
Abstract
In transient forebrain ischemia, sodium orthovanadate as well as insulinlike growth factor-1 (IGF-1) rescued cells from delayed neuronal death in the hippocampal CA1 region. Adult Mongolian gerbils were subjected to 5-minute forebrain ischemia. Immunoblotting analysis with anti–phospho-Akt/PKB (Akt) antibody showed that phosphorylation of Akt at serine-473 (Akt-Ser-473) in the CA1 region decreased immediately after reperfusion, and in turn transiently increased 6 hours after reperfusion. The decreased phosphorylation of Akt-Ser-473 was not observed in the CA3 region. The authors then tested effects of intraventricular injection of orthovanadate and IGF-1, which are known to activate Akt. Treatment with orthovanadate or IGF-1 30 minutes before ischemia blocked delayed neuronal death in the CA1 region. The neuroprotective effects of orthovanadate and IGF-1 were associated with preventing decreased Akt-Ser-473 phosphorylation in the CA1 region observed immediately after reperfusion. Immunohistochemical studies with the anti–phospho-Akt-Ser-473 antibody also demonstrated that Akt was predominantly in the nucleus and was moderately activated in the cell bodies and dendrites of pyramidal neurons after orthovanadate treatment. The orthovanadate treatment also prevented the decrease in phosphorylation of mitogen-activated protein kinase (MAPK). Pretreatment with combined blockade of phosphatidylinositol 3-kinase and MAPK pathways totally abolished the orthovanadate-induced neuroprotective effect. These results suggest that the activation of both Akt and MAPK activities underlie the neuroprotective effects of orthovanadate on the delayed neuronal death in the CA1 region after transient forebrain ischemia.
Keywords
Transient forebrain ischemia induces delayed neuronal death in the CA1 region of the hippocampus (Kirino, 1982). Although the cellular mechanisms underlying delayed neuronal death remain unclear, recent evidence suggests that activation of cysteine proteases, such as members of the interleukin-1 family (caspases), triggers apoptosis seen in delayed neuronal death after forebrain ischemia (Yakovlev et al., 1997; Namura et al., 1998; Chen et al., 1998; Siesjöet al., 1999; Ouyang et al., 1999). In addition, the mechanisms underlying neuronal survival have been extensively characterized and a pivotal role for phosphatidylinositol 3-kinase (PI3-K) and its downstream target, Akt/PKB (Akt), has been established (Ahmed et al., 1997; Datta et al., 1997; Dudek et al., 1997; Kauffmann et al., 1997; Kennedy et al., 1997; Kulik et al., 1997). Stimulation of tyrosine kinase growth factor receptors (Hemmings, 1997) or GTP-binding (G) protein–coupled receptors activates PI3-K (Murga et al., 1998; Krugmann et al., 1999), leading to production of phosphatidylinositol-3,4,5-trisphosphate (PIP3) and phosphatidylinositol-3,4-bisphosphate, which bind to the Akt pleckstrin homology domain and promote dimerization and translocation of Akt to membranes (Datta et al., 1996). Such membrane translocation is an essential step in Akt activation, because addition of an N-terminal myristoylation sequence makes Akt constitutively active (Kohn et al., 1996; Andjelkovic et al., 1997). Akt activation is correlated with phosphorylation of Thr-308 in its catalytic domain and Ser-473 at the C terminus (Alessi et al., 1996). Although it is known that PIP3 -dependent protein kinase-1 (PDK-1) phosphorylates Akt at Thr-308 (Alessi et al., 1997; Stephens et al., 1998), the serine/threonine kinases, including an integrin-linked kinase (Delcommenne et al., 1998), responsible for Ser-473 phosphorylation have not been established. Ca2+ /calmodulin-dependent protein kinase kinase also can phosphorylate Akt at Thr-308 both in vitro and in vivo in a PI3-K–independent manner (Yano et al., 1998).
Receptor tyrosine kinase ligands such as insulinlike growth factor-1 (IGF-1) promote cell survival through Akt activation and concomitant PI3-K activation (Datta et al., 1997; D'Mello et al., 1997; Dudek et al., 1997; Galli et al., 1995; Miller et al., 1997; Párrizas et al., 1997). Indeed, IGF-1 prevents neuronal death after transient forebrain ischemia (Gluckman et al., 1992; Zhu and Auer, 1994; Tagami et al., 1997) and after hypoxic–ischemic injury (Guan et al., 1993, 1996). Topical application of IGF-1 also reduces the infarction volume in rat cortex after transient middle cerebral artery occlusion (Wang et al., 2000). Although the mechanisms underlying neuroprotection by IGF-1 are not fully understood, PI3-K and Akt signaling may be required for IGF-1–induced neuroprotection. Furthermore, the authors recently documented that Akt activation is induced by sublethal ischemic insult in gerbil hippocampus and contributes to neuroprotective ischemic tolerance in CA1 pyramidal neurons (Yano et al., 2001). Protein tyrosine phosphatase (PTP) inhibitors induce protein tyrosine phosphorylation in intact cells. For example, potent PTP inhibitors, such as vanadate and its derivative pervanadate, activate Akt and mitogen-activated protein kinase (MAPK) signaling (Zhao et al., 1996; Wijkander et al., 1997; Meier et al., 1998), thereby mimicking insulin in stimulating glucose transport, lipogenesis, and protein synthesis and in inhibiting lipolysis (Heffetz et al., 1990; Shechter, 1990; Shisheva and Shechter, 1993; Posner et al., 1994; Fantus et al., 1994). The authors hypothesized that treatment with orthovanadate as well as IGF-1 would lead to Akt activation in vivo, thereby rescuing cells from delayed neuronal death after transient forebrain ischemia in gerbil hippocampus. Here, the authors show that intraventricular administration of orthovanadate or IGF-1–activated Akt attenuated the inactivation of Akt, and in turn rescued cells from delayed neuronal death in the hippocampal CA1 region after transient forebrain ischemia. The findings suggest that vanadate compounds and IGF-1 are useful therapeutic agents for ischemic brain damage.
MATERIALS AND METHODS
Induction of ischemia
Adult male Mongolian gerbils (weighing 60 to 80 g) were housed under constant environmental conditions (temperature, 22°C ± 2°C; humidity, 55% ± 5%; and a 12/12 hour light/dark cycle) in the Animal Research Center, Kumamoto University. Gerbils were anesthetized and maintained with 2% halothane in a mixture of 30% O2 and 70% N2 O. The rectal temperature, monitored with a digital thermometer inserted 2 cm into the anus, was maintained at 37°C to 38°C throughout the operation by placing gerbils under a heat lamp and warming them with a blanket. Bilateral common carotid arteries were surgically exposed and quickly occluded with aneurysm clips. After 5-minute occlusion, the clips were removed to restore blood flow. After awakening from anesthesia, gerbils were returned to home cages and allowed to take food and water before experiments. Control animals were sham-operated and received the same experimental procedures except for occlusion of arteries. Ischemic and sham-operated gerbils were killed by decapitation at the indicated times and hippocampal regions were dissected from their brains. All animal experiments were approved by the Committee of Animal Experiments at Kumamoto University School of Medicine.
Administration of wortmannin, sodium orthovanadate, IGF-1, or U0126
For intraventricular injection, animals were anesthetized and placed on a stereotaxic operation frame. After making 1 burr hole at the right parietal skull 2.0 mm posterior from bregma and 3.5 mm lateral, a Hamilton syringe was inserted into the ventricle at a depth of 2.5 mm from the cortical surface. When drug infusion was performed twice, the first injection was made on the left side and the second on the right side. For example, 2 μL of 100 μmol/L wortmannin (Wako Pure Chemical Industries, Osaka, Japan) was injected into the ventricle through a Hamilton syringe at a flow rate of 0.4 μL/min on the left side, and 15 minutes later, 2 μL of 5 mmol/L sodium orthovanadate (Sigma, St. Louis, MO, U.S.A.) was injected on the right side. Thirty minutes later, animals were subjected to 5-minute forebrain ischemia as described above. Sodium orthovanadate (5 mmol/L) and IGF-1 (25 μg/mL; Sigma) were dissolved in phosphate-buffered saline (PBS), and wortmannin (100 μmol/L) was dissolved in PBS with 2% dimethylsulfoxide. U0126 (500 μmol/L; Calbiochem-Novabiochem, San Diego, CA, U.S.A.) was dissolved in PBS with 5% dimethylsulfoxide.
