was expressed at low levels after DIV20. PARP levels peaked at DIV5, and by DIV10 had declined by more than 90%. Inactivation cleavage products of PARP (85 kd) were low under normal culture conditions. Bak expression increased threefold to fourfold between DIV5 and DIV15, and after DIV30 Bak decreased slightly (less than 50%). In contrast, the level of expression of its homologue protein, Bax, was very high at DIV5 and DIV10 and then decreased progressively to low levels at DIV60. The level of expression of the antiapoptotic protein Bcl-xL did not change significantly during the 60 days in culture, while Bcl-2 expression increased fivefold to 10-fold between DIV5 and DIV25 and remained high through DIV60.
Erk42 levels increased threefold to fivefold from DIV5 to DIV10 and remained unchanged through DIV60 (Fig. 2C). Erk44 was only weakly detectable from DIV5 to DIV60. Erk54 protein expression was high at DIV5, and levels decreased slightly over the 60 days in culture.
The progression of apoptosis is different in immature and mature cortical neurons
Internucleosomal fragmentation of DNA was used as an assay for apoptosis in cortical neuron cultures treated with 1, 10, or 100 μmol/L CPT for 4, 8, 24, 48, or 72 hours. Camptothecin induced internucleosomal DNA fragmentation in neuronal cultures treated at DIV5 and DIV30. The timing and magnitude of DNA fragmentation were different in immature and mature neurons (Fig. 3A). The DIV5 neurons showed DNA fragmentation after 24 hours of CPT treatment. With 24 and 48 hours of treatment, long exposures were necessary to visualize a ladder (compare the DNA markers in both gels in Fig. 3A). In contrast, DIV30 neurons treated with the same concentrations of CPT showed prominent internucleosomal DNA fragmentation at 8 hours of treatment, with a peak at approximately 24 hours of treatment (Fig. 3A). Results of gel electrophoresis suggested that concentrations of CPT ranging from 1 to 100 μmol/L did not seem to influence significantly the magnitude of internucleosomal fragmentation of DNA degradation in cultures of different ages. However, light microscopy revealed that 100 μmol/L CPT appeared to induce a more robust and reliable apoptotic response than the lower CPT concentrations in neurons of different ages (data not shown). Therefore, subsequent toxicologic experiments were done using 100 μmol/L CPT.
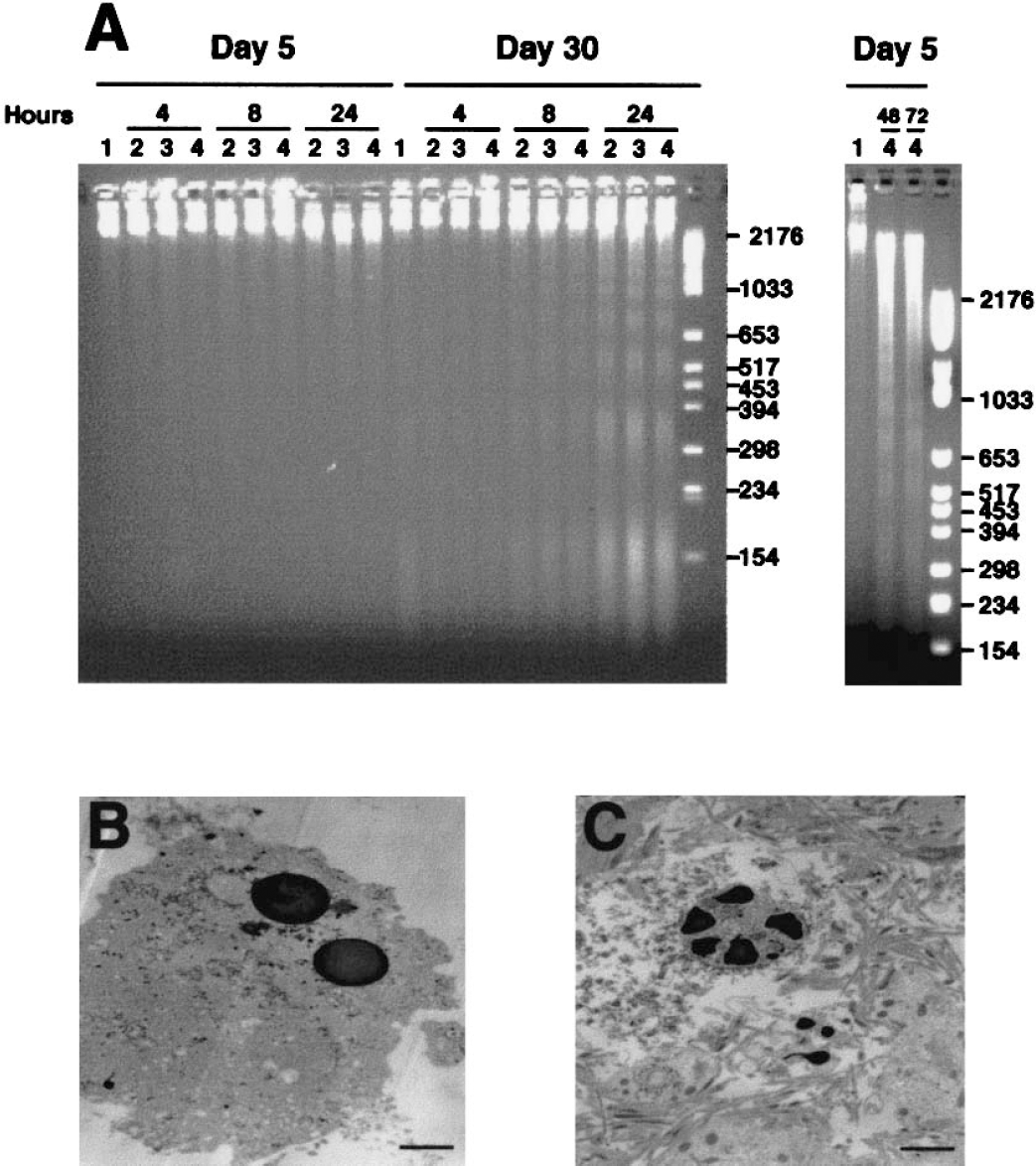
The emergence of DNA fragmentation and chromatin condensation is different in immature and mature cortical neurons treated with camptothecin (CPT).
Electron microscopy confirmed that mouse cortical cultures treated with CPT at DIV5 and DIV30 were morphologically apoptotic (Figs. 3B and 3C). The morphology of the chromatin condensation was different at the two ages. The nuclear morphology in the majority of apoptotic DIV5 neurons was similar to classical apoptosis with the formation of uniformly round chromatin masses (Martin et al., 1998). Apoptosis in DIV30 neurons was different because large, irregularly shaped chromatin masses were formed.
Caspase-3 cleavage is different in immature and mature neurons undergoing camptothecin-induced apoptosis
Caspase-3 was evaluated in subcellular fractions of control neurons and CPT-treated neurons (Fig. 4). The purity of the fractions generated by the subcellular fractionation method has been confirmed (Martin, 1999, 2001;Fig. 8). Caspase-3 proenzyme levels were very different in total extracts of developing and mature neuronal cultures (Fig. 2B). Caspase-3 proenzyme was found in nuclear, pellet, and soluble protein fractions (Fig. 4A to Fig. 4C). The DIV5 cortical neurons had higher levels of proenzyme in nuclear (Fig. 4A), pellet (Fig. 4B), and soluble (Fig. 4C) fractions than DIV30 neurons. In control DIV30 neurons, full-length caspase-3 was highest in the nuclear fraction (Fig. 4A). After exposure to CPT, proenzyme levels were decreased at 24 hours in DIV5 neuron-soluble (Fig. 4C) and pellet (Fig. 4B) fractions, but were unchanged in nuclear (Fig. 4A) fractions. In CPT-treated DIV30 neurons, caspase-3 proenzyme levels were increased modestly in nuclear fractions (Fig. 4A) but were unchanged in pellet and soluble fractions (Figs. 4A and 4B).
