
Abstract
Orthovanadate is a competitive inhibitor of protein tyrosine phosphatases. Some of its reported biologic effects are its insulin mimetic property and its activation of phosphoinositide 3-kinase and extracellular-signal regulated kinase (ERK). The authors previously reported its neuroprotective effect on delayed neuronal death of gerbil hippocampal CA1 neurons via Akt and ERK activation after transient forebrain ischemia. In the present study, the neuroprotective effect of postischemic intraperitoneal administration of sodium orthovanadate (2 mL/kg of 50-mmol/L sodium orthovanadate in saline) was investigated in rats with transient middle cerebral artery occlusion. Ischemic neuronal injury was evaluated 1 day and 28 days after ischemia. The neuroprotective effect of orthovanadate was significant in the cortex but not the caudate putamen (ischemic core) at both 1 and 28 days after ischemia. In orthovanadate group, the activities of Akt and ERK were maintained after reperfusion; they were decreased in saline group. Blood glucose level decreased but within normal range. Regional cerebral blood flow was lower than that of saline group only at 0 hours after reperfusion. These data suggest that orthovanadate has neuroprotective effects in rats with transient middle cerebral artery occlusion and that these effects are mediated by Akt and ERK activation. Furthermore, low blood glucose levels and gradual recovery of regional cerebral blood flow may contribute to neuroprotection.
Rodent models of focal cerebral ischemia after middle cerebral artery occlusion (MCAO) are used to represent ischemic stroke in humans (Ginsberg and Busto, 1989). Intraluminal suture is a relatively noninvasive means of inducing transient MCAO (Koizumi et al., 1986; Longa et al., 1989). It permits reperfusion, which occurs frequently after cerebral infarction, especially in patients with embolism (Ringelstein et al., 1992). After experimental arterial occlusion, the ischemic core is considered necrotic (Garcia et al., 1995; Lipton, 1999). In the penumbra, apoptotic mechanisms play a role (Charriaut-Marlangue et al., 1996; Li et al., 1998) and antiapoptotic agents may be able to rescue these areas (Simon et al., 2001; Yepes et al., 2000).
Akt (protein kinase B) is involved in antiapoptotic signaling downstream of phosphoinositide 3-kinase (PI3-K) in growth factor–mediated signaling cascades. Stimulation of tyrosine kinase growth factor receptors (Hemmings, 1997) or GTP-binding protein–coupled receptors activates PI3-K (Krugmann et al., 1999; Murga et al., 1998), thereby producing phosphatidylinositol-3,4,5-triphosphate (PIP3) and phosphatidylinositol-3,4-biphosphate (PIP2). Both PIP3 and PIP2 lead to dimerization and translocation of Akt to plasma membranes (Datta et al., 1997). In previous studies, phosphorylation of Akt (Thr-308 and Ser-473) by phosphoinositidedependent protein kinase-1 and serine/threonine kinases, including an integrin-linked kinase, was responsible for Akt activation (Alessi et al., 1996, 1997; Andjelkovic et al., 1997; Delcommenne et al., 1998; Kohn et al., 1996; Meier et al., 1997; Stephens et al., 1998). Active Akt phosphorylates Bad, caspase-9, cyclic AMP–responsive element binding protein, nuclear factor-κB, and forkhead transcription factors (Blume-Jensen et al., 1998; Brunet et al., 1999; Cardone et al., 1998; Datta et al., 1997; del Peso et al., 1997; Du and Montminy, 1998; Kops and Burgering, 1999; Romashkova and Makarov, 1999; Tang et al., 1999), thereby inducing antiapoptotic effects.
We have reported that decreased Akt activity is involved in ischemic neuronal cell death (Kawano et al., 2001, 2002) and that activated Akt plays an important role in ischemic tolerance (Yano et al., 2001). Other investigators (Friguls et al., 2001; Janelidze et al., 2001; Noshita et al., 2001) found that Akt activity decreased after transient MCAO. Thus, the induction of Akt activation may ameliorate ischemic neuronal cell injury.
Vanadium, a transition metal found in relative abundance in nature, is of widely varied biologic and physiologic significance (Brichard and Henquin, 1995; Elberg et al., 1994). Because vanadate, the +5 oxidation state of vanadium, appears to act as a phosphate analogue (Bevan et al., 1995; Huyer et al., 1997) mimicking the transition state (Zhang et al., 1997), it behaves as a competitive inhibitor of protein tyrosine phosphatases. Some pharmacologic actions of vanadate have been reported. It mimics many of the intracellular effects of insulin, such as the increase in hexose uptake, glycogen synthesis, glycolysis, and fatty acid synthesis in insulin-sensitive tissues (Shechter, 1990). This effect is probably due to the stimulation of protein tyrosine phosphorylation downstream of the insulin receptor (Shisheva and Shechter, 1993). It was recently shown that vanadate affected the phosphorylation levels of intracellular proteins via inhibition of nonselective protein phosphatases (Morinville et al., 1998). The activation of PI3-K by vanadate has been shown in rat adipocytes (Molero et al., 1998) and vanadate stimulated extracellular-signal regulated kinase (ERK) in Chinese hamster ovary cells (Pandey et al, 1995) and mouse epidermal C141 cells (Zhang et al., 2002). Similarly, intraventricular administration of orthovanadate (H2VO4) activates Akt through PI3-K activation and ERK (Kawano et al., 2001). Therefore, orthovanadate may exert beneficial effects on cell survival via Akt and ERK activation; in fact, we previously documented that it has neuroprotective effects in the delayed neuronal death of gerbil hippocampal CA1 neurons (Kawano et al., 2001).
Therefore, we speculated that orthovanadate exerts neuroprotection via Akt and ERK activation in experimental transient MCAO, such as the forebrain ischemia model. In the current study, we administered orthovanadate intraperitoneally to rats after the induction of 1.5-hour transient MCAO and evaluated the extent of ischemic injury at 1 (acute phase) and 28 days (delayed phase). We also examined the mechanisms underlying the protective effects by evaluating Akt and ERK activation and changes in physiologic parameters.
MATERIALS AND METHODS
Experimental animals
The Animal Care and Use Committee of Kumamoto University approved all experiments. Male Wistar rats weighing 330 to 400 g were kept under constant environmental conditions (temperature, 22° ± 2°C; humidity, 55% ± 5%; 12/12-hour light/dark cycle) in the Animal Research Center of Kumamoto University with free access to food and water before and after all procedures. Anesthesia was induced with 4% halothane and maintained with 2–2.5% halothane, 30% oxygen, and 70% nitrous oxide via a facemask. The left femoral artery was cannulated to measure arterial blood gases. Samples were taken before ischemia, at 45 minutes after MCAO, and immediately (0), 2, and 24 hours after reperfusion, and analyzed in a blood gas analyzer (SL Blood Analysis System Series 2000, Diametrics Medical, MN, U.S.A). The rectal temperature was monitored and kept at 38.0° ± 0.5°C by using a feedback-regulated heating system (Small Animals Heat Controller, Unique Medical, Tokyo, Japan) during surgery. Intermittently, the blood pressure was monitored via a tail artery (TK-340 Rat-Mouse Manometer-Tachometer, Unicom, Chiba, Japan), and blood glucose levels were measured via a tail vein (Precision Q.I.D.™ System, Abbott Laboratories, Illinois, U.S.A).
Regional cerebral blood flow measurements
Changes in regional cerebral blood flow (rCBF) were recorded on the dura of the left parietal cortex using a laser-Doppler flowmeter (ALF21, Advance, Tokyo, Japan) attached to a hollow plastic tube. After the rats were placed in a stereotaxic frame, craniectomy (2 mm lateral and 1 mm caudal to the bregma) was performed using a drill and extreme care. After the tube was fixed to the bone with resin, the flowmeter probe was inserted. Changes in rCBF were expressed as a percentage of baseline rCBF.