Tissue preparation for double immunofluorescence staining
At 0, 2, 6 hours, and 7 days after 5-minute ischemia, gerbils were anesthetized with pentobarbital (50 mg/kg intraperitoneally) and perfused through the left ventricle of the heart with ice-cold PBS followed by 4% paraformaldehyde in 0.1 mol/L phosphate buffer (PB). For immunohistochemical studies using anti–phospho-Ser-473 antibody, animals were perfused with PBS containing 30 mmol/L sodium pyrophosphate and 50 mmol/L NaF, followed by 4% paraformaldehyde in 0.1 mol/L PB containing 30 mmol/L sodium pyrophosphate and 50 mmol/L NaF to prevent dephosphorylation. Brains were removed and postfixed overnight at 4°C in the same fixative solution. Coronal brain sections (30-μm-thick) at the level of the hippocampus were prepared using a vibratome. Sections were incubated by floating in the following media: 30 minutes in 0.1 mol/L PB containing 0.01% Triton-X100; 1 hour in PBS with 3% bovine serum albumin (blocking solution); overnight with polyclonal anti–phospho-Ser-473 antibody (diluted 1:50; Upstate Biotechnology, Lake Placid, NY, U.S.A.) and monoclonal anti-Akt1 antibody (diluted 1:500; Transduction Lab., Lexington, KY, U.S.A.) in blocking solution. Sections then were labeled for 2 hours with both Alexa 594-labeled anti-sheep IgG (Molecular Probes, Eugene, OR, U.S.A.) and fluorescein-labeled anti-mouse IgG (Leinco Technologies, Ballwin, MO, U.S.A.) in PBS (diluted 1:200). After several washes with PBS, the sections were mounted on glass slides with coverslips and analyzed using a confocal laser microscope (Fluoview, Olympus, Tokyo, Japan). To normalize immunoreactivity with anti–phospho-Ser-473 antibody, the authors analyzed hippocampal slices without changing the confocal laser intensity and laser aperture. Furthermore, sections were double-stained with anti–phospho-Ser-473 and anti-Akt1 antibodies. The authors confirmed that the intensity of immunoreactivity with anti-Akt1 antibody was the same between control and ischemic hippocampal slices.
Gel electrophoresis and immunoblotting
Gerbils were killed by decapitation immediately after reperfusion or at the indicated times after reperfusion. After brains were removed and rinsed once with cold PBS, CA1 and CA3 regions of the hippocampus were dissected out in cold PBS under a microscope. Each tissue sample was stored in a test tube and kept at −80°C before use. Frozen tissues were homogenized with a hand homogenizer and sonicated with a Sonifier-250 (Branson, Danbury, CT, U.S.A.) at 0°C in 0.2 mL of the homogenization buffer containing 50 mmol/L Tris-HCl (pH 7.5), 0.5% Triton X-100, 4 mmol/L EGTA, 10 mmol/L EDTA, 0.5 mol/L NaCl, 1 mmol/L Na3 VO4, 30 mmol/L sodium pyrophosphate, 50 mmol/L NaF, 50 μg/mL leupeptin, 25 μg/mL pepstatin A, 50 μg/mL trypsin inhibitor, and 1 mmol/L dithiothreitol (DTT). Insoluble materials were removed by a 10-minute centrifugation at 15,000 g. After determination of protein content in each supernatant fraction using Bradford's solution, each sample containing equivalent amounts of protein was applied to 10% acrylamide denaturing gel (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) (Laemmli, 1970). After electrophoresis, proteins were transferred for 2 hours at 70 V to an Immobilon PVDF membrane (Millipore Corp., Bedford, MA, U.S.A.) (Towbin et al., 1979). Blotting membranes were incubated for 1 hour in PBS containing 3% bovine serum albumin (blocking solution) at room temperature and incubated overnight at 4°C with a 1:200 dilution of anti–phospho-Ser-473 antibody (New England Bio Labs, Beverly, MA, U.S.A.) or a 1:1000 dilution of anti-Akt1 antibody in blocking solution. After several washes with PBS containing 0.1% Tween-20 (PBS-T), the membranes were incubated for 1 hour with horseradish peroxidase (HRP)-conjugated anti-rabbit antibody (Amersham Pharmacia Biotech, Arlington Heights, IL, U.S.A.) or anti-sheep IgG antibody (Leinco Technologies) diluted 1:10000. Membranes then were processed with enhanced chemiluminescence (ECL) Western blotting detection reagents (Amersham Pharmacia Biotech). The images were scanned and analyzed semiquantitatively using the NIH image (public domain software developed at the United States National Institutes of Health and available on the internet at http://rsb.info.nih.gov/nih-image). Similar methods were used for immunoblotting with anti–phospho-MAPK (Promega, Madison, WI, U.S.A.) and anti-MAPK (pan ERK) antibodies (Transduction Lab).
Histopathologic analysis
After 7 days, gerbils were anesthetized and perfusion-fixed with 4% paraformaldehyde as described. Hippocampal serial sections (30 μm) were stained with 5 μmol/L propidium iodide (Molecular Probes) diluted in PBS and were inspected under a confocal laser microscope. Viable and nonviable neurons in the pyramidal cell layer of hippocampal CA1 regions were counted from sections 1.4 to 1.8 mm posterior to bregma. Viable neurons were defined as cells with normal morphologic properties exhibiting round nuclei stained with propidium iodide. Four sections were obtained from 5 to 8 animals in each condition. The total number of the viable cells from the left and right hippocampi for each animal was calculated as an average of 2 randomly chosen 1350 × 1000-μm area from the 4 different slices counted by 2 independent observers without knowledge of treatment conditions. Sections also were stained with cresyl violet to count viable neurons, and the number of viable cells was the same as that stained with propidium iodide.
Akt kinase assay
Akt activity was measured with an Akt assay kit (New England Bio Labs) according to the manufacturer's protocol with minor modifications. Dissected hippocampal tissues were homogenized with a hand homogenizer in lysis buffer. Extracts containing equivalent amounts of protein were incubated with a slurry of immobilized Akt antibody for 2 hours at 4°C. Immunoprecipitates were washed twice with lysis buffer and twice with kinase buffer containing 25 mmol/L Tris-HCl (pH 7.5), 5 mmol/L glycerophosphate, 2 mmol/L DTT, 0.1 mmol/L Na3 VO4, and 10 mmol/L MgCl2. Immunoprecipitates then were subjected to an in vitro kinase assay in kinase buffer with 400 μmol/L ATP and 1 μg GSK-3α/β crosstide corresponding to residues surrounding GSK-3α/β (Ser-21/9) (CGPKGPGRRGRRRTSSFAEG) (New England Bio Labs) for 30 minutes at 30°C. The reaction was stopped by adding sodium dodecyl sulfate sample buffer. Phosphorylation of Ser-21 or Ser-9 of GSK-3α/β crosstide was analyzed by immunoblotting with anti-phospho-GSK-3α/β (Ser-21/9) antibody.
Statistical analysis
All values reported here were expressed as mean ± SD. Overall statistical significance for differences among groups was tested by one-way analysis of variance, followed by multiple comparisons between each group or between control and other groups using Dunnett's multiple comparison test. P < 0.05 was considered significant.