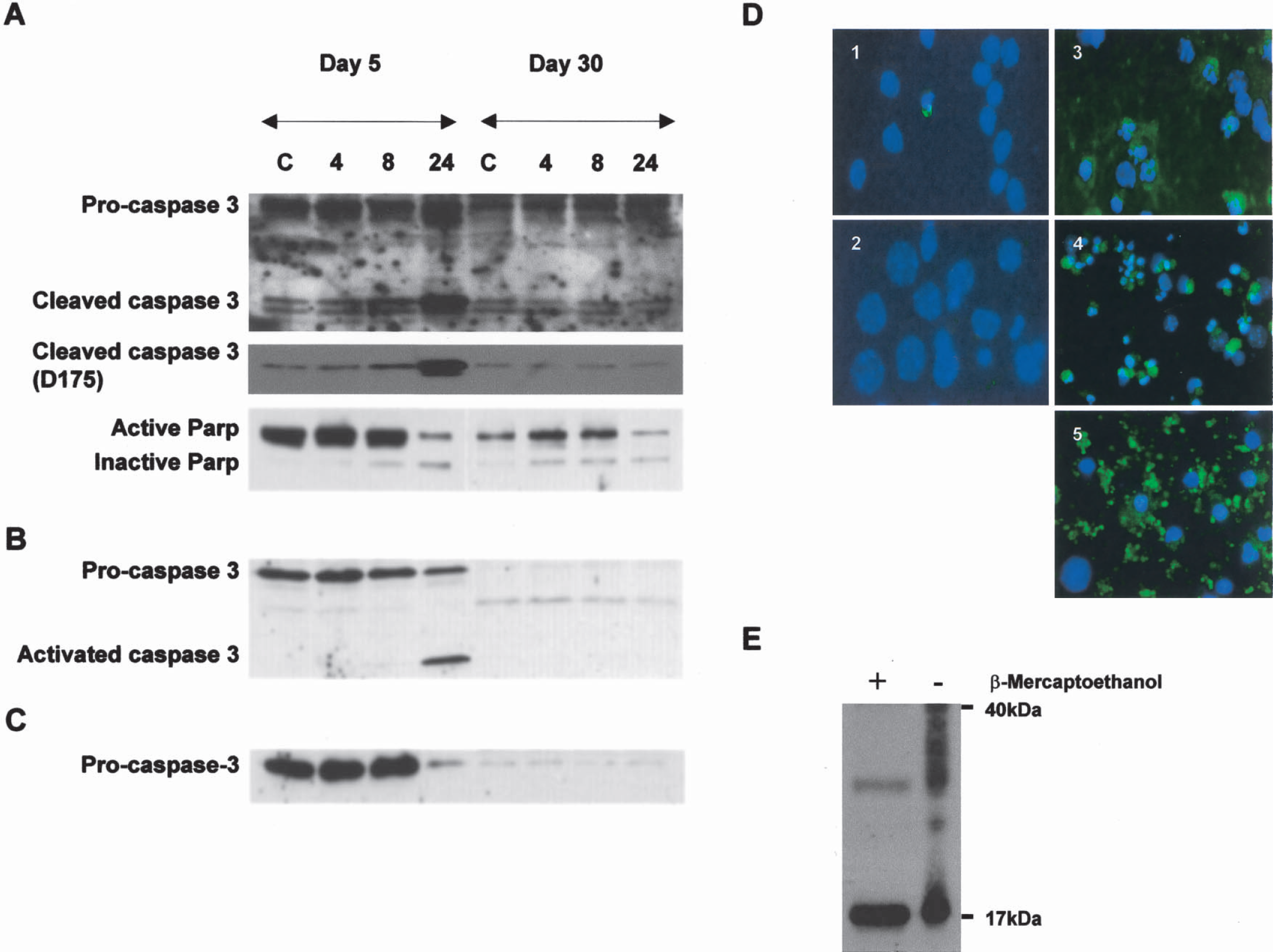
Caspase-3 cleavage is different in immature and mature cortical neurons undergoing camptothecin (CPT)-induced apoptosis.
Cleaved caspase-3 was evaluated in subcellular fractions of control neurons and CPT-treated neurons (Fig. 4). In control neurons, cleaved caspase-3 was found primarily in the nuclear fraction with two different antibodies (Figs. 4A and 4B). Control neurons at DIV5 and DIV30 had similar low levels of constitutively cleaved caspase-3 in nuclear fractions. In DIV5 neurons exposed to CPT, the levels of cleaved caspase-3 subunits (17–19 kd) progressively increased in nuclear fractions between 4 and 24 hours (Fig. 4A), and abruptly increased between 8 and 24 hours in pellet fractions (Fig. 4B). In contrast, the levels of cleaved caspase-3 did not change in nuclear and pellet fractions of DIV30 neurons after CPT treatment.
Cortical neurons were examined for cleaved caspase-3 using immunocytochemistry. Cleaved caspase-3 immunoreactivity (detected with D175 antibody) was rarely observed in control cells, but was seen infrequently in isolated apoptotic cells in control cultures at DIV5 and DIV30 (Figs. 4D1, 2). After CPT exposure, DIV5 and DIV30 neurons showed different immunolocalization patterns for cleaved caspase-3. In DIV5 neurons with 24-hour CPT exposure, cleaved caspase-3 was observed in the perikaryal cytoplasm and nucleus and in neuronal processes in the surrounding matrix (Fig. 4D3). In DIV30 neurons with 8-hour CPT exposure, cleaved caspase-3 immunoreactivity was prominent in the perikaryal cytoplasm and nucleus (Fig. 4D4), and with 24-hour CPT exposure cleaved caspase-3 was localized to cellular debris of apoptotic neurons (Fig. 4D5). Apoptotic cells incubated with secondary antibody without prior treatment with cleaved caspase-3 antibody did not show labeling (data not shown).
Immunofluorescence due to nonspecific secondary antibody binding to degenerating neurons in vitro has not been observed with the concentration used in the present study (Lesuisse and Martin, 2002). Similar patterns of specific labeling for cleaved caspase-3 have been seen in apoptotic striatal neurons in vivo (Lok and Martin, 2002) and apoptotic thalamic neurons in vivo (Natale et al., 2002).
The levels of PARP were examined by immunoblotting because this DNA repair enzyme is cleaved and inactivated by active caspase-3 (Lazebnik et al., 1994). The levels of intact PARP are much higher in DIV5 neurons compared with DIV30 neurons. PARP was inactivated in CPT-treated DIV5 and DIV30 neurons, as seen by the formation of an 85-kd cleavage product (Fig. 4A), indicating the activation of caspase-3. The cleavage of PARP occurred earlier in DIV30 neurons compared with DIV5 neurons.
Cleaved caspase-3 interacts with many proteins
To understand the seemingly discrepant immunoblotting and immunolocalization results on cleaved caspase-3 during CPT-induced apoptosis, we hypothesized that cleaved caspase-3 forms complexes with other proteins that could serve as a pool of constitutively cleaved, inactive caspase-3. The DSP crosslinking and immunoprecipitation showed that cleaved caspase-3 in cortical neurons binds many proteins of different sizes (Fig. 4E, right lane). As a negative control, protein interactions involving cleaved caspase-3 were reversed by β-mercaptoethanol, resulting in a single major 17-kd band of cleaved caspase-3 (Fig. 4E, left lane).