Induction of focal ischemia
After removing the rats from the stereotaxic apparatus, reversible focal ischemia was induced using a modification of the method described by Koizumi et al. (1986). Briefly, under an operating microscope, the left common carotid artery (CCA) was exposed through a midline incision in the neck. About 20 mm of 3-0 nylon suture coated with poly-L-lysine (Belayev et al., 1996) were introduced into the left internal carotid artery through the CCA. Immediately (0 hr) after occlusion, either 2 mL/kg of 50 mmol/L sodium orthovanadate (Wako Pure Chemical Industries, Osaka, Japan) in saline or saline alone was administered IP. After 1.5 hours the suture was withdrawn to allow middle cerebral artery reperfusion. Neurologic examinations were performed 10 minutes before reperfusion using a modification of the neurologic score of Bederson et al. (1986). Accordingly, grade 0 was recorded in the absence of observable deficits; grade 1 in the presence of forelimb flexion; grades 2 and 3 when there was decreased resistance to lateral push in the absence or presence of circling, respectively; and grade 4 was assigned to comatose animals. Rats with grades 0 and 4 were excluded from further experiments.
Measurement of the area of early ischemic brain injury
Animals were decapitated 1 day after ischemia induction (n = 8). Their brains were quickly removed, placed in cold saline solution for 10 minutes, and then cut into 2 mm-thick coronal slices using a rodent brain matrix. Six selected sections (slice a, +5 mm; b, +3 mm; c, +1 mm; d, −1 mm e, −3 mm; f, −5 mm from bregma) were stained for 30 minutes in a 2% solution of 2,3,5-triphenyltetrazolium chloride (TTC) at 37°C. The area of ischemic brain injury was measured using the NIH image program. Infarct areas were corrected to compensate for edema formation by subtracting the area of the intact ipsilateral hemisphere from the area of the intact contralateral hemisphere. Areas of the cortex and caudate putamen on each slice were calculated separately, and the infarct areas of total, cortex, and caudate putamen on each slice were added together and multiplied by slice thickness to give the infarct volume.
Measurement of the area of delayed ischemic brain injury
Animals were allowed to survive for 28 days and then their brains were fixed by cardiovascular perfusion with an aldehyde fixative (4% formaldehyde in 0.1-mol/L phosphate buffer) and immersed in fixative overnight (n = 8). Each brain was cut and six sections corresponding to those obtained for early injury studies were embedded in paraffin, cut into 5-μm sections, and stained with hematoxylin and eosin. The areas and volume of ischemic brain injury were measured as described above. The ipsilateral was smaller than the contralateral hemisphere, and there was atrophy in both the cortex and lateral part of the caudate putamen. We calculated the tissue reduction (contralateral hemisphere area minus ipsilateral hemisphere area) plus the area of the remaining infarct scar, and we regarded the results as total delayed infarct tissue damage (Persson et al., 1989).
Gel electrophoresis and immunoblotting
Samples were obtained from the periinfarct cortex and caudate putamen (ischemic core) (Fig. 1, X and Y) on the ipsilateral side. After decapitation at 0, 2, 6, or 24 hours after reperfusion and at preischemic time points (n = 4, respectively), the brains were removed and sections from the indicated regions were cut in cold saline under a microscope. Each sample was kept at −80°C until use. Frozen tissues were homogenized and sonicated with a Biorupture instrument (UCD-200TM, Cosmo Bio, Tokyo, Japan) at 0°C in 0.2 mL of homogenization buffer containing 50-mmol/L Tris-HCl (pH 7.5), 0.5% Triton X-100, 4-mmol/L EGTA, 10-mmol/L EDTA, 0.5-mol/L NaCl, 1-mmol/L Na3VO4, 30-mmol/L sodium pyrophosphate, 50-mmol/L NaF, 50-μg/mL leupeptin, 25-μg/mL pepstatin A, 50-μg/mL trypsin inhibitor, and 1-mmol/L dithiothreitol. Insoluble materials were removed by 15-minute centrifugation at 15,000 g. The protein content in each supernatant fraction was determined using Bradford's solution, each sample was applied to a 10% acrylamide denaturing gel, subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis, and then the proteins were transferred for 1 hour at 70 V to an Immobilon PVDF membrane (Millipore Corp., Bedford, MA, USA). Blotting membranes were incubated for 1 hour with 5% nonfat milk in phosphate-buffered saline (PBS) containing 0.1% Tween-20 (PBST) at room temperature and incubated overnight at 4°C with the first antibody solutions, such as a 1:1,000 dilution of anti-Akt antibody (Cell Signaling Technology, Beverly, MA, U.S.A), a 1:200 dilution of anti–phospho-Akt (Ser473) antibody (Cell Signaling Technology), a 1:3,000 dilution of anti–mitogen-activated protein kinase (pan ERK) antibodies (Transduction Lab., Lexington, KY, U.S.A), or a 1:500 dilution of anti-MAP kinase, activated (diphosphorylated ERK-1&2) antibody (Sigma-Aldrich, St. Louis, MO, U.S.A.) in nonfat milk in PBST. After several washes with PBST, the membranes were incubated for 1 hour with horseradish peroxidase–conjugated antirabbit antibody (Vector Laboratories, Burlingame, CA, U.S.A.) diluted 1:5,000 and processed with enhanced chemiluminescence Western blotting detection reagents (Amersham Pharmacia Biotech, Arlington Heights, IL, USA). The images were scanned and analyzed semiquantitatively using the NIH image software. Changes in total Akt and ERK expression, and the phosphorylation of Akt-Ser-473, and ERK were expressed as a percentage of the preischemia level.
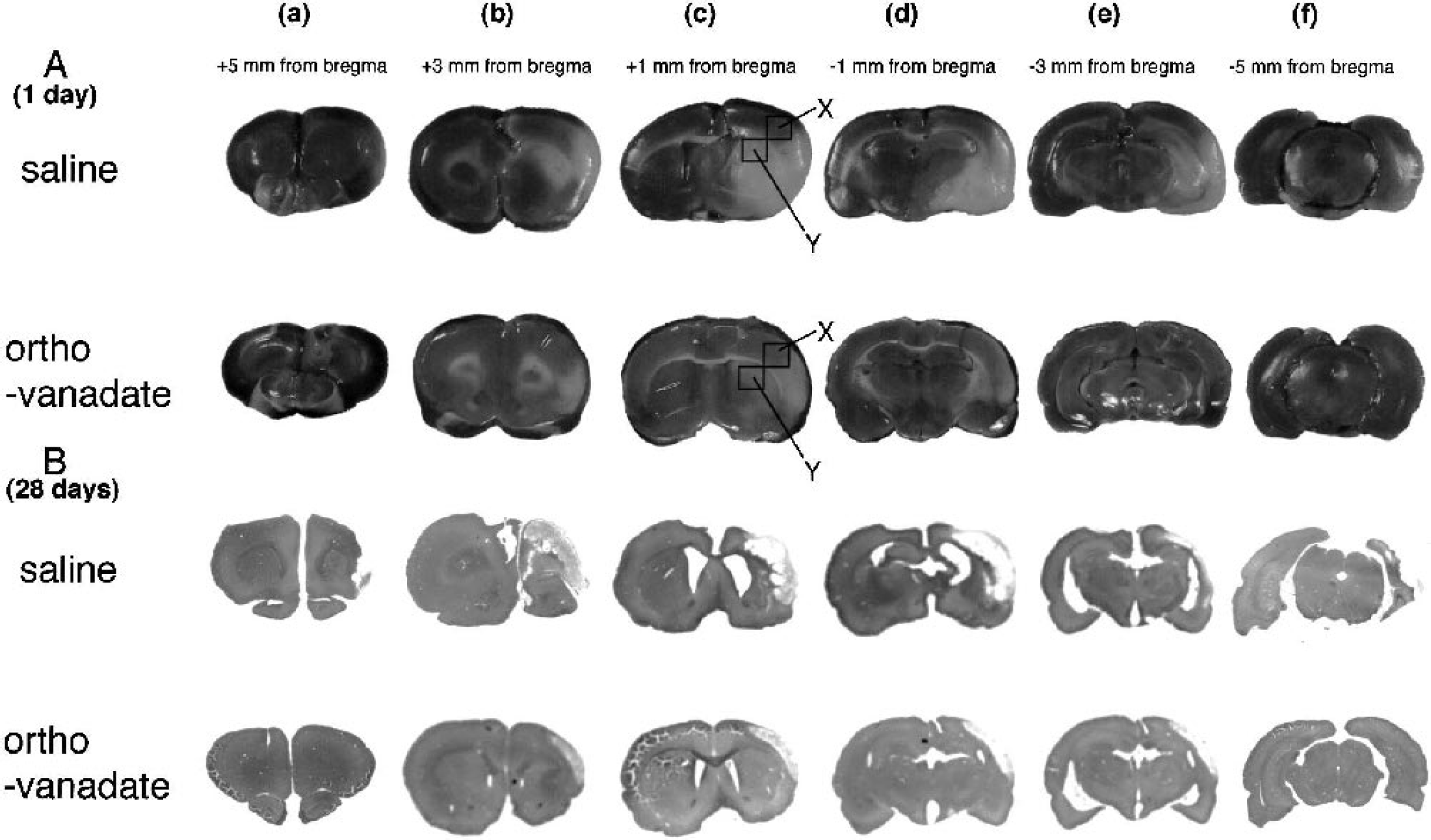
Representative ischemic lesions in six coronal sections from saline and orthovanadate groups at 1 (TTC) and 28 days (H&E) after ischemia. Slices a, b, c, d, e, and f were cut at +5, +3, +1, −1, −3, and −5 mm from bregma, respectively. The plus symbol indicates anterior direction. X and Y identify the area from which the sections were cut and evaluated by Western blotting and immunofluorescence staining as cortex and caudate putamen in Figs. 4, 5, 6, 7, and 8. H&E, hematoxylin and eosin; TTC, 2,3,5-triphenyltetrazolium chloride.