RESULTS
Changes in activity of Akt after transient forebrain ischemia
Changes in activity of Akt were examined after transient forebrain ischemia. To evaluate Akt activities after 5-minute transient forebrain ischemia, the authors measured phosphorylation of Akt at Ser-473, which is required for Akt activation. Lethal ischemia, which caused death of approximately 90% of the pyramidal cells in the CA1 region, resulted in dephosphorylation of Akt-Ser-473 in the CA1 region immediately (0 hour) after reperfusion without changes in Akt protein levels (Fig. 1A). Akt phosphorylation then transiently increased 6 hours after reperfusion and returned to the basal level observed in sham-operated animals within 1 day of reperfusion. The results of biphasic changes in Akt phosphorylation after ischemia were consistent with the previous observations using rat ischemic model (Ouyang et al., 1999). Akt activity was determined by an immunocomplex kinase assay using GSK-3α/β crosstide as substrate. Akt kinase activity after 5-minute ischemia decreased to 12% of that of sham-operated gerbils and increased to 240% 6 hours after reperfusion (Fig. 1B). Changes in Akt activity were correlated with changes in Akt-Ser-473 phosphorylation. In contrast with the CA1 region, decreased Akt-Ser-473 phosphorylation was not evident in the CA3 region immediately after reperfusion (Fig. 1C). Total Akt protein levels were unchanged in the CA3 region in samples from both sham-operated and ischemic animals (data not shown).
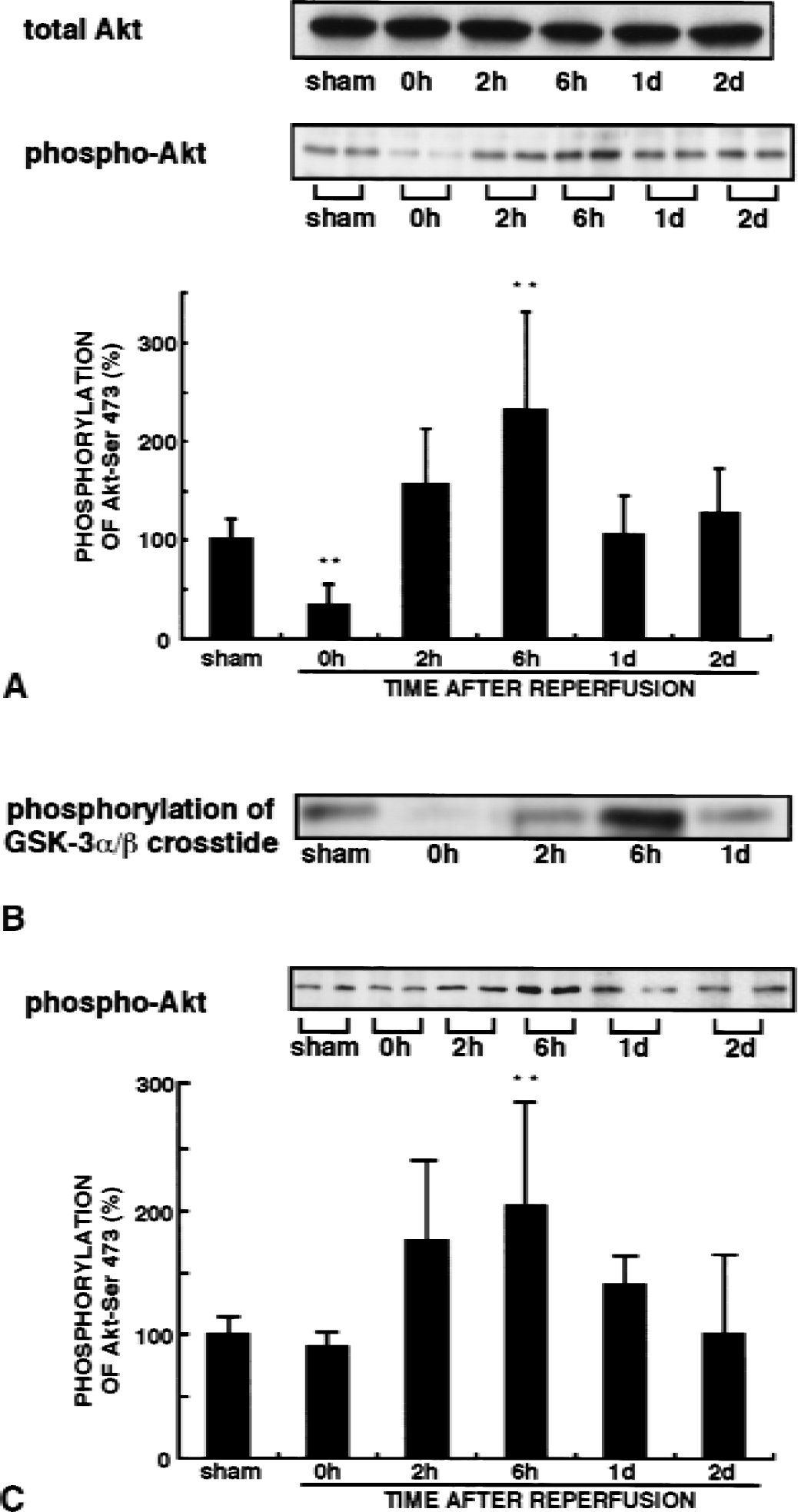
Changes in Akt-Ser-473 phosphorylation and Akt activity after transient forebrain ischemia. Extracts were obtained from the hippocampal CA1 or CA3 region from sham-operated animals or from 5-minute ischemic animals at the indicated times.
Akt activation after intraventricular administration of orthovanadate and IGF-1
Vanadate is a general protein tyrosine phosphatase inhibitor (Swarup et al., 1982). Vanadate and IGF-1 can activate Akt in nonneuronal cells (Alessi et al., 1996; Meier et al., 1998). The authors next examined whether treatments with orthovanadate or IGF-1 in vivo increase in both Akt phosphorylation and its activity in hippocampal neurons. Before in vivo studies, the authors tested effects of orthovanadate and IGF-1 on Akt activity in cultured hippocampal neurons. Fifty μmol/L orthovanadate and 50 ng/mL IGF-1 maximally stimulated Akt activity (data not shown). The maximal Akt activation was observed within 15 to 30 minutes after treatments with orthovanadate and IGF-1 (data not shown).
After intraventricular administration of 2 μL of 5 mmol/L orthovanadate or 25 μg/mL IGF-1, extracts from the CA1 regions were subjected to immunoblotting analysis with anti–phospho-Akt-Ser-473 antibody and immunocomplex kinase assay for Akt. Fifteen minutes after intraventricular administration of orthovanadate and IGF-1, Akt-Ser-473 phosphorylation significantly increased to 194% and 154%, respectively, of levels seen in sham-operated animals that received the vehicle. Increased Akt phosphorylation was transient and returned to a basal level within 30 minutes of administration (Fig. 2A). Consistent with Akt phosphorylation, Akt kinase activity as assessed by GSK-3α/β crosstide phosphorylation significantly increased (187% of that of sham-operated animals) 15 minutes after administration of orthovanadate (Fig. 2B). Intraventricular administration of selective PI3-K inhibitor, wortmannin, 15 minutes before orthovanadate treatment prevented the orthovanadate-induced Akt phosphorylation (Fig. 2C); thus, orthovanadate activates Akt through PI3-K activation.