Mitogen-activated protein kinase signaling is different in immature and mature cortical neurons during camptothecin-induced apoptosis
Components of the MAP kinase pathway were evaluated in subcellular fractions of control and CPT-treated DIV5 and DIV30 neurons. In control neurons, Erk54 was higher in nuclear fractions of mature neurons, but Erk54 was higher in the pellet and soluble fractions of immature neurons (Fig. 5A). Changes in Erk after CPT treatment were different in neuronal cultures at different ages. In DIV5 neuron nuclei, Erk54 levels were increased approximately fivefold after 24 hours of CPT exposure, whereas in DIV30 neuron nuclei only a 1.5-fold increase in Erk54 occurred after 24-hour CPT exposure (Fig. 5A). Erk54 levels in the pellet fraction increased slightly (< 50%) in DIV5 neurons after 24-hour exposure to CPT, but no change occurred in DIV30 neurons (Fig. 5A). Erk54 levels in the soluble compartment decreased by more than 50% in DIV5 neurons between 8 and 24 hours of CPT treatment, but Erk54 levels did not change in soluble fractions of DIV30 neurons (Fig. 5A).
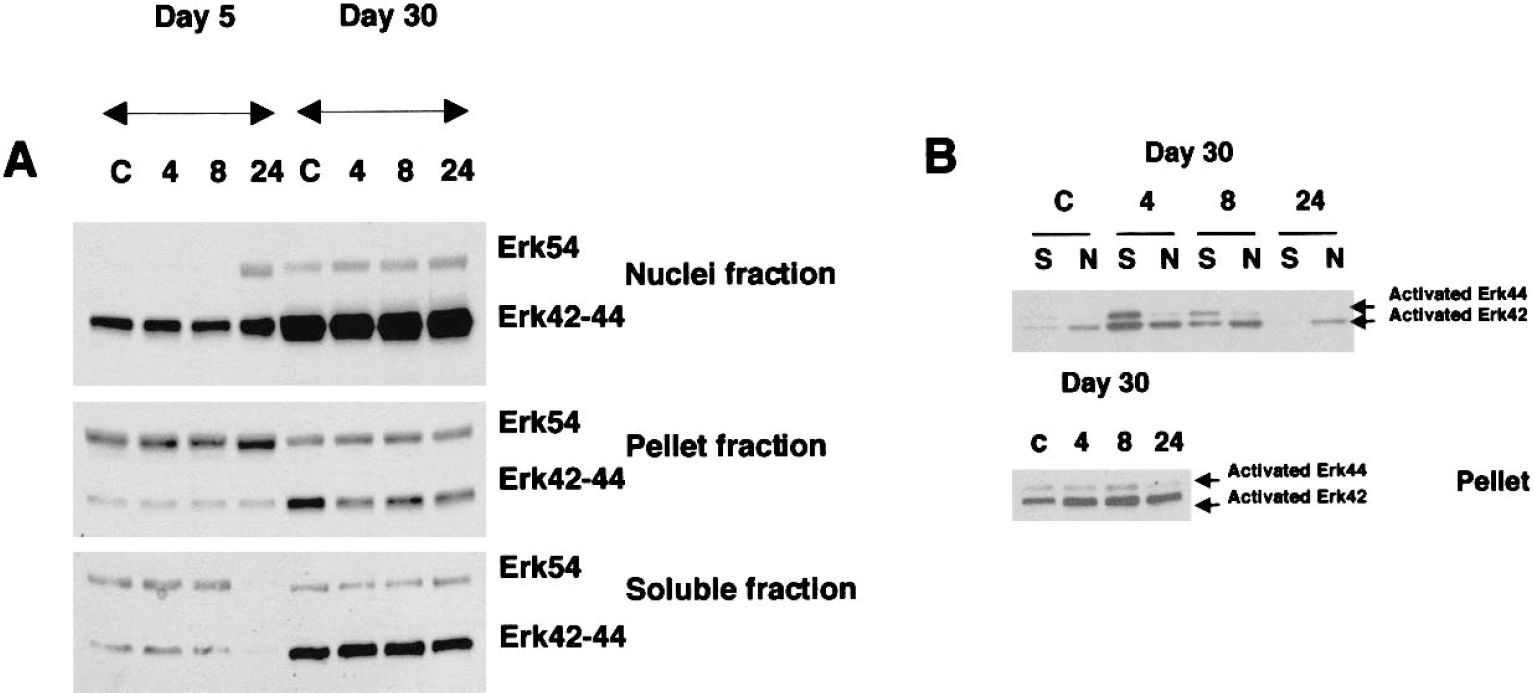
Extracellular signal-regulated kinase (Erk) signaling is different in immature and mature cortical neurons undergoing camptothecin (CPT)-induced apoptosis. DIV5 (days in vitro: 5) and DIV30 mouse cortical neurons were treated with vehicle (control, C) or with 100 μmol/L CPT for 4, 8, or 24 hours (18 wells per treatment) and collected for subcellular fractionation into nuclear, pellet, and soluble fractions and subsequent immunoblot analysis with pan-Erk antibody
Erk42/44 levels in nuclear, pellet, and soluble fractions were lower in DIV5 neurons compared with DIV30 neurons (Fig. 5A). Total Erk42/44 levels in DIV5 and DIV30 neurons during CPT-induced apoptosis did not change in the different subcellular fractions, except for a loss in the soluble fraction of DIV5 neurons at 24 hours (Fig. 5A).
Active Erk levels were different in immature and mature control neurons and in neurons responding to DNA damage. Phosphorylated Erk42/44 was not detected in the different subcellular fractions of control DIV5 neurons, and no phosphorylation of Erk42/44 occurred in DIV5 neurons after CPT exposure (data not shown). In contrast, phospho-Erk was detected in control DIV30 neurons (Fig. 5B). Activated Erk42 levels were generally higher than activated Erk44 levels in nuclear, soluble, and pellet fractions. The pellet fraction had the highest level of activated Erk42 (Fig. 5B). Phospho-Erk42/44 levels in nuclear and soluble fractions increased dramatically (> 10-fold for soluble Erk44) after CPT after 4 hours of treatment (Fig. 5B) and remained high until 8 hours of treatment, after which levels decreased. Activated Erk42/44 levels in the pellet fraction were approximately double control levels at 4 and 8 hours (Fig. 5B).
A reversible caspase-3 inhibitor blocks apoptosis in immature neurons and MEK inhibition blocks apoptosis in immature and mature neurons
Caspase-3 is activated in cortical neurons undergoing CPT-induced apoptosis, based on immunoblotting or immunolocalization for cleaved caspase-3 and on PARP cleavage. Furthermore, Erk42/44 is activated prominently in DIV30 neurons, but not in DIV5 neurons. To determine whether these pathways are participating in the mechanisms of apoptosis in cortical neurons induced by CPT, cells were treated either with inhibitors of caspase-3 or with MEK. Caspase-3 inhibition blocked CPT-induced apoptosis in DIV5 but not DIV25 cortical neurons (Fig. 6). The MEK1/2 inhibitor U0126 blocked apoptosis in both DIV5 and DIV25 cortical neurons (Fig. 6). The neuroprotection was more dramatic in DIV5 neurons. In control cells not exposed to CPT, U0126 induced apoptosis in DIV25 neurons but not in DIV5 neurons (Fig. 6).