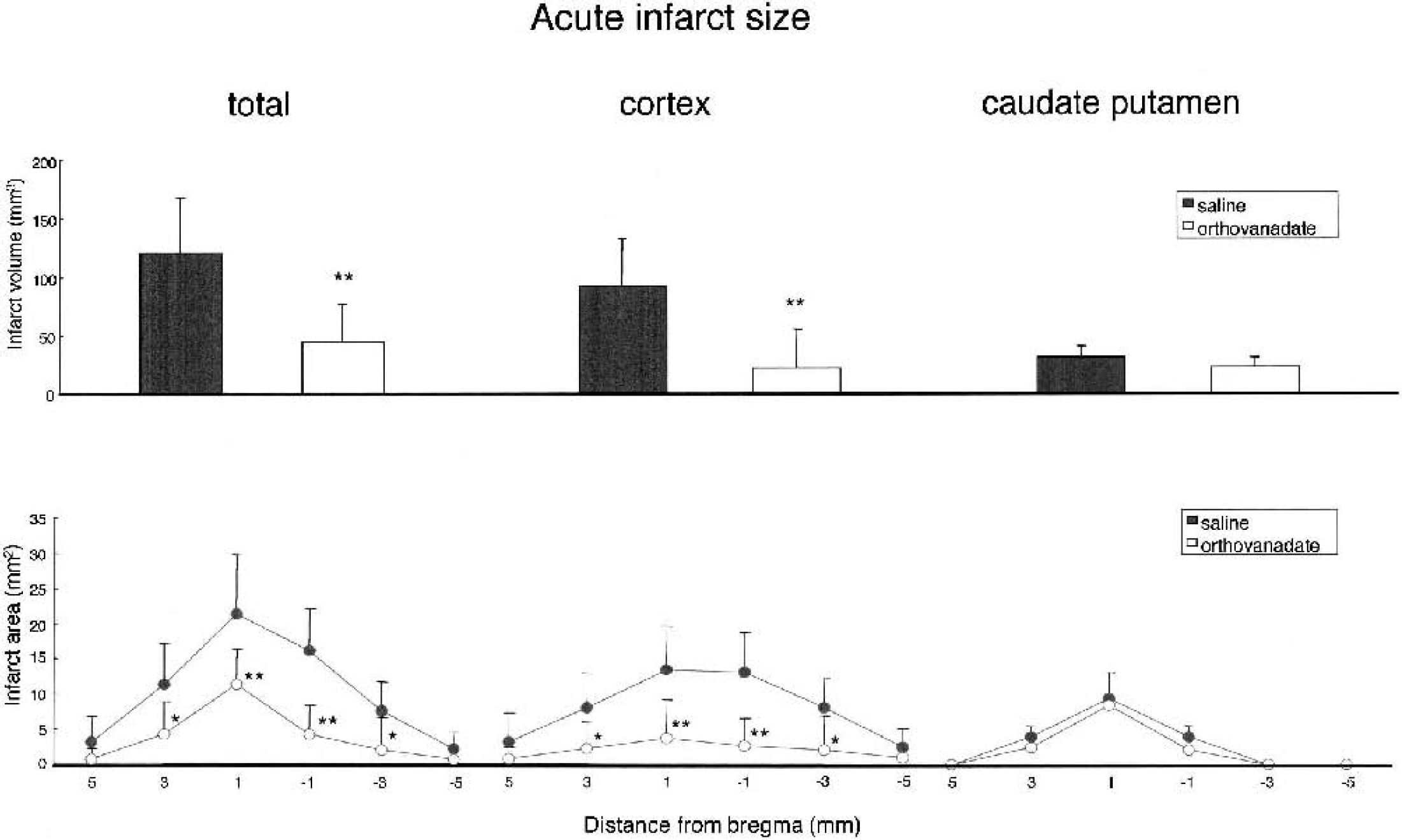
Quantitative analysis of infarct areas in six coronal sections and infarct volume from saline (n = 8) and orthovanadate (n = 8) groups in the early phase (1 day) after ischemia induction. Infarct areas and volume of the total, the cortex, and the caudate putamen were evaluated. Values are the mean ± SD; asterisks denote significant differences between saline and orthovanadate groups (*P < 0.05; **P < 0.01).
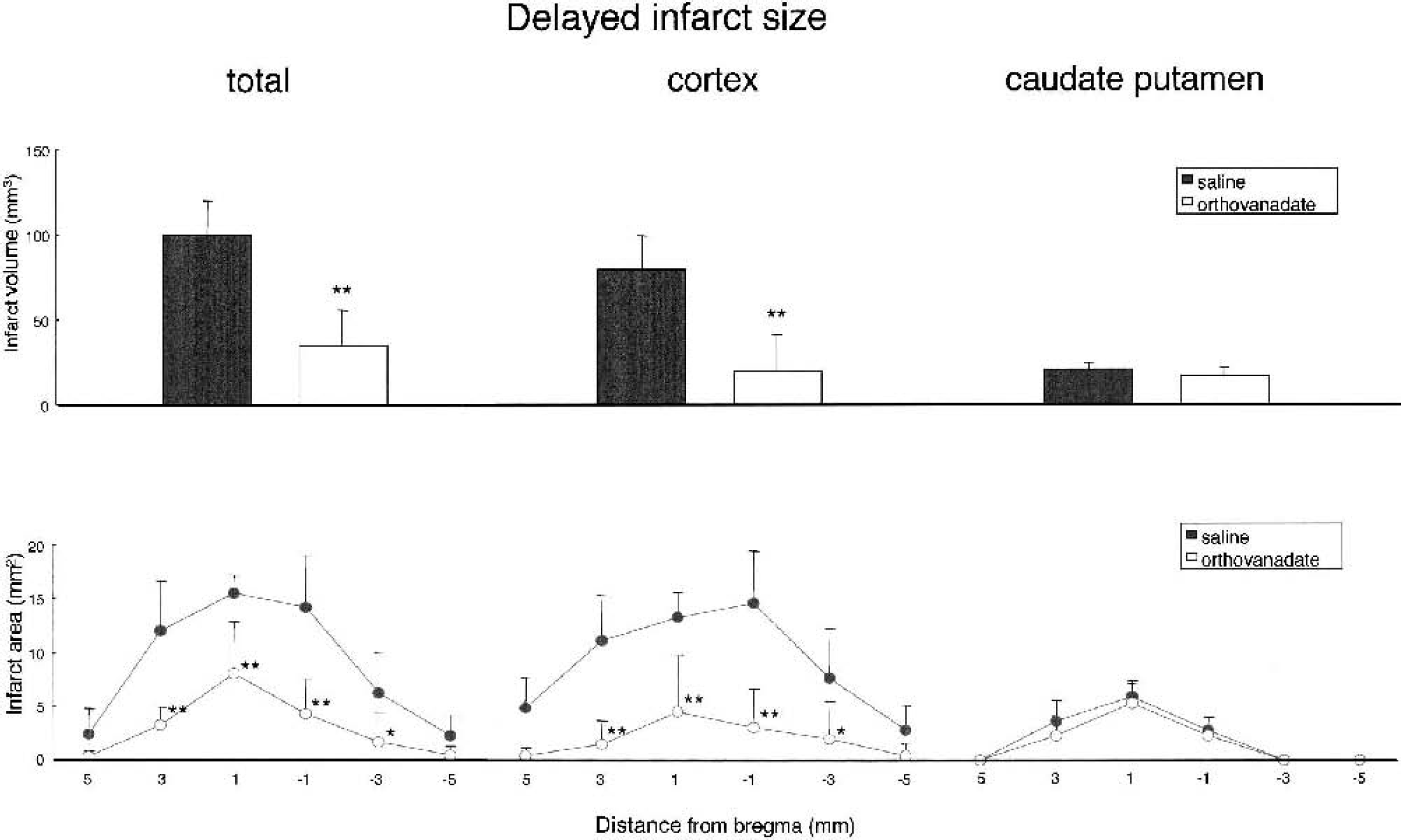
Quantitative analysis of infarct areas in six coronal sections and infarct volume from saline (n = 8) and orthovanadate (n = 8) groups in the delayed phase (28 days) after ischemia induction. Infarct areas and volume of the total, the cortex, and the caudate putamen were evaluated. Values are the mean ± SD; asterisks denote significant differences between saline and orthovanadate groups (*P < 0.05; **P < 0.01).