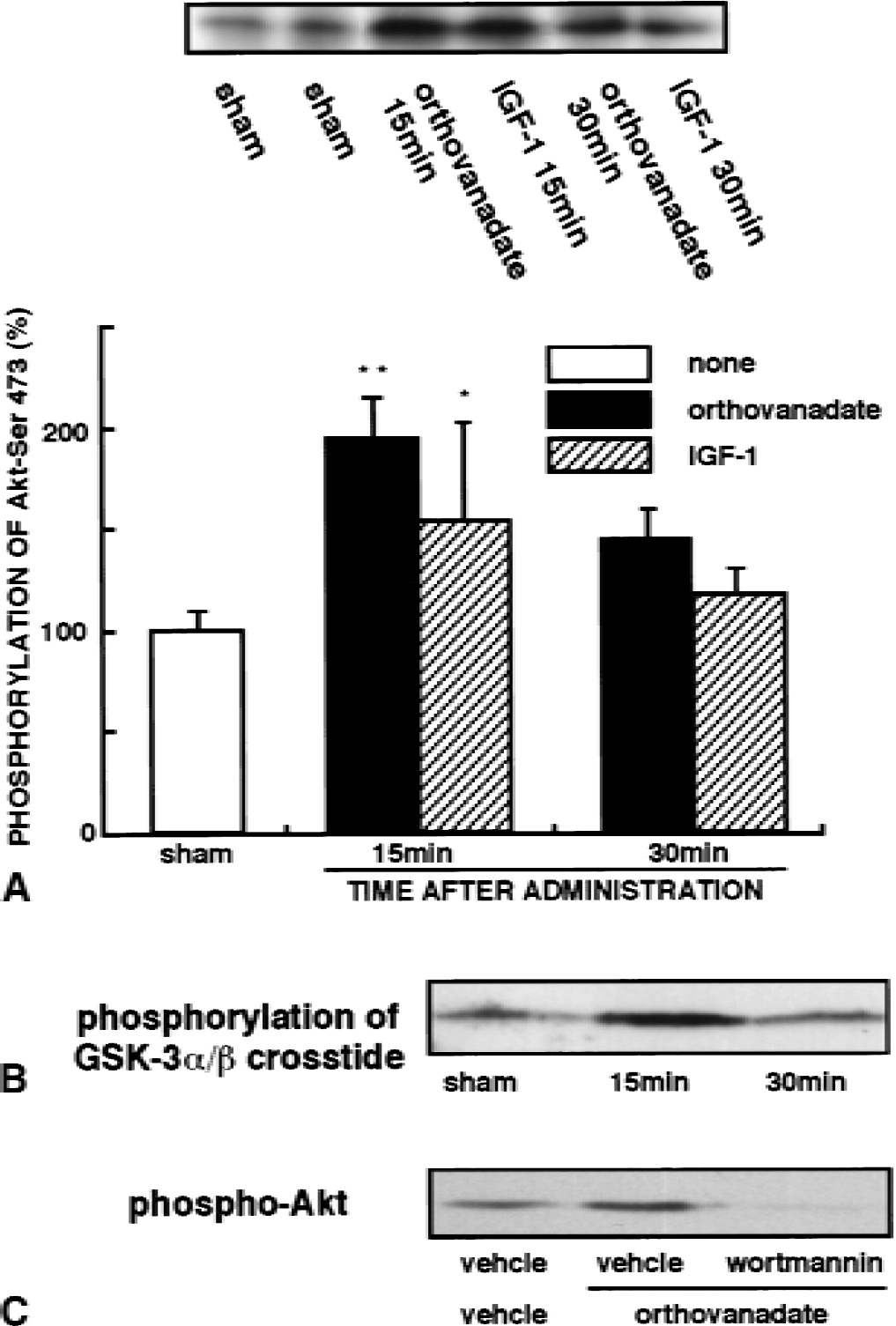
Effects of IGF-1 and orthovanadate on phosphorylation and activity of Akt in the hippocampal CA1 region without ischemia.
Immunohistochemical localization of activated Akt after administration of orthovanadate
The authors next addressed whether intraventricular administration of orthovanadate induces Akt activation in pyramidal neurons. Hippocampal slices obtained from sham-operated and orthovanadate-treated gerbils were double-stained with conventional anti-Akt antibody and anti–phospho-Akt-Ser-473 antibody. In sham-operated animals, the immunoreactivity for conventional Akt was observed mainly in cell bodies and dendrites of pyramidal neurons and was weak in surrounding nonneuronal cells (Fig. 3A). By contrast, active Akt was predominantly localized in cell bodies of pyramidal neurons in the hippocampus (Fig. 3C). In these cells, immunostaining was observed in a punctate pattern and was localized to the submembrane area (Fig. 3C). After orthovanadate treatment, immunoreactivity seen with anti–phospho-Akt antibody markedly increased in dendrites as well as in the cell bodies of pyramidal neurons (Fig. 3D) without changes in immunoreactivity against conventional Akt antibody (Fig. 3B). In the cell bodies, active Akt was evident in the nucleus and in the cytoplasm (Fig. 3D and 3H).
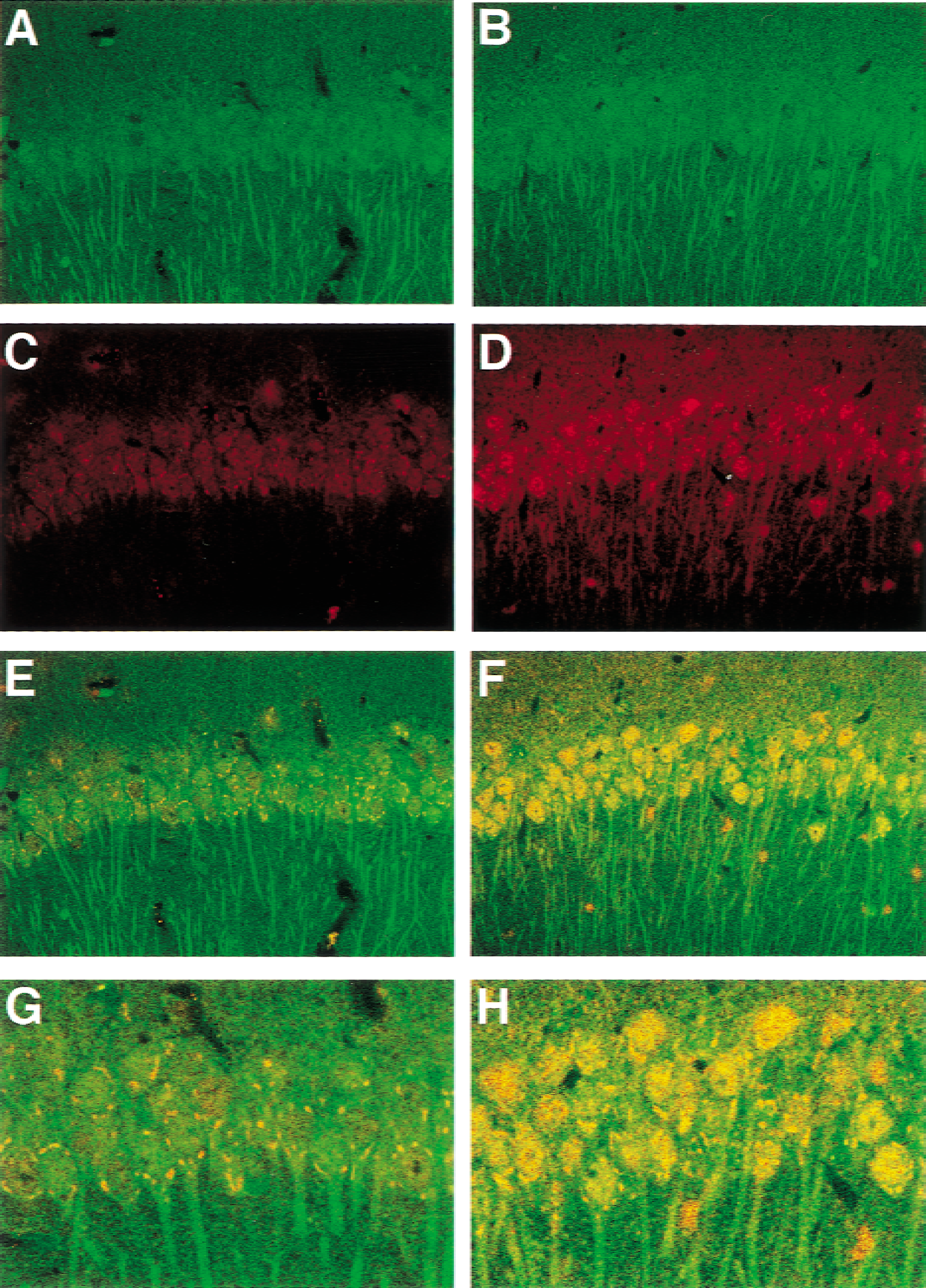
Immunohistochemical localization of active Akt in hippocampal CA1 region. Gerbils were perfused and fixed with 4% paraformaldehyde after treatment with or without orthovanadate. In cases of sham-operated
Prevention of Akt inactivation with orthovanadate and IGF-1 in CA1 region
The authors next examined levels of Akt-Ser-473 phosphorylation following ischemia after treatment with orthovanadate or IGF-1. Intraventricular administration of orthovanadate 30 minutes before ischemia prevented the dephosphorylation of Akt-Ser-473 observed immediately after 5-minute ischemia in the CA1 region (Fig. 4A and 4C). Levels of total Akt protein were unaffected (Fig. 4A). In addition, increased Akt-Ser-473 phosphorylation after orthovanadate treatment was observed 2 and 6 hours after ischemia, whereas the significant increase was observed after 6 hours in ischemic animals without orthovanadate. Similarly, administration of IGF-1 eliminated the ischemia-induced dephosphorylation of Akt (Fig. 4B and 4C). Using the immunocomplex kinase assay, the authors also confirmed that pretreatment with orthovanadate rescued the ischemia-induced decrease in Akt activity observed after 5-minute ischemia (Fig. 4D).