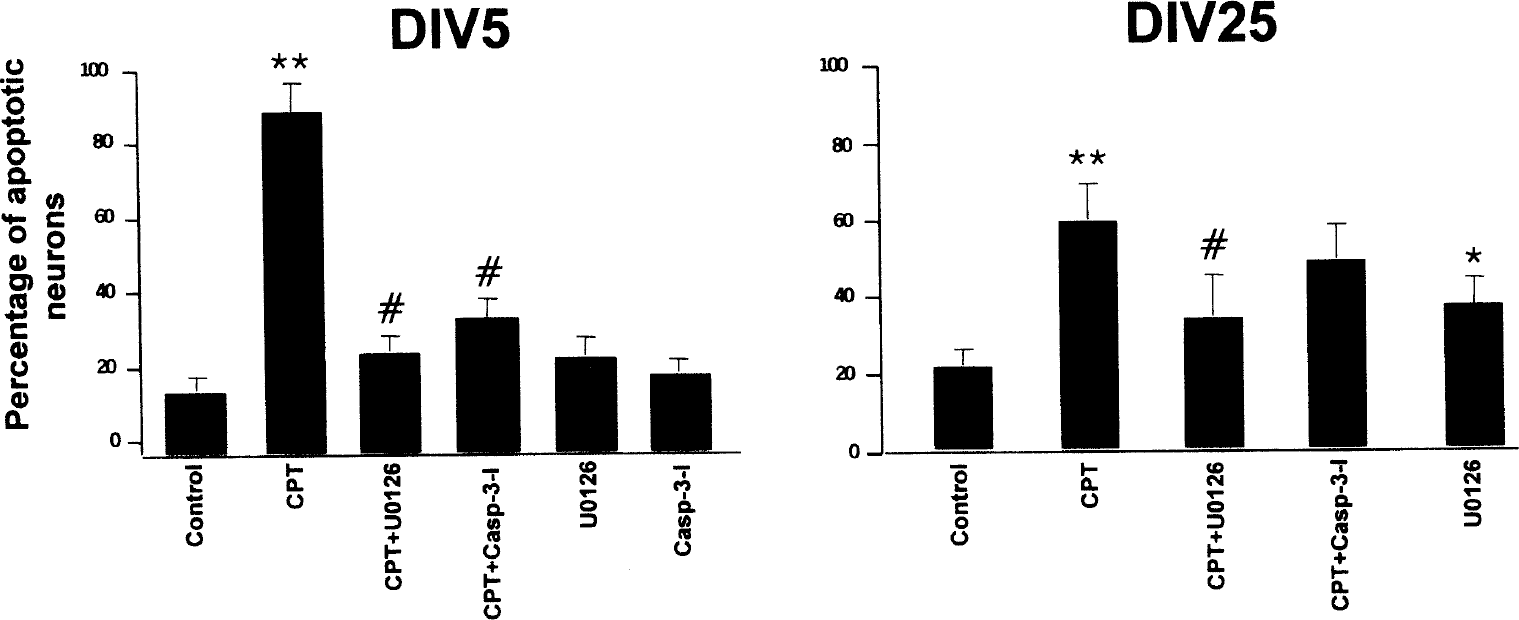
The effects of drugs on camptothecin (CPT)-induced apoptosis in immature and mature cortical neurons are different. Immature cortical neuron cultures at DIV5 (days in vitro: 5; left) and mature cortical neurons cultures at DIV25 (right) were treated as controls or were exposed to 100 μmol/L CPT for 24 hours alone or with prior exposure (2-hour pretreatment) to cell-permeable reversible caspase-3 inhibitor (Casp-3-I) or the MEK1/2 inhibitor U0126, or were treated with only U0126 or caspase-3 inhibitor. Hoechst 33258 staining was used to determine the percentages of apoptotic neurons. The values are mean ± SD. Results shown are from experiments using inhibitors at 100-μmol/L concentrations. Similar results were obtained using inhibitors at 10-μmol/L concentrations. These results were reproduced in three different platings. In DIV5 neurons, CPT induced significant (** P < 0.001) apoptosis compared with the control. The U0126 and caspase-3 inhibitor significantly (#P < 0.001) attenuated the CPT-induced apoptosis in DIV5 neurons. In DIV25 neurons, CPT induced significant (** P < 0.01) apoptosis compared with the control. U0126 significantly (#P < 0.05) attenuated the CPT-induced apoptosis in DIV25 neurons. In DIV25 neurons not exposed to CPT, U0126 caused a significant (* P < 0.05) induction of apoptosis compared with the control. MEK, MAP kinase kinase.
We evaluated if the antiapoptotic effects of U0126 were associated with a blockade of alterations in the MAP kinase pathway in CPT-treated neurons (Fig. 7). In CPT-treated DIV5 neurons, U0126 blocked the subcellular translocation of Erk54 from soluble to nuclear compartments and the nuclear accumulation of cleaved caspase-3 (Fig. 7A). Evidence for Erk42/44 activation and translocation in DIV5 neurons during apoptosis was not observed (Fig. 5A); thus, these events were not studied in cells treated with CPT and U0126. In CPT-treated DIV25 neurons, U0126 blocked the activation of Erk42/44 early in the process of apoptosis, and this block in Erk42/44 activation was sustained (Fig. 7B).
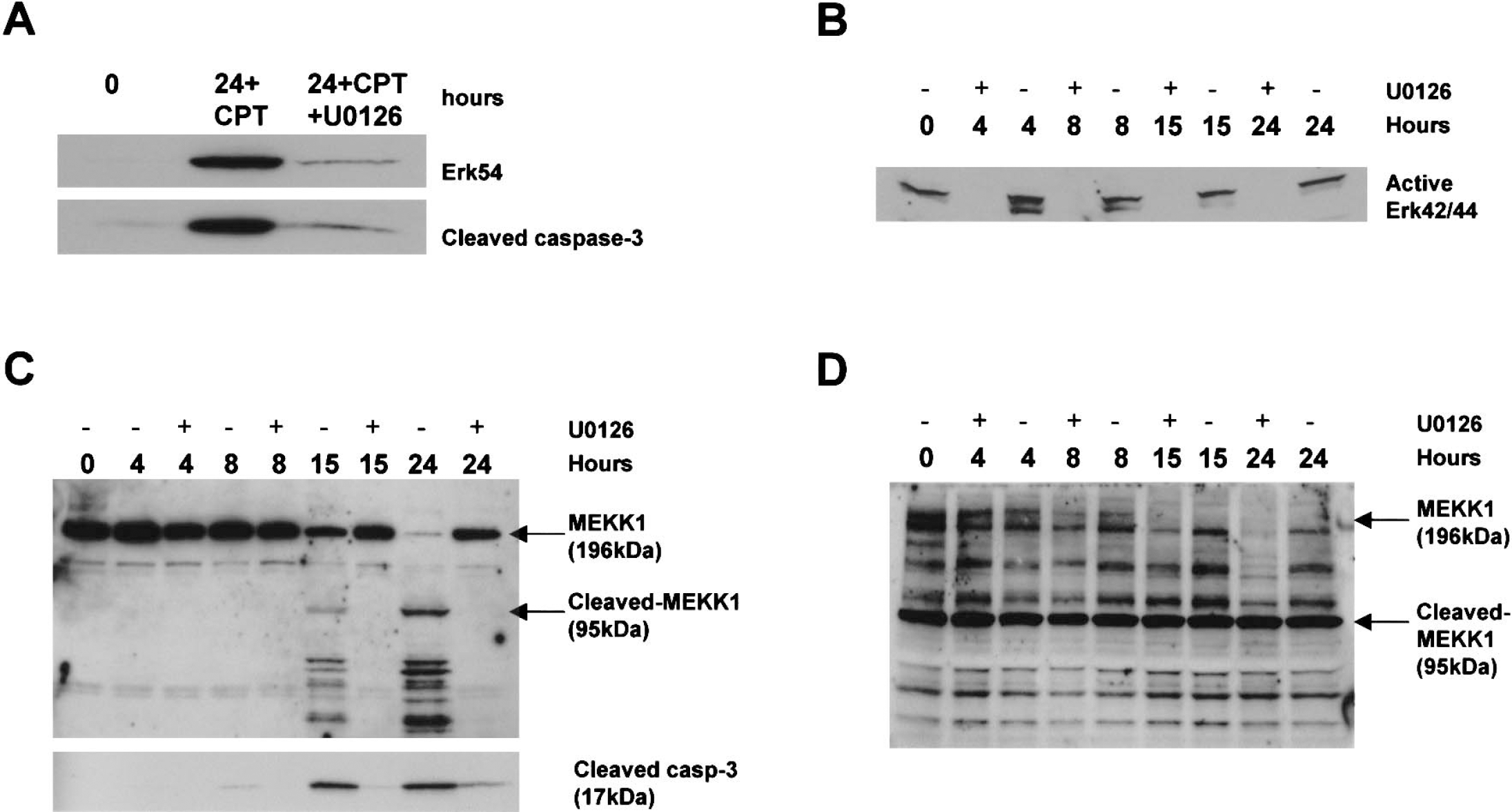
Different components of the MAP kinase pathway are activated in immature and mature cortical neurons undergoing apoptosis and are blocked by an inhibitor of MEK (U0126).