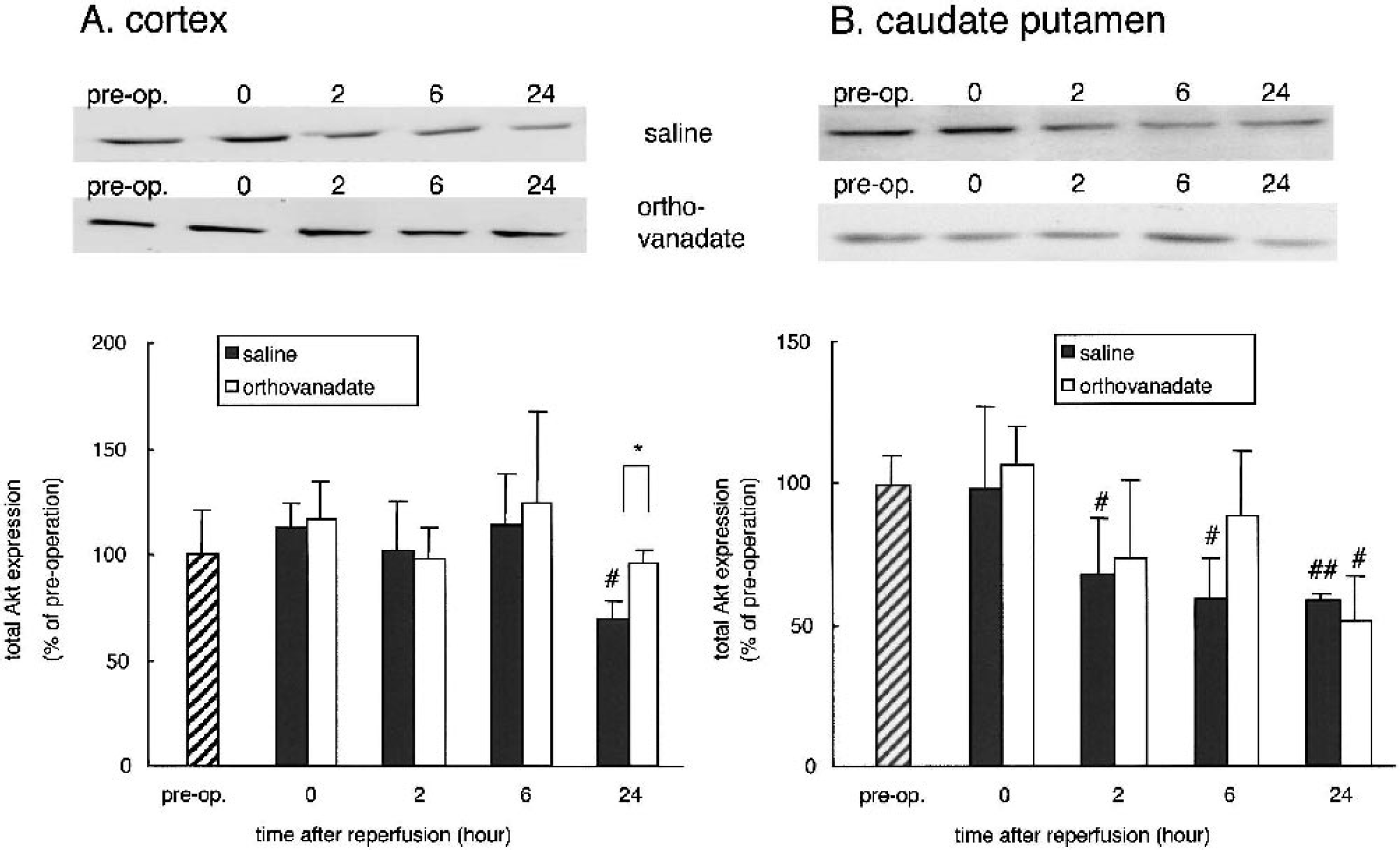
Changes in total Akt expression in the cortex (
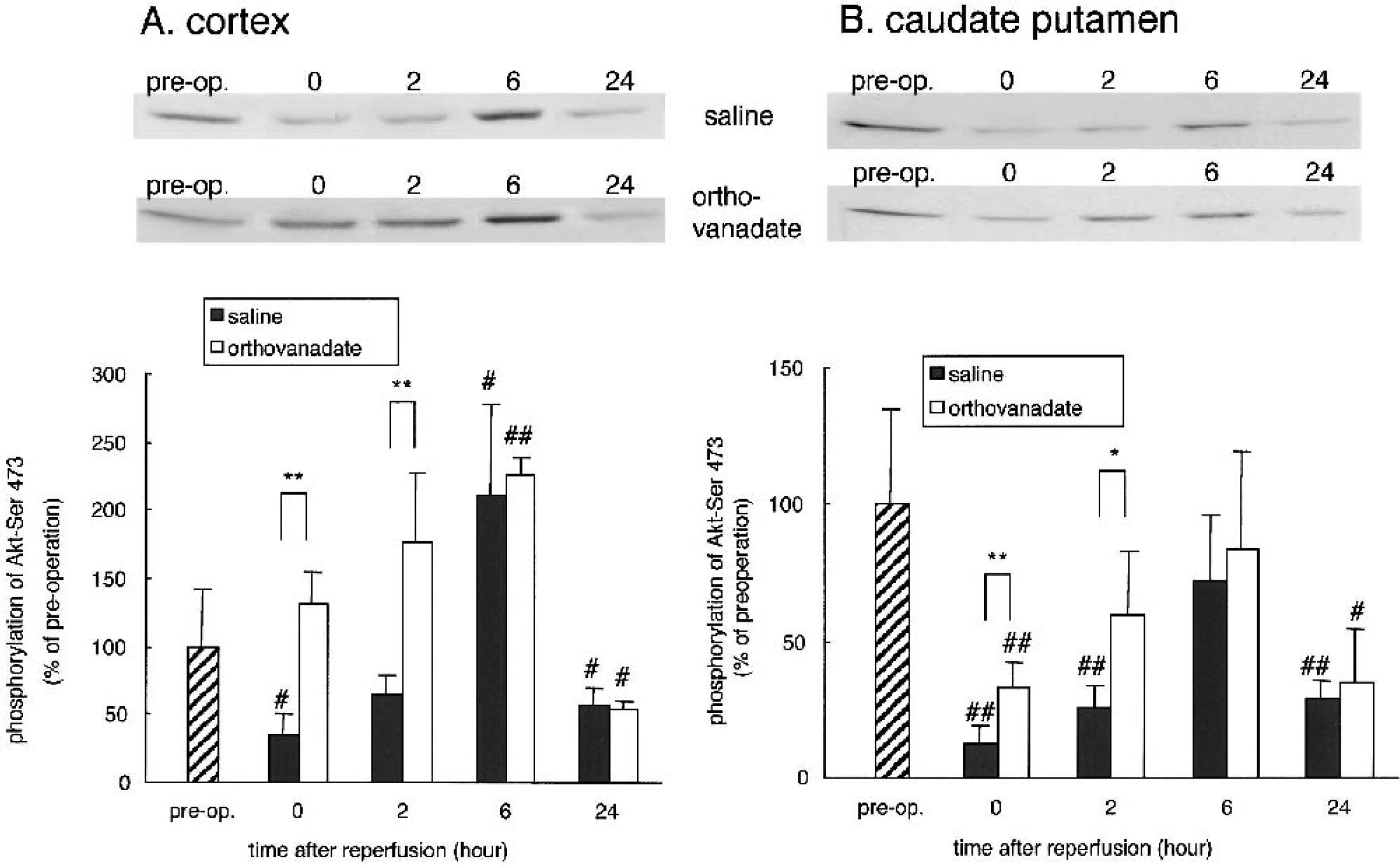
Changes in Akt-Ser-473 phosphorylation in the cortex (
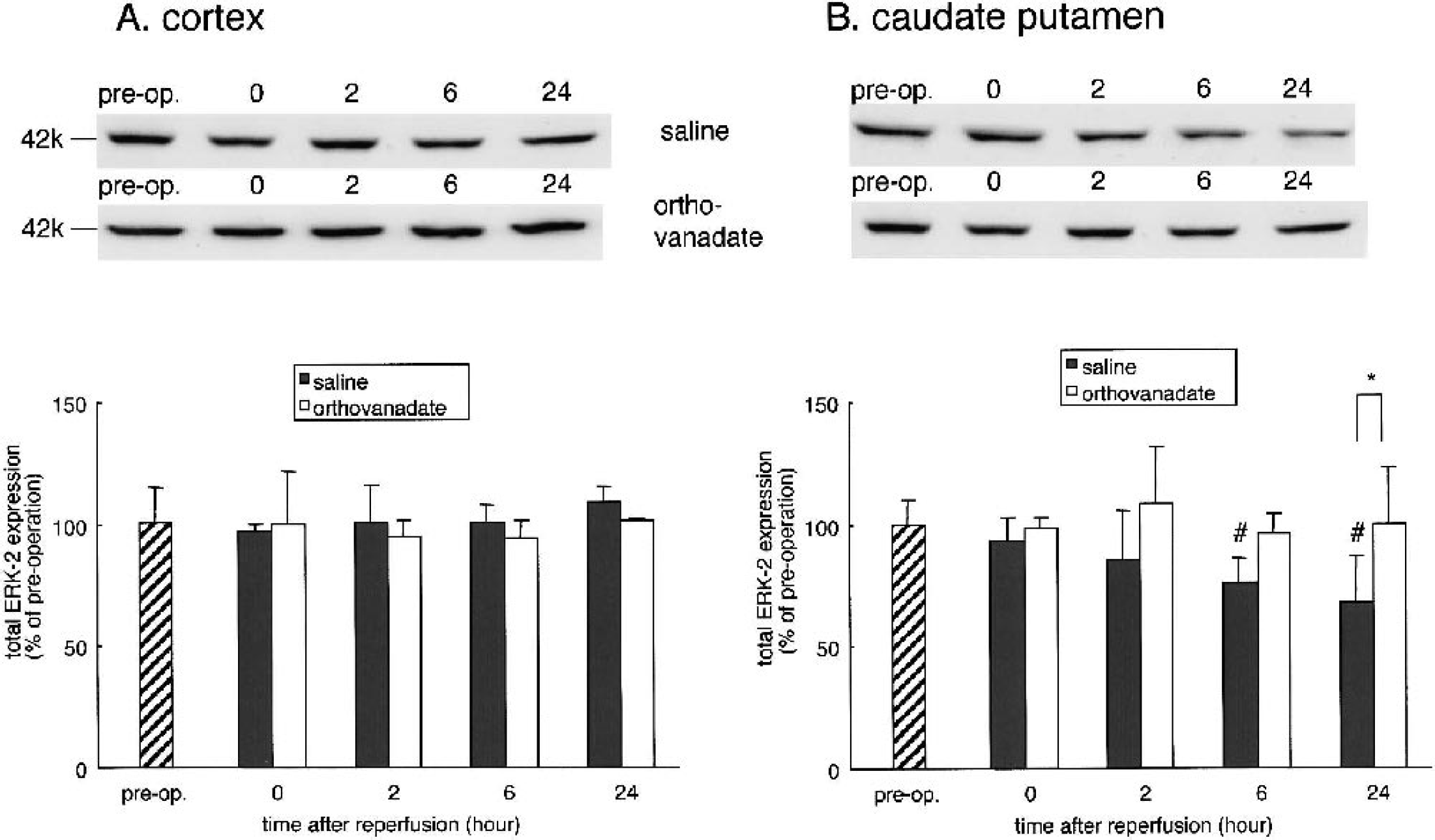
Changes in total ERK expression in the cortex (
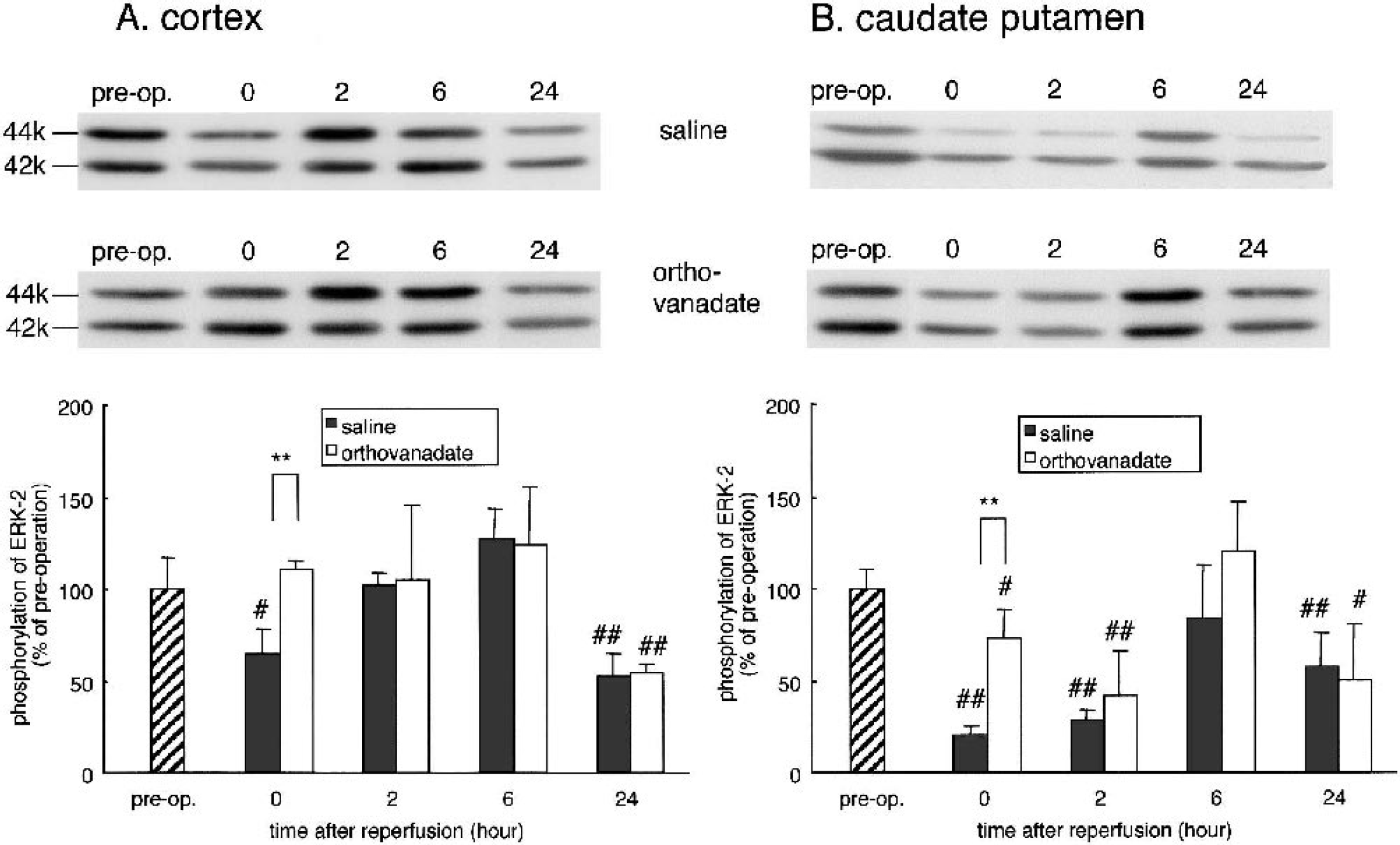
Changes in ERK phosphorylation in the cortex (
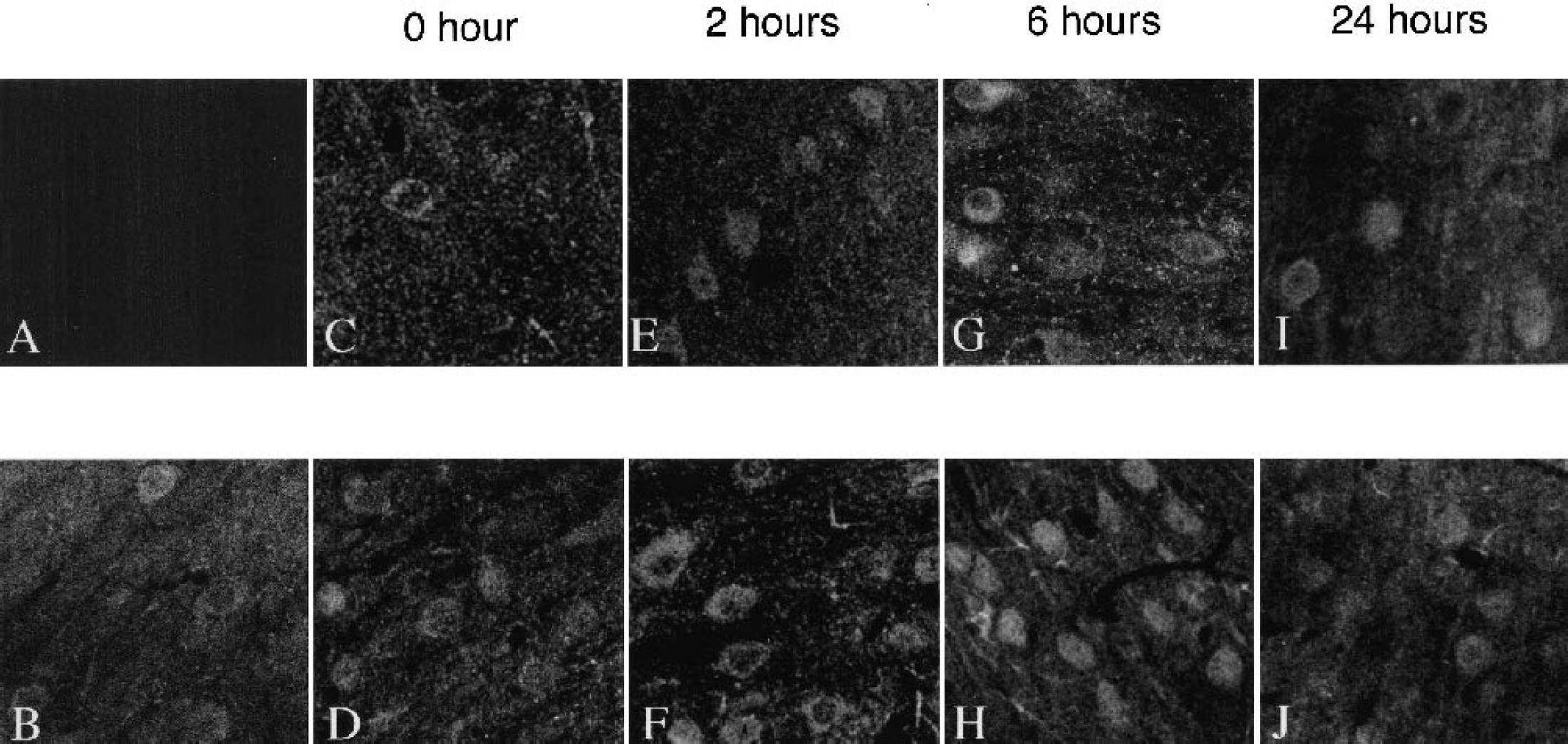
Immunohistochemical localization of activated Akt in the periinfarct area corresponding to area X in Fig. 1 after reperfusion. Immunostaining was performed with anti–phospho-Akt-Ser-473 antibody. There was no staining in the absence of primary antibody (
Tissue preparation for immunofluorescence staining
At 0, 2, 6, or 24 hours after reperfusion and at preischemic time points (n = 4, respectively), the rat brains were fixed by cardiovascular perfusion with an aldehyde fixative (4% formaldehyde in 0.1-mol/L phosphate buffer containing 30-mmol/L sodium pyrophosphate and 50-mmol/L NaF to prevent dephosphorylation) and immersed in fixative overnight. Coronal sections (30 μm) at the level of section c (Fig. 1) were cut with a vibratome, washed twice in PBS for 5 minutes at room temperature, and then with PBS in 0.1-mol/L containing 0.01% Triton-X100 (30 minutes). Nonspecific binding sites were blocked for 1 hour in PBS with 3% bovine serum albumin (blocking solution). The sections were incubated overnight at 4°C with primary antibody for anti–phospho-Akt (Ser473) antibody IHC-specific (diluted 1:100 for immunohistochemistry; Cell Signaling Technology) in blocking solution. After three 5-minute PBS washes, the sections were labeled for 2 hours with biotinylated anti–rabbit immunoglobulin G (Vector) diluted to 1:200 in PBS and then incubated for 1 hour with FITC-labeled avidin (Vector). After several washes with PBS, they were mounted on glass slides with coverslips and analyzed under a confocal laser microscope (Fluoview, Olympus, Tokyo, Japan). To normalize immunoreactivity with anti–phospho-Akt (Ser473) antibody, the brain slices were analyzed without changing the confocal laser intensity and laser aperture.
Statistical analysis
All values are expressed as the mean ± SD. All repeatedmeasures data, including blood gases, blood pressure, blood glucose levels, rCBF, infarct areas, and infarct volume, were analyzed by one-way analysis of variance. For statistical comparison of groups at each time point and to analyze total Akt and ERK expression and phosphorylation of Akt (Ser473) and ERK, an unpaired t-test was used. Differences of P < 0.05 were considered significant.
RESULTS
Physiologic parameters
The observed changes in physiologic parameters after MCAO including blood gases, blood pressure, blood glucose (n = 8), and rCBF (n = 5) are shown in Table 1. Rats in the saline group (n = 5) showed mild alkalosis at 0 and 2 hours after reperfusion. Orthovanadate group (n = 5) manifested increased oxygenation and CO2 accumulation resulting in mild acidosis at 45 minutes after occlusion and at 0 hours after reperfusion; these changes recovered at 2 hours after reperfusion. There were no statistically significant differences in hematocrit and serum Na+ levels (data not shown).