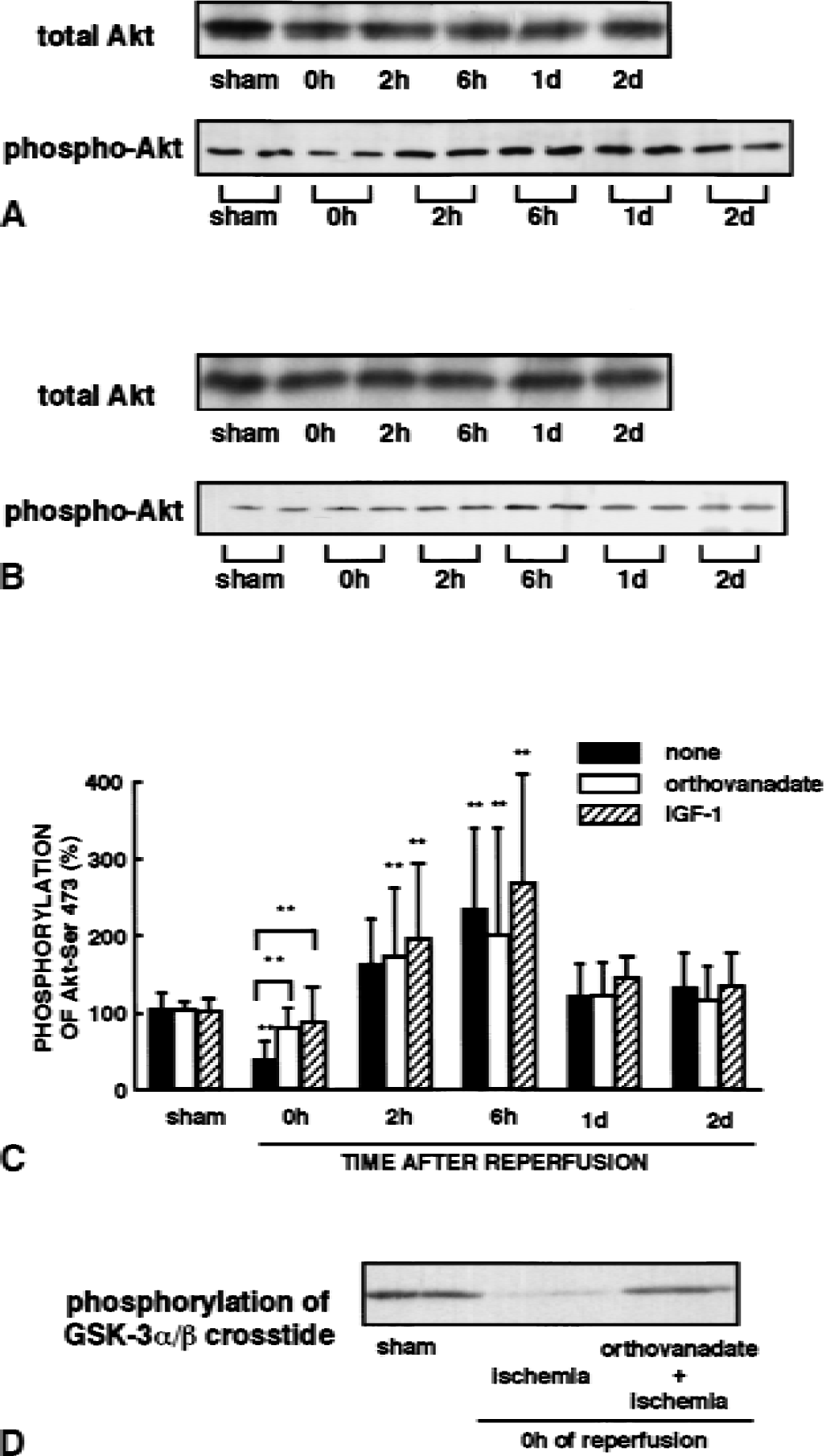
Effects of orthovanadate and IGF-1 treatment on phosphorylation of Akt after transient forebrain ischemia. Gerbils were intraventricularly administered with
Immunohistochemical localization of activated Akt after ischemia
To confirm that changes in Akt activity and Ser-473 phosphorylation occurred in neurons of the CA1 region, active Akt was visualized using the anti–phospho-Akt-Ser-473 antibody. Consistent with decreased Akt activity after ischemia, the intensity of immunofluorescence against anti–Akt-Ser-473 markedly decreased in the pyramidal neurons of ischemia-induced animals, but weak and punctate staining remained in the cell bodies (Fig. 5B) compared with the negative control staining (Fig. 5E). Increased Akt-Ser-473 phosphorylation observed 6 hours after ischemia was evident in the cell bodies of pyramidal neurons (Fig. 5C). In addition, increased immunostaining also was observed in stratum lacunosum, stratum radiatum, and stratum oriens in the CA1 region (Fig. 5C), suggesting that Akt may be activated in the surrounding nonneuronal cells. Furthermore, treatment with orthovanadate prevented the decrease in immunoreactivity induced by ischemia (Fig. 5D).