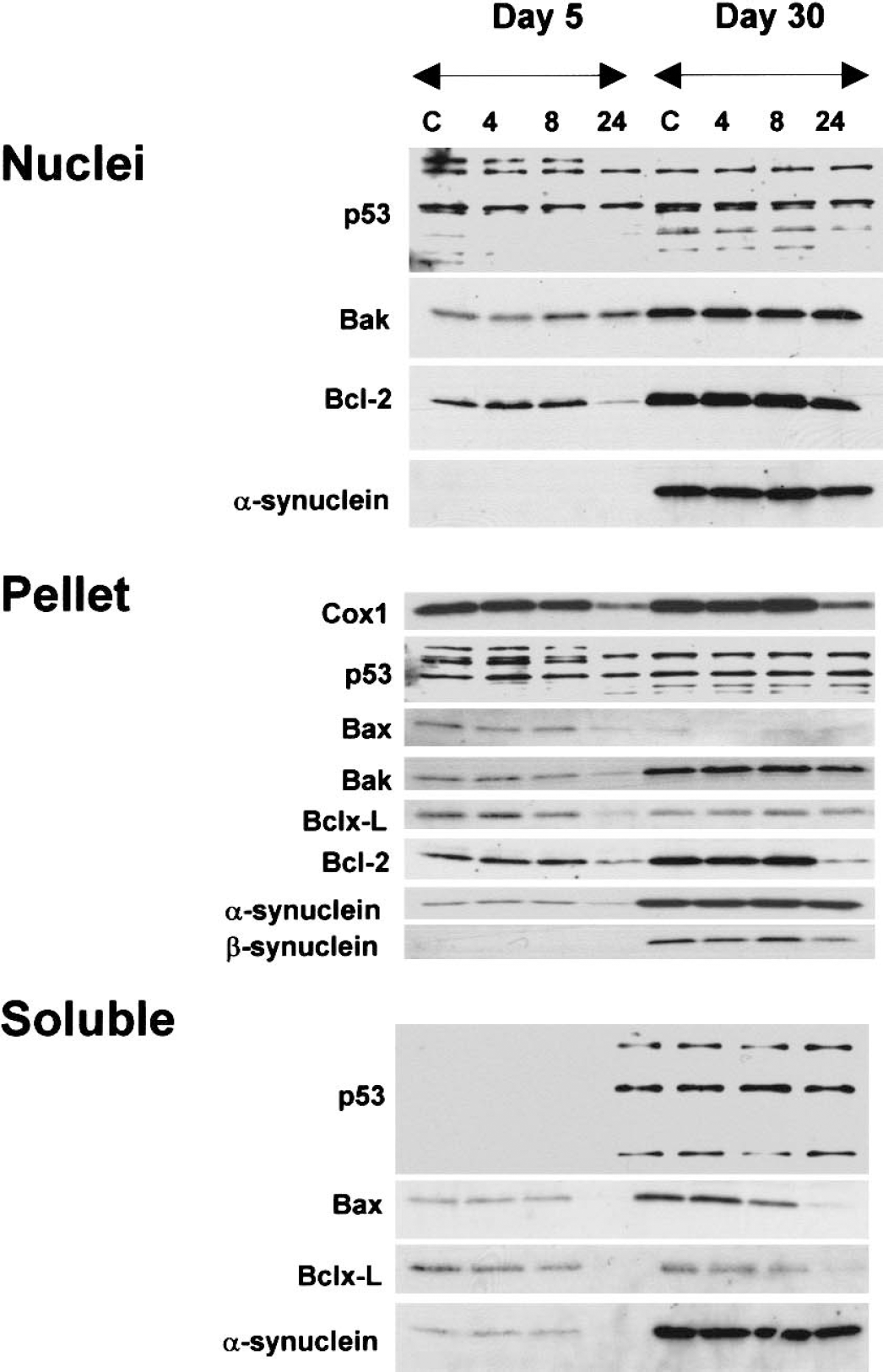
Subcellular levels of p53, Bcl-2 family members, and synuclein proteins during camptothecin (CPT)-induced neuronal apoptosis. DIV5 (days in vitro: 5) and DIV30 mouse cortical neurons were treated with vehicle (control, C) or with 100 μmol/L CPT for 4, 8, or 24 hours (18 wells per treatment) and collected for subcellular fractionation into nuclear, pellet, and soluble fractions and subsequent analysis by immunoblotting. Nuclear fractions were evaluated for p53, Bak, Bcl-2, and α-synuclein. Mitochondrial-enriched pellet fractions were analyzed for Cox1, p53, Bax, Bak, Bcl-xL, Bcl-2, and α/β-synucleins. The mitochondrial enrichment of this fraction was confirmed by the high levels of Cox1. Bax, Bak, Bcl-xL, and Bcl-2 were reduced at 24 hours in DIV5 neurons. Bcl-2 was lost in DIV30 neurons. Soluble fractions were evaluated for p53, Bax, Bcl-xL, and α-synuclein. p53 was not detected in soluble fractions. DIV5 neurons showed loss of Bax, Bcl-xL and α-synuclein at 24 hours. DIV30 neurons showed a loss of Bax and Bcl-xL at 24 hours. These results were reproduced in three different platings and at least three different immunoblots per antibody.
The MAP kinase pathway can function in DNA damage-induced apoptosis through MEKK1 in nonneuronal cells. Full-length MEKK1 (∼160 to 197 kd) promotes cell survival, whereas cleaved MEKK1 (∼72 to 95 kd) promotes apoptosis (Cardone et al., 1997;Widmann et al., 1998). Caspase-3 generates MEKK1 C-terminal fragments that have constitutively active proapoptotic kinase activity (Cardone et al., 1997). We examined the levels of full-length MEKK1 and putative proapoptotic MEKK1 fragments in nontreated and CPT-treated cortical neuron cultures. In control DIV5 neurons, a single major immunoreactive band of approximately 196 kd was detected (Fig. 7C), corresponding to full-length MEKK1. In CPT-treated neurons at 4 and 8 hours, only one major band of approximately 196 kd was still detected, and the levels were similar to control (Fig. 7C). In contrast, in CPT-treated neurons at 15 and 24 hours, several cleavage products of MEKK1 were observed. MEKK1 cleavage was greater at 24 hours compared with 15 hours (Fig. 7C). These cleavage products had sizes (∼70 to 95) that were similar to the reported sizes of the active kinase fragments of MEKK1 that induce apoptosis (Cardone et al., 1997). The detection of these immunoreactive bands with the MEKK1 antibody was blocked by competition with a synthetic peptide corresponding to the C-terminal domain of MEKK1 (data not shown). There was an inverse relationship between the levels of MEKK1 fragments and full-length MEKK1, supporting the conclusion that the immunoreactive proteins at approximately 70 to 95 kd are derived from cleavage of MEKK1 holoenzyme. The levels of full-length MEKK1 decreased correspondingly at 15 hours, and were very low at 24 hours, at a time when the level of immunoreactive MEKK1 fragments was greatest (Fig. 7C). The formation of MEKK1 C-terminal fragments coincided with the formation of cleaved caspase-3 in DIV5 neurons (Fig. 7C, lower blot). U0126 blocked MEKK1 degradation and the formation of the putative proapoptotic MEKK1 fragments and active caspase-3 in immature neurons (Fig. 7C). Mature neurons had lower levels of full-length MEKK1 and greater amounts of constitutively cleaved MEKK1 C-terminal fragments compared with immature neurons. The level of MEKK1 fragments at approximately 70 to 95 kd remained unchanged during apoptosis (Fig. 7D). However, the level of full-length MEKK1 decreased during apoptosis (Fig. 7D). Treatment with U0126 further decreased the level of full-length MEKK1 in CPT-exposed mature neurons (Fig. 7D).