Physiologic parameters
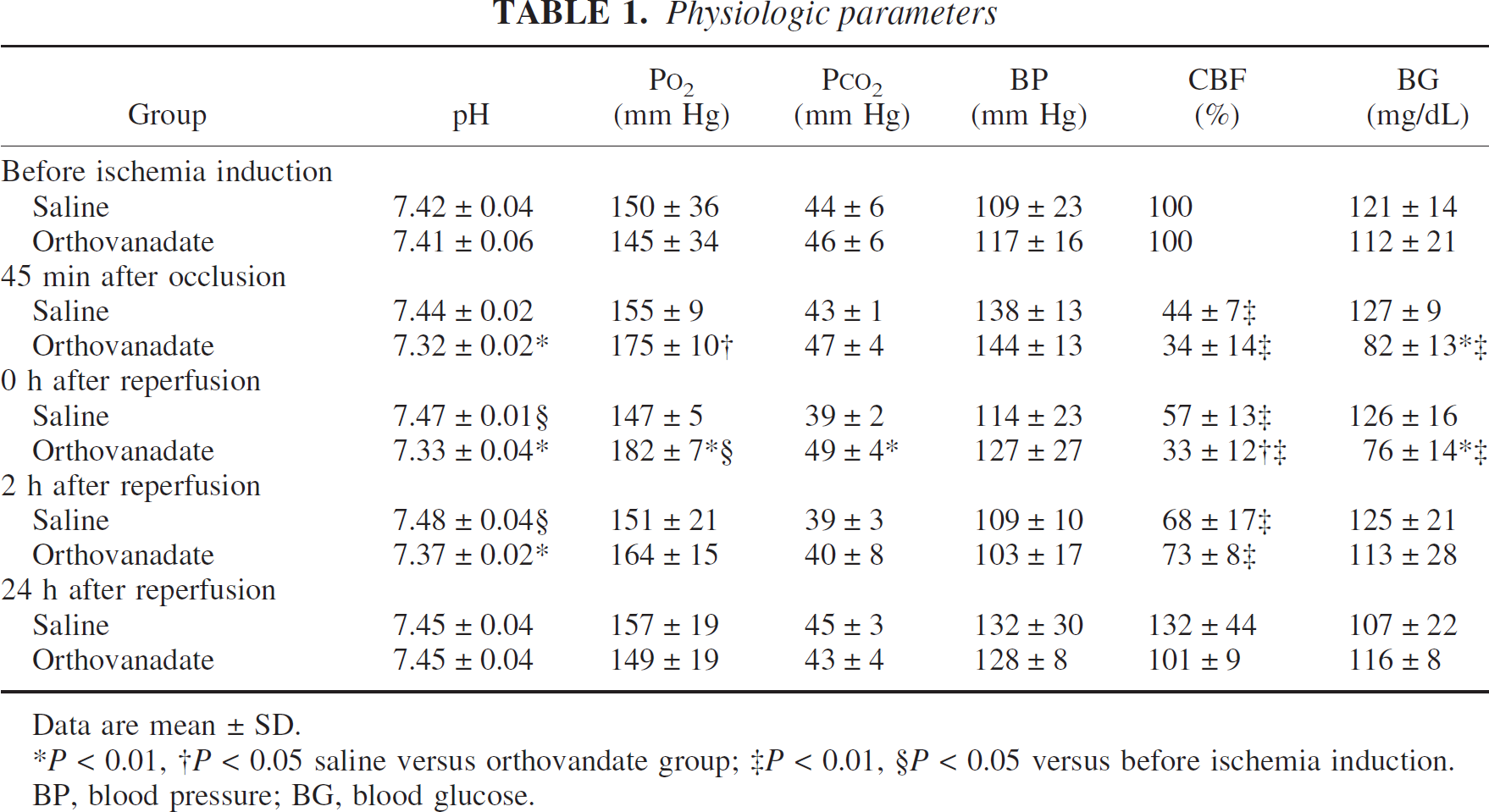
Data are mean ± SD.
P < 0.01,
P < 0.05 saline versus orthovandate group;
P < 0.01,
P < 0.05 versus before ischemia induction.
BP, blood pressure; BG, blood glucose.
Compared with the preischemia baseline values, the blood glucose level did not change significantly in saline group (n = 8). However, blood glucose levels in orthovanadate group (n = 8) decreased significantly at 45 minutes after occlusion (82 ± 13 mg/dL) and at 0 hours after reperfusion (76 ± 14 mg/dL) and subsequently recovered to the preischemia baseline.
Compared with the baseline values, rCBF decreased to 15 ± 14% and 21 ± 14% of preischemic values in saline and orthovanadate groups at 0 hours after occlusion; however, the difference was not statistically significant. During occlusion, rCBF was not significantly different between the two groups. However, at 0 hours after reperfusion, rCBF was significantly lower in the orthovanadate group than in the saline group (33 ± 12% vs. 57 ± 13%).
The neurologic scores were not significantly different between saline and orthovanadate groups (1.7 ± 0.6, 1.8 ± 0.5, respectively; n = 11). None of the animals tested were hypertensive during the experiments (n = 5).
Ischemic brain injury
Representative ischemic lesions in six coronal sections (a–f) at 1 and 28 days after ischemia induction are shown in Fig. 1. The infarct volume in the acute and delayed phases is summarized in Figs. 2 and 3. The total infarct volume in orthovanadate group was significantly smaller than in the saline group at both the acute (44.4 ± 32.5 mm3 vs. 120.4 ± 48.2 mm3) and delayed (34.8 ± 21.2 mm3 vs. 99.2 ± 20.1 mm3) phases. The infarct volume of the cortex was significantly smaller in the orthovanadate group than in the saline group at both the acute (21.1 ± 32.2 mm3 vs. 88.3 ± 39.7 mm3) and delayed (18.7 ± 22 mm3 vs. 79.2 ± 19.4 mm3) phases. However, in the caudate putamen, there was no significant difference between the two groups with respect to the infarct volume at both the acute and delayed phases.
Western blot analysis of total Akt expression and phosphorylation after transient MCAO
We previously reported a decreased Akt activity after transient ischemia in the gerbil hippocampus that underlies the neuronal death in the hippocampal pyramidal neurons. To address the question of whether Akt activity also underlies neuronal death in the MCAO model, we first measured total postreperfusion Akt expression in the cortex (Fig. 1, X) and caudate putamen (Fig. 1 Y). In the cortex (Fig. 4A), total Akt expression did not change significantly until 6 hours after reperfusion. However, at 24 hours, it decreased to 69.2 ± 8.5% in saline group, whereas in orthovanadate group this value was significantly higher 95.5 ± 7% (P < 0.05). In the caudate putamen (Fig. 4B) of both groups it gradually decreased until 24 hours. We posit that the decrease in total Akt expression is due to ischemic injury detected by TTC staining (Fig. 1). Figure 5 shows the postischemic changes in Akt-Ser-473 phosphorylation in the cortex (Fig. 1, X) and caudate putamen (Fig. 1, Y). In the cortex of orthovanadate group, Akt-Ser-473 phosphorylation was not decreased and was significantly higher at 0 and 2 hours compared with the saline group (131.8 ± 23% vs. 34.7 ± 16%; 176.3 ± 51.7% vs. 65 ± 13.9%, respectively). In both groups, it was transiently increased at 6 hours and decreased at 24 hours. In the caudate putamen of both groups, Akt-Ser-473 phosphorylation decreased at 0, 2, and 24 hours; the time course was similar to that seen in the cortex of the saline group. It should be noted that the decrease in phosphorylation of Akt-Ser-473 was more pronounced in the saline group at least within 2 hours after reperfusion.
Western blot analysis of ERK expression and phosphorylation after transient MCAO
We also previously reported a decrease in ERK activity after transient ischemia in the gerbil hippocampus that underlies neuronal death in the hippocampal pyramidal neurons. Because the phosphorylation-related alterations were similar in ERK-1 (44 kd) and ERK-2 (42 kd) after ischemia (Kawano et al., 2001), we focused on expression (Fig. 6) and phosphorylation (Fig. 7) of ERK-2 to address the question whether ERK activity also underlies neuronal death in the MCAO models. In the cortex (Fig. 6A), total ERK-2 expression did not change significantly until 24 hours after reperfusion in both the saline and orthovanadate groups. In the caudate putamen (Fig. 6B) of the saline group, the decrease in total ERK-2 at 6 (76 ± 10%) and 24 hours (68 ± 18.4%) was more pronounced than in the orthovanadate group. Phosphorylation of ERK-2 in the cortex (Fig. 7A) of saline group was significantly lower at 0 hours than in orthovanadate group (65.5 ± 13.2% vs. 111 ± 5%). ERK-2 phosphorylation in both groups manifested normal levels at 2 and 6 hours, and decreased at 24 hours after reperfusion; there were no significant differences between these groups. In the caudate putamen of both groups, ERK-2 phosphorylation decreased at 0, 2, and 24 hours. The time course was similar to that of Akt phosphorylation seen in the caudate putamen.
Immunohistochemical localization of Akt-Ser-473 phosphorylation after orthovanadate administration
We previously demonstrated that the phosphorylation of Akt was associated with its translocation to the nucleus after transient ischemia in gerbil hippocampal neurons. We next defined the location of the phosphorylated Akt in the MCAO model. The periinfarct cortex (Fig. 1, X) and caudate putamen (Fig. 1, Y) of slice c from both groups were immunostained with anti–phospho-Akt (Ser473) antibody (n = 4, Fig. 8). Immunofluorescence in the absence of primary antibody was negligible (Fig. 8A). At 0 and 2 hours after reperfusion, the cortex of saline group showed weak immunoreactivity for Akt-Ser-473 phosphorylation, primarily in the cytoplasm of neuronal cells (Figs. 8C and 8E). Immunoreactivity became transiently stronger at 6 hours (Fig. 8G). At 24 hours, immunoreactivity was weak but present in both the cytoplasm and nuclei (Fig. 8I). However, in orthovanadate group, strong immunoreactivity in the cytoplasm was observed at 2 hours (Fig. 8F), whereas strong nuclear localization was present at 6 hours (Fig. 8H) and decreased at 24 hours (Fig. 8J). In the caudate putamen of both groups, the immunoreactive changes were similar to those seen in the cortex of saline group, albeit weaker (data not shown).