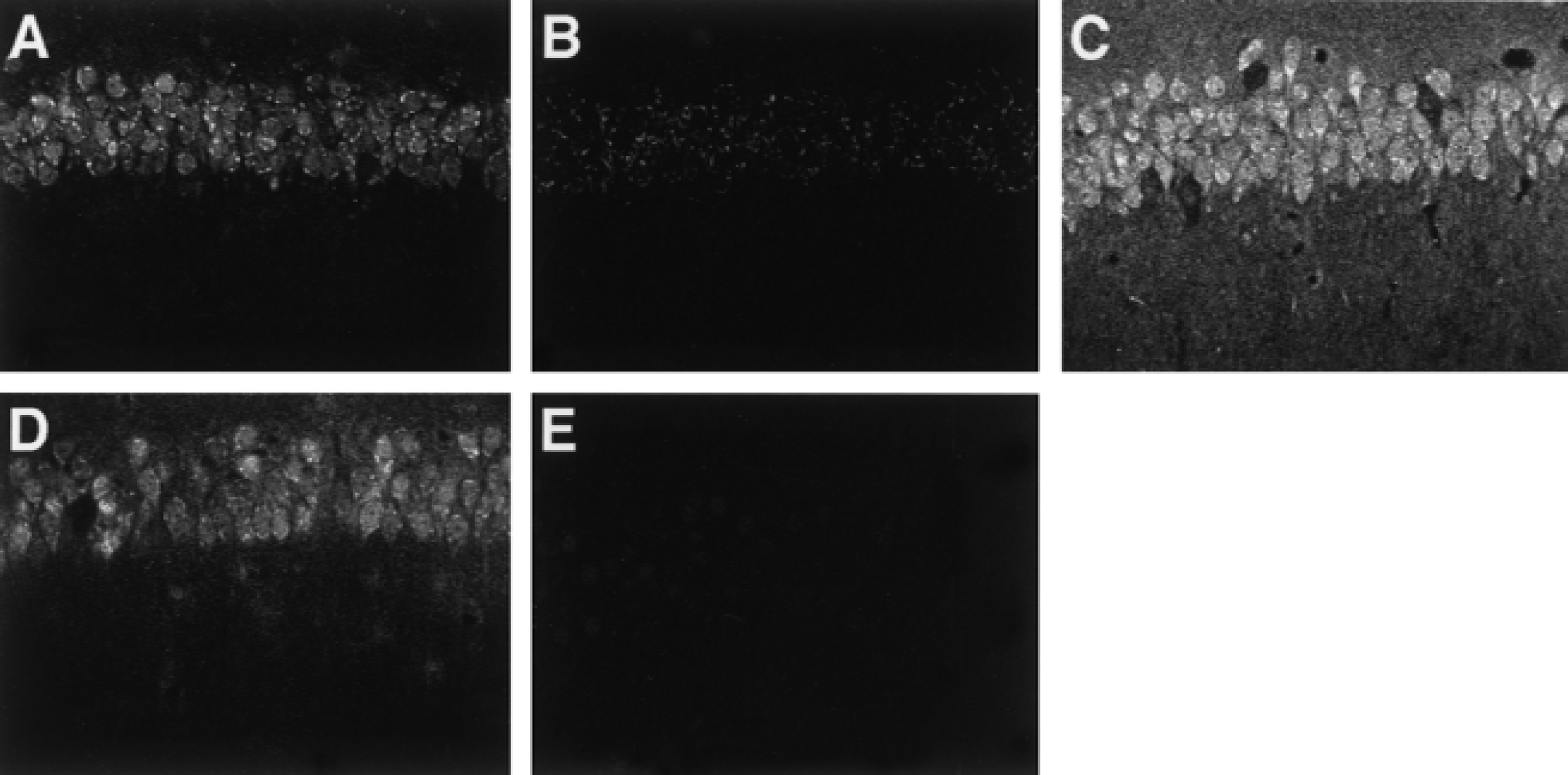
Immunohistochemical localization of active Akt in the hippocampal CA1 region. Gerbils were perfused and fixed with 4% paraformaldehyde with or without ischemia. Confocal laser scanning light microscopic analysis of the hippocampal CA1 pyramidal cell layer was performed after immunostaining with anti–phospho-Akt-Ser-473 antibody. Hippocampal slices were obtained from sham-operated gerbils
Orthovanadate and IGF-1 protection against delayed neuronal death in the CA1 hippocampus after transient forebrain ischemia
Because orthovanadate and IGF-1 prevented the decrease in Akt activity in CA1 pyramidal cells, the authors examined their neuroprotective effects on ischemia-induced delayed neuronal death. Seven days after transient forebrain ischemia, severe neuronal losses of greater than 90% compared with sham-operated animals occurred in the CA1 region (Fig. 6C and 6D). By contrast, treatment with orthovanadate (Fig. 6E and 6F) or IGF-1 (Fig. 6G and 6H) protected neurons from delayed neuronal death, resulting in survival of 80% and 90% of neurons relative to controls, respectively. Furthermore, pretreatment with wortmannin, a selective PI3-K inhibitor, significantly reduced the neuroprotective effect of orthovanadate (Fig. 6I, 6J, and 6K). The authors confirmed that intraventricular administration of wortmannin (2 μL of 100 μmol/L in PBS) significantly inhibited both basal and orthovanadate-induced in vivo phosphorylation of Akt-Ser-473 (Yano et al., 2001) (Fig. 2C). Thus, treatment with orthovanadate or IGF-1 rescues pyramidal cells from delayed neuronal death partly through PI-3K-Akt activation.
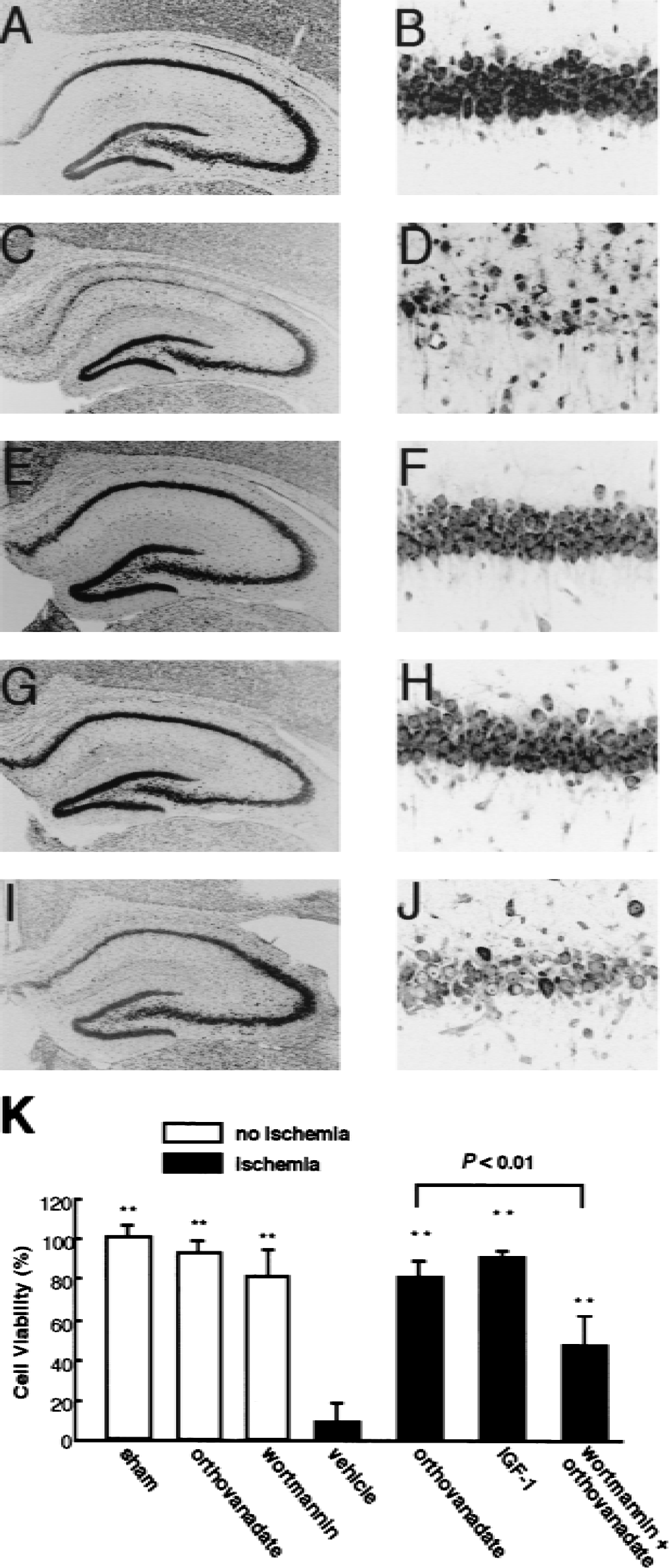
Histologic changes in the hippocampal CA1 pyramidal cell layer after transient forebrain ischemia. The vehicle or drugs were injected into the right cerebral ventricle 30 minutes before ischemia. Histology was performed 7 days after sham operation or reperfusion in gerbils subjected to 5-minute ischemia. Hippocampal slices were stained with propidium iodide and observed using a confocal laser microscope. Representative histologic sections of hippocampus
Neuroprotective effects of posttreatment with orthovanadate
The authors next examined time courses of neuroprotective effects of posttreatment with orthovanadate after ischemia. As shown in Fig. 6K, pretreatment with orthovanadate largely rescued cells from delayed neuronal death. Postischemic intraventricular administration of orthovanadate also significantly rescued cells by posttreatment at 30 minutes and 1 hour, but not at 2 hours after reperfusion (Fig. 7).