Subcellular levels of p53, Bcl-2 family members, and synuclein proteins in cortical neurons and changes during camptothecin-induced apoptosis
We evaluated p53 in three subcellular fractions of cortical neurons. p53 was detected in nuclear and pellet fractions of DIV5 and DIV30 neurons, but was not present in soluble fractions of DIV5 neurons (Fig. 8). Overall no major changes in the levels occurred early during apoptosis.
The levels of three Bcl-2 family members were evaluated in nuclear fractions of cortical neurons (Fig. 8, nuclear). Bak and Bcl-2, but not Bax, were detected in nuclear fractions of mouse cortical neurons. The presence of Bcl-2 in the nucleus is consistent with its localization to the nuclear envelope (Lithgow et al., 1994), but the presence of Bak in the nuclear fraction is a novel finding. Both Bak and Bcl-2 levels were higher in mature neurons compared with immature neurons. The level of Bcl-2 in DIV5 neurons decreased markedly with CPT for 24 hours, but Bcl-2 levels did not change in CPT-treated DIV30 neurons (Fig. 8, nuclei). Bak levels were unchanged in apoptotic DIV5 and DIV30 neurons (Fig. 8, nuclei).
The insoluble pellet fraction of mouse cortical neurons was identified as the mitochondrial-enriched fraction based on the high level of Cox1 (Fig. 8, pellet). The mitochondrial fraction contained Bax, Bak, Bcl-xL, and Bcl-2, consistent with the localization of some of these proteins in mitochondrial membranes in nonneuronal cells (Lithgow et al., 1994) and neurons (Martin and Liu, 2002). Bak and Bcl-2 were higher in mitochondrial fractions of mature neurons, but Bax and Bcl-xL were higher in mitochondrial fractions of immature neurons. A dramatic loss of Cox1 in DIV5 and DIV30 neurons with 24-hour exposure to CPT showed that mitochondrial integrity was compromised between 8 and 24 hours during apoptosis (Fig. 8, pellet). Electron microscopy confirmed the degeneration of mitochondria by 24 hours of treatment (Figs. 3B and 3C). Late changes occurred in the levels of Bcl-2 family members in the mitochondrial-enriched membrane fraction during cortical neuron apoptosis (Fig. 8, pellet). In DIV5 neurons exposed to CPT, the levels of Bax and Bak decreased dramatically (> 50%). In contrast, in DIV30 neurons, Bax and Bak levels did not change conspicuously during apoptosis (Fig. 8, pellet). The level of Bcl-xL decreased by more than 50% in DIV5 neurons after 24 hours of treatment with CPT, but remained unchanged in DIV30 neurons treated similarly. Bcl-2 levels decreased in neurons at both ages between 8 and 24 hours of treatment.
In the soluble fraction of control cortical neurons, Bax and Bcl-xL were detected, but Bak and Bcl-2 were not detected (Fig. 8, soluble). Bax was more enriched in soluble fractions of neurons at DIV30 compared with DIV5 neurons. Bax levels decreased by more than 90% by 24 hours of CPT treatment at both ages. The level of Bcl-xL was similar in control cortical neurons at DIV5 and DIV30, and decreased by more than 50% between 8 and 24 hour of treatment with CPT.
Synuclein proteins function in apoptosis (Ostrerova et al., 1999). Major changes in their levels were not detected early during apoptosis of mouse cortical neurons (Fig. 8; nuclei, pellet, and soluble fractions). More interesting was the finding of differential localization in subcellular fractions. β-synuclein was not present in nuclear or soluble fractions of neurons at DIV5 and DIV30 or in the pellet fraction of DIV5 neurons; however, β-synuclein was present selectively in the pellet fraction of mature neurons.
DISCUSSION
The degeneration of neurons in vivo induced by excitotoxicity and axotomy-target deprivation appears to be influenced by CNS maturity (Martin, 2001;Martin et al., 2001;Natale et al., 2002;Portera-Cailliau et al., 1997). Thus, neuronal death might be different in immature and mature neurons. Here, we tested the hypothesis that signaling mechanisms for DNA damaged-induced apoptosis in immature and mature neurons are different. We found that immature and mature neurons engage different apoptotic mechanisms during apoptosis in vitro. To classify the cell death in our study as apoptosis, we used DNA fragmentation patterns, morphology, caspase-3 and MEKK1 cleavage, and sensitivity to caspase inhibition. Caspase and MAP kinase pathways have different contributions to CPT-induced apoptosis in immature and mature cortical neurons in vitro. Apoptotic immature and mature neurons were different in terms of structure and fragmentation of DNA, cleavage of caspase-3 and MEKK1, nuclear translocation of Erk54, and phosphorylation of Erk42/44 (Fig. 9). A caspase-3 inhibitor blocked apoptosis in immature neurons but not in mature neurons, while a MEK inhibitor blocked apoptosis in neurons at both ages. These results show for the first time that young and older cortical neurons use different molecular mechanisms for apoptosis.

Diagram contrasting some of the different and similar molecular mechanisms that are engaged during camptothecin (CPT)-induced apoptosis of immature and mature mouse cortical neurons. Erk, extracellular signal-regulated kinase; MAP, mitogen-activated protein; MEK, MAP kinase kinase; MEKK1, MAP kinase kinase kinase-1; PARP, poly-ADP ribose polymerase.
Development of a new long-term cell culture model to study apoptosis in neurons at different ages
To evaluate the molecular mechanisms of apoptosis in neurons at different ages, a healthy long-term neuronal culture system was required. We established a long-term primary neuronal culture system using mouse cortical neurons and conducted a thorough morphologic and biochemical characterization to demonstrate the legitimacy of this neuronal system. Dendritic, synaptic, and metabolic markers revealed the relative immaturity or maturity of these neurons. These cortical neuron cultures undergo robust dendritic maturation as shown by the expression of NMDA and AMPA receptors, prominent synapse formation as revealed by the expression of synpatophysin and synucleins, and metabolic maturation as shown by the expression of the glycolytic enzyme GAPDH. These cells remain healthy for at least 60 days, as evidenced by a sustained expression of glutamate receptors, synaptic proteins, Erk, and GAPDH. Morphologic assessments by light microscopy and EM confirmed the maturation and sustained viability of these neurons. The ability to maintain these cells to DIV60 attests to the healthy condition of these long-term neuronal cultures and strengthens our confidence that neuronal cell death experiments of fully mature but younger cultures (e.g., DIV30 neurons) will be representative of initially healthy cells that are undergoing a stress-induced or toxin-induced death process.