DISCUSSION
The current study shows that orthovanadate, administered after ischemia induction, ameliorates ischemic neuronal damage after transient MCAO in rats. This protective effect was correlated with the prevention of a decrease in Akt and ERK phosphorylation during the early phase. We previously reported that orthovanadate stimulated Akt and ERK activity in vitro. Moreover, we demonstrated that pretreatment wortmannin and U0126, inhibitors of PI3-K and mitogen-activated protein kinase/ERK kinase, totally suppressed orthovanadate-induced neuroprotection in gerbil hippocampus (Kawano et al., 2001). Therefore, we thought that orthovanadate activated both Akt and ERK, resulting in neuroprotection. We also found that orthovanadate had a significant effect on blood gases, maintained blood glucose levels within the normal range, and led to a gradual recovery of rCBF after reperfusion.
Akt activation is a principal factor in the prevention of apoptosis. Changes in Akt phosphorylation were reported in animal models of transient forebrain ischemia (Kawano et al., 2001; Yano et al., 2001) and transient focal ischemia (Friguls et al., 2001; Janelidze et al., 2001; Noshita et al., 2001) and neuronal injury was correlated with Akt activity. In gerbils with transient forebrain ischemia, orthovanadate protected against delayed hippocampal neuronal death by Akt activation; activated Akt phosphorylated FKHR, resulting in suppression of Fas ligand expression (Kawano et al., 2001, 2002). We also showed that in the penumbra after MCAO, orthovanadate induced Akt activation and translocation to the nucleus in the early stage. This finding suggests that orthovanadate induces activated Akt-phosphorylated FKHR concomitant with a reduction in its DNA-binding activity, resulting in suppression of Fas ligand expression (Rosenbaum et al., 2000). Other investigators reported that the Akt signaling cascade activates cyclic AMP–responsive element binding protein and nuclear factor-κB (Du and Montminy, 1998; Romashkova and Makarov, 1999) and that they are related to antiapoptotic signals in cerebral ischemia (Walton and Dragunow, 2000; Yu et al., 1999). We speculate that orthovanadate stimulates the Akt-related antiapoptotic transcription factor pathway and rescues penumbra regions affected by apoptotic mechanisms (Li et al., 1995a,b, 1997), and that Akt activation underlies the effect of orthovanadate observed in this study.
Mitogen-activated protein kinase, including ERK, c-Jun N-terminal kinase (JNK), and p38, is involved in the survival and apoptotic responses in certain cell death paradigms in some types of cells (Ichijo et al., 1997; Kummer et al., 1997; Xia et al., 1995). ERK is normally activated in response to growth and differentiation factors, whereas JNK and p38 are usually activated in response to inflammatory cytokines and cellular stresses. Increased ERK phosphorylation has been reported in rodents after transient MCAO (Alessandrini et al., 1999; Irving et al., 2000; Namura et al., 2001). Whether activation of ERK is protective or detrimental to neurons is controversial. We previously demonstrated that orthovanadate activated ERK, resulting in neuroprotection against forebrain ischemia in gerbils (Kawano et al., 2001). In the present study, although orthovanadate activated ERK only at 0 hours after reperfusion, the ERK activation closely correlated with neuroprotective action of orthovanadate. Taken together, the Akt and ERK activation may contribute to the orthovanadate-induced neuroprotection. The neuroprotective effect of orthovanadate was not present in the caudate putamen (ischemic core), where Akt and ERK activity levels were below the preischemic baseline level. Although vanadium passes through the blood–brain barrier at least 30 minutes after intraperitoneal administration (Roshchin et al., 1980), we speculate that the blood supply to the brains of our experimental animals was severely disturbed, thus preventing orthovanadate from reaching these areas.
We found orthovanadate to have effects other than Akt and ERK activation. In this study, orthovanadate decreased blood glucose levels significantly at 45 minutes after occlusion and 0 hours after reperfusion. Hyperglycemia adversely affects energy metabolism in patients with cerebral ischemia due to severe lactic acidosis and results in poor clinical outcomes (Gardiner et al., 1982; Nedergaard, 1987; Siemkowicz, 1981; Welsh et al., 1980; Yip et al., 1991). Moreover, hypoglycemic ischemia leads to major metabolic derangement and increases brain tissue damage (Vannucci et al., 1980; Wass and Lanier, 1996). Normoglycemic patients with MCAO suffered less brain damage and had lower death rates due to acute hemispheric edema and small infarcts than did hypoglycemic patients (de Courten-Myers et al., 1994). Furthermore, lowering the blood glucose levels, even to within the physiologic range, decreased the size of infarction in rats (Hamilton et al., 1995). Our results show that in rats with transient MCAO, orthovanadate maintains blood glucose levels at the lower end of the normal range, suggesting that one of its neuroprotective effects is the reduction of blood glucose to within the normal range.
We also demonstrated the gradual, orthovanadate-induced recovery of rCBF after middle cerebral artery recanalization. The intravenous administration of vanadate produced marked hemodynamic changes in rats (Carmignani et al., 1995). Lopez-Novoa and Garrido (1986) reported that in rats, vanadate induced a decrease in cardiac output and blood flow through several organs, increased peripheral vascular resistance, and led to hypertension resulting in a decrease of CBF. Despite their lower rCBF, the neurologic score of orthovanadate group was not significantly different from the saline group at 10 minutes before reperfusion. Moreover, their rCBF returned to the level of the saline group at 2 hours after reperfusion. Hypothermia, regarded as neuroprotective, inhibits postischemic hyperperfusion and delayed or sustained hypoperfusion in ischemic perifocal regions (Huang et al., 1998). Thus, the gradual, orthovanadate-induced recovery of rCBF may play a role in its observed neuronal protective effect.
Furthermore, slight acidosis and CO2 accumulation were found in orthovanadate group at 45 minutes after occlusion and at 0 hours but not at 2 hours after reperfusion. Vanadate-induced effects on the respiratory system have been reported (World Health Organization, 1988). Vanadium compounds induced contraction of the airway smooth muscle in guinea pigs (Nayler and Sparrow, 1983), irritation of the respiratory mucosa, an asthmatic-type bronchitis, and expiratory difficulty on acute exposure (Roshchin, 1967). When these side effects occurred, they persisted for more than 2 hours. We have no experience with this phenomenon, and at present we are unable to identify the causes of the transient blood gas perturbations seen in our study.
Our results illustrate the neuroprotective effect of orthovanadate; however, cytotoxic effects of vanadium compounds have been reported (Bay et al., 1997; Capella et al., 2000, 2002; Colin et al., 1994; Cruz et al., 1995; Figiel and Kaczmarek, 1997; Ye et al., 1999). These studies reported the cytotoxicity in tumor and cultured proliferative cells, but not in neuronal cells. Figiel and Kaczmarek (1997) were alone in reporting the neurotoxicity of orthovanadate; they incubated cells from dentate gyrus for 48 hours in the presence of high doses of orthovanadate. We subjected our rats to a single injection of sodium orthovanadate and found no evidence of neuronal cell injury in the contralateral hemisphere, including the dentate gyrus. Thus, we conclude that our injection method is safe and neuroprotective.
In conclusion, we suggest that orthovanadate exerts neuroprotective effects via Akt and ERK activation. The orthovanadate-induced gradual recovery of rCBF and the lowering of the blood glucose level to within the normal range may contribute to its neuroprotective effect. Previously, we reported that neuroprotection of orthovanadate was inhibited by wortmannin and U0126 (Kawano et al., 2001). We posit that Akt and ERK activation was sufficiently effective to protect neuronal cells against ischemia in our MCAO model. The gentle recovery of rCBF and normalized blood glucose levels may have provided additional protection. Orthovanadate has been used to treat patients with diabetes mellitus, and further studies will show whether it may also have a place in the treatment of patients at risk for cerebral ischemia.
Footnotes
Acknowledgments:
The authors thank Mrs. Masayo Obata for technical assistance.