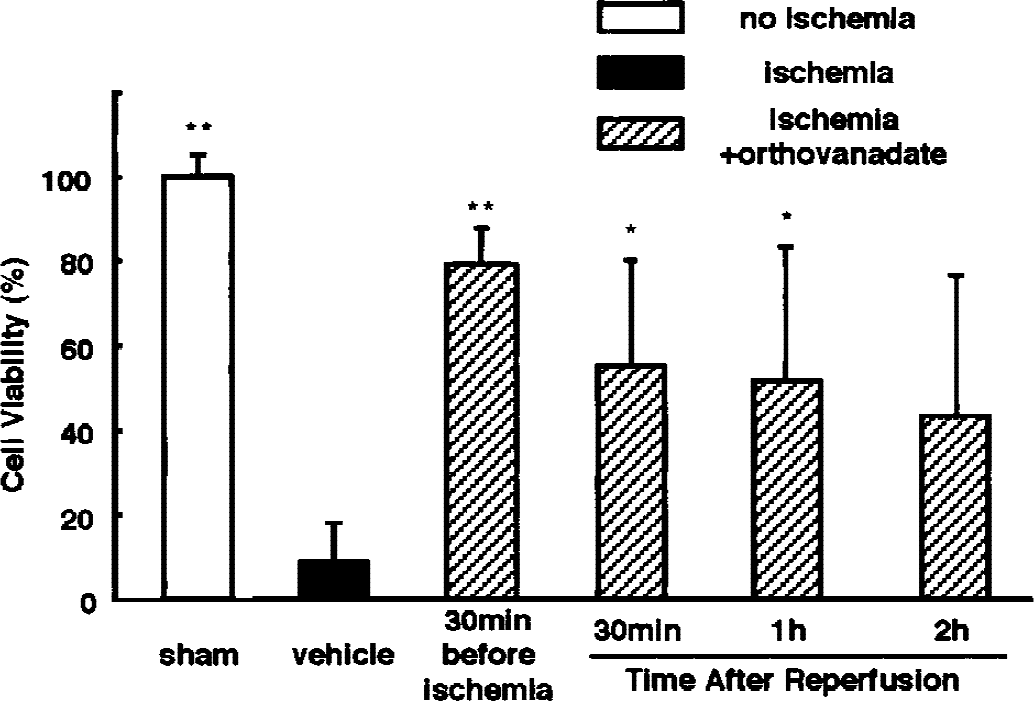
Effects of posttreatment with orthovanadate on ischemia-induced delayed neuronal death in the hippocampal CA1 region. Two microliters orthovanadate was administrated into the right cerebral ventricle of ischemic animals 30 minutes, 1, or 2 hours after reperfusion. Surviving neurons in the CA1 region were quantitatively analyzed as in Fig. 6. Data from sham-operated and ischemic animals treated with the vehicle and pretreated with orthovanadate were the same as those shown in Fig. 6. Results are mean ± SD (n = 5) and are expressed as percentage of sham-operated animals. Statistically significant differences are indicated by asterisks (* P < 0.05, ** P < 0.01).
To eliminate the possibility that the neuroprotective effect of orthovanadate could be caused by hypothermia, body temperature was monitored after 5-minute ischemia with or without orthovanadate treatment. No difference in body temperatures was observed at any time point recorded up to 24 hours before or after ischemia between the vehicle-treated and orthovanadate-treated groups (data not shown).
Involvement of the MAPK pathway in neuroprotection with orthovanadate
As shown in Fig. 6K, orthovanadate-induced neuroprotection was partially inhibited by wortmannin treatment, suggesting that Akt activation is partly responsible for such neuroprotection. However, wortmannin treatment completely inhibited orthovanadate-induced Akt activation (Fig. 2C), suggesting that other signaling mediating survival such as MAPK pathway is activated by orthovanadate treatment. Treatment with 50 to 100 μmol/L orthovanadate stimulated MAPK up to 200% of controls in cultured hippocampal slices (data not shown). The maximal activation of MAPK was obtained up to 300% of control with 200 μmol/L orthovanadate 30 minutes after stimulation. In addition, phosphorylation of MAPK required for its activation decreased in the CA1 region immediately after reperfusion without changes in MAPK protein levels (Fig. 8A and 8C). Intraventricular administration of orthovanadate prevented the decrease in phosphorylation of MAPK (Fig. 8B and 8C). To verify involvement of MAPK in the orthovanadate-induced neuroprotection, the authors analyzed the effects of U0126, a specific MAPK kinase inhibitor, on neuroprotective effects of orthovanadate. Intraventricular administration of 2 μL of 500 μmol/L U0126 or a combination of U0126 plus wortmannin had no apparent effect on cell viability without ischemia (Fig. 8D). Pretreatment with U0126 significantly reduced the neuroprotective effects of orthovanadate and a combination of both wortmannin and U0126 totally suppressed orthovanadate-induced neuroprotection (Fig. 8D). These results suggest that, in addition to the Akt pathway, the MAPK pathway is involved in orthovanadate-induced neuroprotective effects.
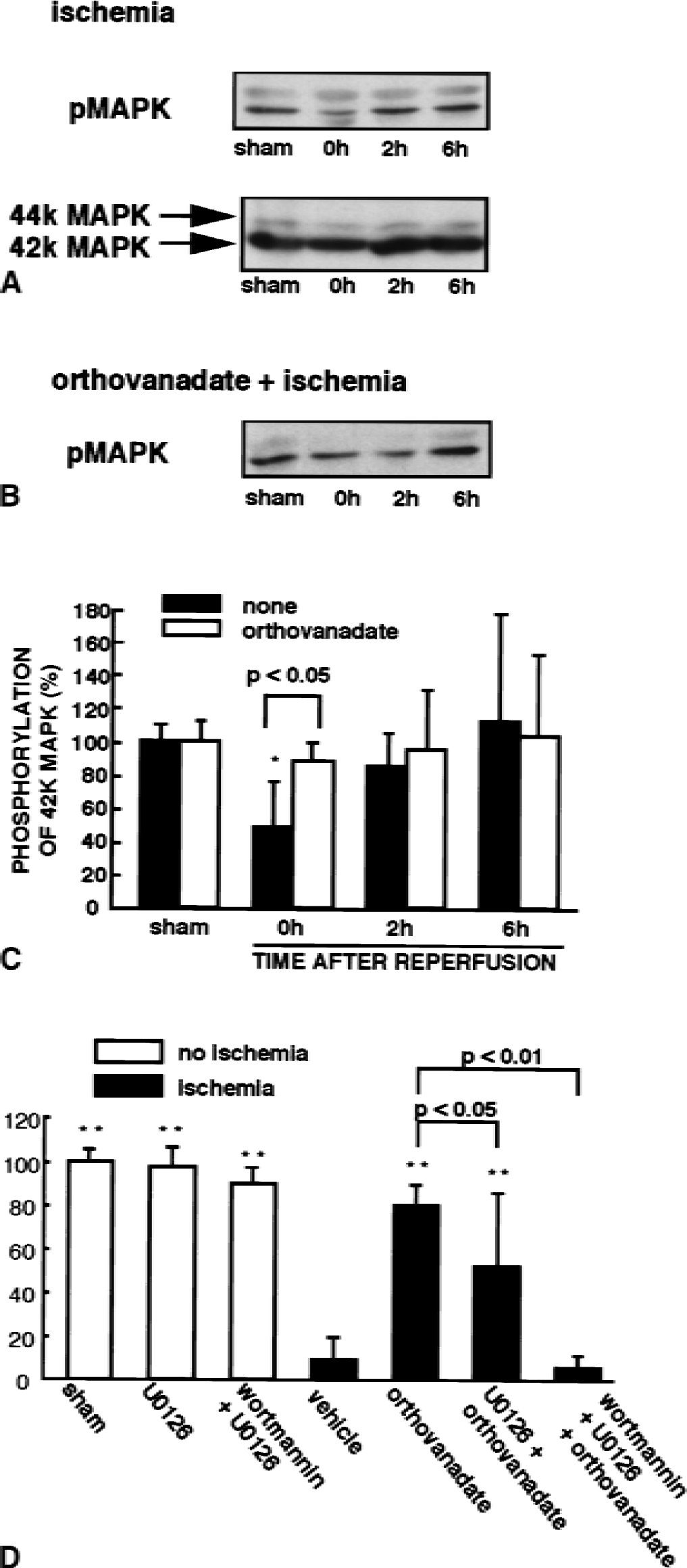
Changes in MAPK phosphorylation and effects of U0126 and wortmannin on orthovanadate-induced neuroprotection after forebrain ischemia.