We used CPT to induce apoptosis in cortical neurons at different stages of maturation. A DNA-damaging agent was used as an apoptotic stimulus because DNA lesions contribute to the pathogenesis of CNS injury in children and adults (Kohji et al., 1998;Martin, 2001). Furthermore, upper cortical motor neuron degeneration in amyotrophic lateral sclerosis is a form of apoptosis that may be mediated by p53 mechanisms triggered by DNA damage (Martin, 2001). The induction of neuronal apoptosis after exposure to CPT has been shown previously (Morris and Geller, 1996;Park et al., 1997;Stefanis et al., 1999;Xiang et al., 1998). In nonneuronal cells, this poisoning may involve the stabilization of topoisomerase—DNA complexes and premature termination of transcription (Bendixen et al., 1990;Hsiang et al., 1985) as well as the formation of DNA single-strand breaks (Nieves-Neira and Pommier, 1999).
The progression of DNA fragmentation is different in immature and mature neurons undergoing apoptosis
DNA fragmentation was different in immature and mature cortical neurons undergoing apoptosis. Internucleosomal fragmentation of DNA occurred earlier and more prominently in DIV30 neurons than in DIV5 neurons (Fig. 9). The differences in DNA degradation patterns may reflect the participation of different endonucleases in the death of immature and mature neurons or different types of chromatin degradation early during apoptosis. Immature neurons may generate larger fragments (> 50 kb) early during apoptosis that are undetectable by conventional agarose gel electrophoresis (MacManus et al., 1997). Other studies have shown that apoptosis can occur in some cells without internucleosomal fragmentation of DNA (Tomei et al., 1993). The finding of a study of muscle cells that internucleosomal cleavage of DNA during apoptosis varies depending on the degree of differentiation (Fimia et al., 1996) is consistent with our results. Inactivation of the DNA repair enzyme PARP also occurred more rapidly in older neurons compared with young neurons. It is interesting that both DNA fragmentation and PARP cleavage were observed earlier in older neurons, despite more apoptosis in CTP-treated young neurons. The explanation for this observation is uncertain because of insufficient information on quantitative relationships between the fragmentation of DNA and death of single cells. In CTP-treated neurons, EM revealed that the morphology of the chromatin condensation into discrete aggregates were different in immature and mature neurons, with the immature neuron morphology being identical to that found in classical apoptosis (Martin et al., 1998). These results support the conclusion that mechanisms of DNA degradation and condensation are different in young and mature neurons. These data extend earlier structural studies suggesting that the apoptotic process is different in immature and mature neurons (Martin et al., 1998;Portera-Cailliau et al., 1997).
Caspase-3 cleavage is more prominent in immature neurons than in mature neurons during apoptosis
The processing of caspase-3 during apoptosis of cortical neurons is maturation dependent. Low constitutive levels of cleaved caspase-3 were detected by immunoblotting in control and CPT-treated immature and mature neurons. Caspase-3 cleavage products increased dramatically in immature neurons but not in mature neurons undergoing apoptosis. Cleaved caspase-3 accumulated in nuclear and mitochondrial-enriched fractions. A nuclear and mitochondrial translocation of cleaved caspase-3 in apoptotic immature cortical neurons is supported by findings in apoptotic nonneuronal cells in vitro (Zhivotovsky et al., 1999) and in striatal neurons undergoing excitotoxic death in newborn brain (Lok and Martin, 2002). In apoptotic mature cortical neurons, we did not find increased caspase-3 cleavage products in immunoblots of different subcellular fractions. However, immunolocalization showed a prominent presence of cleaved caspase-3 in apoptotic mature neurons that was not seen in control neurons. In both CPT-treated immature and mature neurons, PARP was inactivated, indicating activation of caspases (Lazebnik et al., 1994). It is therefore possible that apoptosis of mature neurons does not require formation of additional caspase-3 subunits via proteolysis of proenzyme, and that preexisting low levels of cleaved caspase-3 are sufficient to execute the death process. The rapid and robust internucleosomal fragmentation of DNA and the early PARP inactivation in apoptotic mature neurons (Fig. 9) are consistent with this possibility. Constitutively cleaved caspase-3 may be regulated by other proteins that function as endogenous inhibitors of cleaved caspase-3. This idea is in part supported by our crosslinking results. Some of these proteins that bind cleaved caspase-3 are likely to be substrates, but some could be negative regulators. In unstressed normal cells, cleaved caspase-3 might interact with possible inhibitor proteins or the subunits may be folded or assembled as inactive enzyme. This could explain the lack of immunoreactivity in control cells because the epitope that is recognized by the cleaved caspase-3 antibody is masked. In this state, constitutive cleaved caspase-3 would be inactive and could not execute the apoptotic process. Thus, activation of caspase-3 in mature neurons may occur by mechanisms other than proteolytic cleavage of proenzyme. It is also possible that apoptosis in young and older neurons has varying contributions of caspase-3–independent mechanisms.
Immature and mature neurons were different in their sensitivity to caspase-3 inhibitor. A reversible caspase-3 inhibitor had significant antiapoptotic actions in DIV5 neurons but not in DIV30 neurons. To our knowledge, this is the first demonstration of antiapoptotic actions of a reversible caspase-3 inhibitor in neurons (other studies have used irreversible inhibitors). This finding suggests that apoptosis in immature neurons may be caspase-3-dependent, whereas apoptosis of mature neurons is caspase-3-independent. However, the robust presence of cleaved caspase-3 observed directly in apoptotic mature neurons and the PARP cleavage would argue against this conclusion or in favor of the involvement of other caspases. It is not yet evident where caspase inhibitors act in intact cells to inhibit caspase activity. Inhibition of caspase-3 may occur in the cytoplasm, but the main action of active caspase-3 may be in the nucleus in mature neurons. This peptide inhibitor might not “see” existing cleaved caspase-3 in the nucleus. Furthermore, a peptide that is a reversible caspase-3 inhibitor would have to compete with endogenous protein inhibitors. Alternatively, caspases other than caspase-3 may be activated in apoptotic cortical neurons. Different types of caspase inhibitors need to be used to further evaluate the role of caspase-3 and other caspases in apoptosis of mature cortical neurons.
Previous studies have not clearly identified the role of caspases in CPT-induced apoptosis of immature neurons. Others have reported protection against CPT-induced apoptosis with 100 μmol/L caspase-3 inhibitor in rat embryonic cortical neurons at DIV1 (Stefanis et al., 1999) and mouse telencephalic neuron cultures at DIV4 (Johnson et al., 1999). Caspase inhibition partially protected against and delayed CPT-induced apoptosis in mouse embryonic cortical neurons at DIV2 (Keramaris et al., 2000). However, other experiments have failed to block CPT-induced neuronal apoptosis with caspase inhibitor (Park et al., 1997). Moreover, caspase-3 deletion in caspase-3−/− mice appears to only delay the neuronal death by 24 hours (Keramaris et al., 2000). We found with a caspase-3 inhibitor a near complete block of CPT-induced apoptosis of immature cortical neurons at 24 hours.
Mitogen-activated protein kinase pathway participates in the mechanisms of neuronal apoptosis
The MAP kinase pathway functions in the regulation of cellular proliferation, differentiation, survival, and death (Cardone et al., 1997;English et al., 1999), although in postmitotic cells (such as neurons) the functions of this pathway are much less understood. Our study shows that different downstream (Erk42/44 and Erk54) and upstream (MEKK1) components of the MAP kinase pathway are enlisted during CPT-induced apoptosis in mature and immature neurons.
In CPT-treated mature neurons, phosphorylated Erk42/44 was sequestered in the nucleus early (4 hours) after exposure. A parallel decrease in the level of phosphorylated Erk42/44 in the soluble fraction suggests translocation from the cytoplasm to the nucleus. The phosphorylation and nuclear accumulation of Erk42/44 preceded the internucleosomal fragmentation of DNA (detected robustly at 8 hours). When activated, Erk42/44 translocates to the nucleus to phosphorylate target proteins, including transcription factors (Elk, Sap, and Sp1) and histone proteins (English et al., 1999), many of which are involved in apoptosis. This nuclear translocation of active Erk42/44 may function as a preapoptotic signal or a transient, compensatory survival signal in mature neurons. There is precedence for Erk activation functioning proapoptotically in mesangial cells (Ishikawa and Kitamura, 1999) and neuronal cells (Satoh et al., 2000;Stanciu and DeFrance, 2002;Stanciu et al., 2000) after oxidative stress. We observed that blocking Erk activation by MEK inhibition protects cortical neurons against apoptosis, supporting a proapoptotic role for Erk activation in mature neurons.
Phosphorylation of Erk42/44 was not observed during the progression of apoptosis in immature neurons at DIV5. However, U0126 also blocked apoptosis in CPT-treated DIV5 neurons. The increased level of Erk54 could explain the apoptotic activity of the MAP kinase pathway in DIV5 neurons after exposure to CPT. This 54-kd protein detected with a pan-Erk antibody may be related to the Erk family (Keel et al., 1995). Erk54 has kinase activity and may be an isoform of Erk44 (Keel et al., 1995); however, Erk54 is distinct from Erk42/44 based on size, chromatographic properties, and substrate specificity (Kyriaks and Auruch, 1990). Because the level of Erk54 decreased dramatically (> 80%) in the soluble fraction, Erk54 (rather than Erk42/44) appears to be translocated to the nucleus in immature neurons but not mature neurons. This translocation of Erk54 coincided with the prominent nuclear accumulation of cleaved caspase-3. Both of these molecular events were blocked by MEK inhibition, which strongly protected DIV5 neurons against apoptosis.
MEKK1 could also account for the proapoptotic activity of the MAP kinase pathway in cortical neurons. This upstream kinase in the MAP kinase cascade also has dual functions. Full-length MEKK1 promotes cell survival, whereas MEKK1 C-terminal fragments promote apoptosis (Cardone et al., 1997;Widmann et al., 1998). Caspases act as switches to convert MEKK1 from a survival signal to a proapoptotic effector. MEKK1 fragments have constitutive kinase activity that can activate caspases, comprising a positive-feedback loop for driving apoptosis (Cardone et al., 1997). MEKK1 activity is required for apoptosis in nonneuronal cells after DNA damage (Widmann et al., 1998). We show for the first time that MEKK1 appears to have a role in neuronal apoptosis induced by DNA damage. This role might be different in immature and mature neurons stimulated to undergo apoptosis as well as in unstressed normal neurons. For instance, cleaved MEKK1 levels were constitutively higher in mature neurons than in immature neurons, suggesting that C-terminal fragments of MEKK1 are inactive as proapoptotic kinases in mature neurons. Major C-terminal fragments of MEKK1 were not present constitutively in immature neurons. However, during CPT-induced apoptosis of immature cortical neurons, the formation of C-terminal fragments of MEKK1 coincided with caspase-3 cleavage and decreased full-length MEKK1. In contrast, during CPT-induced apoptosis of mature cortical neurons, levels of major C-terminal fragments of MEKK1 did not appear to change, but levels of full-length (survival) MEKK1 declined, suggesting different mechanisms for MEKK1 processing and stabilization in mature and immature neurons. The loss of full-length MEKK1 may be sufficient to engage apoptosis in mature neurons. The antiapoptotic actions of U0126 could be mediated by the prevention of MEKK1 cleavage in immature neurons, but this is unlikely the case in mature neurons because MEK inhibition enhanced the loss of full-length MEKK1 during apoptosis. Thus, the primary antiapoptotic action of U0126 in mature neurons may be through blockade of Erk42/44 activation with a feedback modulation of MEKK1.
The duality of the MAP kinase pathway in regulating cell death or survival has been realized previously. In nonneuronal cells, basal constitutive activity of Erk functions in cell survival, whereas a transient upregulation of Erk induces apoptosis (Ishikawa and Kitamura, 1999). Depending on the cell type and context, Erk signaling can participate in neuronal survival (Hetman et al., 1999) or neuronal death (Satoh et al., 2000;Stanciu et al., 2000). A neuronal study revealed that Erk activation by neurotrophin is important for survival of rat cortical neurons in vitro (Hetman et al., 1999). However, activation of Erk42/44 might contribute to neuronal apoptosis in some in vitro models of neurotoxicity (Jiang et al., 2000;Stanciu et al., 2000), although the mechanisms are not known. We found that the MAP kinase pathway has a survival function in unstressed mature DIV30 neurons but not in unstressed immature DIV5 neurons. Specifically, MEK inhibition induced apoptosis in healthy, unstressed mature neurons but not in immature neurons (Fig. 6). This finding is consistent with the observation that DIV5 neurons do not require constitutive phosphorylation of Erk42/44. Therefore, Erk42/44 phosphorylation is less important as a survival signal in unstressed immature neurons compared with mature neurons. However, in mature and immature neurons undergoing genotoxic stress, the MAP kinase pathway assumes a proapoptotic function (Figs. 6 and 9). The proapoptotic activity of Erk in immature neurons with DNA damage did not appear to require phosphorylation of Erk42/44. Our study is the first to reveal a proapoptotic function of Erk during DNA damage-induced neuronal apoptosis that may be unrelated to Erk42/44 phosphorylation.
Roles of p53 and Bax during camptothecin-induced cortical neuronal apoptosis require further examination
Camptothecin neurotoxicity may involve p53 and Bax. Neuronal death induced by CPT in granule neurons is blocked in cells deficient in p53 and Bax (Xiang et al., 1998). We did not, however, observe increased p53 and Bax protein levels in different subcellular fractions (nuclear, insoluble/pellet, and soluble) of DIV5 and DIV30 cortical neurons exposed to CPT. An early change in the intracellular localization of Bax has been found in apoptotic nonneuronal cells, with a redistribution of soluble Bax to mitochondria promoting cell death (Wolter et al., 1997). In human colon adenocarcinoma cells treated with CPT, Bax redistributes from the cytosol to organelle membranes within 1 or 2 hours after drug exposure (Gajkowska et al., 2001). During striatal neuron apoptosis in newborn rat brain, Bax translocates to mitochondrial membranes within 2 hours after the stimulus (Lok and Martin, 2002). In cultured cortical neurons, the levels of Bax in the mitochondrial-enriched cell fraction and soluble fraction decreased dramatically between 8 and 24 hours after exposure to CPT. However, an early subcellular translocation of Bax could have been missed because our earliest time point was 4 hours after CPT; moreover, p53 activation (phosphorylation) needs to be evaluated in our system. Additional experiments are necessary to define the role of Bax and p53 in CPT-induced cortical neuron apoptosis.
CONCLUSIONS
Apoptotic cell death mechanisms in immature and mature cortical neurons are different (Fig. 9). Young and old neurons may use different caspase-3 activation mechanisms and enlist different components of the MAP kinase pathway during apoptosis. Caspase-3 activation by proenzyme cleavage occurs more strongly in apoptotic immature neurons compared with older neurons. The cleavage of full-length MEKK1 into putative proapoptotic fragments also occurs more prominently in immature neurons versus mature neurons. In contrast, Erk 42/44 is strongly activated in mature neurons undergoing apoptosis but not in immature neurons, while Erk54 increases in immature but not mature neurons. Our experiments show for the first time that the developmental maturity or age of neurons influences the mechanisms of apoptosis induced by DNA damage and that phamacologic therapies for the protection of young and older neurons might be different.
Footnotes
Acknowledgments
The authors are grateful for the expert technical assistance of Frank Barksdale and Ann Price.