DISCUSSION
Consistent with neuroprotective effects of IGF-1 in transient forebrain ischemia (Gluckman et al., 1992; Zhu and Auer, 1994; Tagami et al., 1997), IGF-1 blocked delayed neuronal death after a global cerebral ischemia in gerbil. Interestingly, orthovanadate, a nonselective phosphotyrosine phosphatase inhibitor, had potent neuroprotective effect on the cell death. This may imply that nonspecific inhibition of phosphotyrosine phosphatases is a potential target for stroke treatments. The orthovanadate-induced neuroprotection was completely ineffective when animals were pretreated with both wortmannin and U0126. Wortmannin and U0126 are potent inhibitors of PI3-K (Arcaro and Wymann, 1993) and MAPK kinase (Favata et al., 1998), respectively. Intracerebral ventricular injection of wortmannin completely inhibited the orthovanadate-induced Akt activation in the current study. The authors speculate that orthovanadate exert neuroprotective action through Akt and MAPK activation. Because decreased Akt activity observed immediately after reperfusion was seen only in the CA1 but not the CA3 region, the authors suggest that decreased Akt activity underlie the vulnerability of CA1 pyramidal neurons after ischemia. Because the authors recently reported that Akt activation had an essential role for induction of ischemic tolerance (Yano et al., 2001), the authors here primarily analyze the Akt pathway in the orthovanadate-induced neuroprotection.
Akt was inactivated in vivo by dephosphorylation immediately after ischemia, probably due to relative activation of protein phosphatases because of lack of ATP during ischemia (Ouyang et al., 1999). Among the protein phosphatases, protein phosphatase 2A possibly accounts for the dephosphorylation of Akt; because protein phosphatase 2A, but not protein phosphatase 1, inactivates Akt in vitro (Andjelkovic et al., 1996). Ca2+ mobilization through N-methyl- d -aspartate receptors in ischemic conditions possibly activate Ca2+ /calmodulin-dependent protein phosphatase, calcineurin. However, using recombinant Akt protein, the authors tested this hypothesis by assaying dephosphorylation of Akt-Ser-473 by calcineurin in vitro and found that calcineurin did not dephosphorylate Akt-Ser-473 (data not shown). In addition, the authors recently reported decreased calcineurin activity immediately after forebrain ischemia in a four-vessel occlusion model of rat, in which protein phosphatase 2A activity did not change (Morioka et al., 1999). Thus, protein phosphatase 2A, but not protein phosphatase 1 and calcineurin, may be the major protein phosphatase that accounts for the dephosphorylation of Akt.
In contrast to the decrease in Akt activity observed after 5-minute ischemia, phospho-Akt increased above baseline levels 6 hours after reperfusion following ischemia. After ischemia and reperfusion, changes in levels of several cytokines and growth factors also have been reported. Tumor necrosis factor α (TNFα) is induced 1.5 hours after reperfusion in mouse hippocampus after transient global ischemia (Uno et al., 1997). Vascular endothelial growth factor (VEGF) mRNA and its protein increase within 24 hours in rat hippocampus after transient forebrain ischemia (Lee et al., 1999) or middle cerebral artery occlusion (Hayashi et al., 1997). Insulinlike growth factor-1 also accumulates in blood vessels of the damaged hemisphere within 5 hours after hypoxia–ischemia injury (Beilharz et al., 1998). Like IGF-1, TNFα and VEGF activate PI3-K (Pastorino et al., 1999) and Akt (Gerber et al., 1998; Fujio and Walsh, 1999), respectively. Taken together, increased Akt phosphorylation observed 6 hours after reperfusion may be caused by induction of multiple factors including TNFα, VEGF, and IGF-1.
The characteristic distribution of active Akt suggests several downstream targets. L-type, voltage-dependent Ca2+ channels in the plasma membranes of the cell body and dendrites of pyramidal cells could represent a potential target, because Akt-dependent Ca2+ channel activation is required for IGF-1–induced neuroprotection in cerebellar granule cells (Blair et al., 1999). Bad and caspase 9 may also function in Akt-induced neuroprotection. Bad, a proapoptotic protein of the Bcl-2 family, is phosphorylated by Akt with concomitant dissociation from Bcl-XL, which in turn prevents mitochondrial damage, such as release of cytochrome c (Datta et al., 1997; Kennedy et al., 1999; Zha et al., 1996).
Similarly, phosphorylation and subsequent inactivation of caspase 9 inhibits apoptotic signaling (Cardone et al., 1998). Release of cytochrome c from mitochondria and activation of a caspaselike protease with a con comitant decrease in Akt activity was documented in a two-vessel occlusion rat model (Ouyang et al., 1999). Furthermore, activation of Akt in nuclei of pyramidal neurons may play a pivotal role in expression of antiapoptotic proteins through activation of transcription factors, such as the cyclic adenosine monophosphate–responsive element binding protein (CREB) and Forkhead family transcription factors. For example, activation of Akt signaling pathways leads to CREB phosphorylation (Du and Montminy, 1998), and IGF-1 induces CREB phosphorylation through the Akt pathway (Pugazhenthi et al., 1999). Activation of CREB also stimulates IGF-1–induced Bcl-2 expression through the PI3-K pathway (Pugazhenthi et al., 2000). A Forkhead family transcription factor also is directly phosphorylated by Akt and inhibits expression of proapoptotic proteins such as the Fas ligand (Brunet et al., 1999). Furthermore, IGF-1 could phosphorylate Forkhead family transcription factor, FKHRL1, through PI3-K/Akt pathway in rat pheochromocytoma (PC12) cells (Zheng et al., 2000).
Although IGF-1 attenuates apoptosis of hippocampal neurons induced by cerebral ischemia (Gluckman et al., 1992; Guan et al., 1993, 1996; Zhu and Auer, 1994; Tagami et al., 1997; Wang et al., 2000), the penetration of peptides such as growth factors into the hippocampus is generally slow when administered into the ventricle and very inefficient when administrated to peripheral tissues. Vanadate, vanadium complexes, and vanadyl compounds can produce insulinomimetic effects in the liver, adipocytes, and muscle when administered in peripheral tissues (McNeill et al., 1995; Bevan et al., 1995). The biologic activity of these vanadate and vanadium complexes is not, however, restricted to nonneuronal cells. Differentiation and neurite outgrowth of PC12 cells and neurite outgrowth of human neuroblastoma SH-SY5Y cells are stimulated by treatment with orthovanadate (Rogers et al., 1994). Although vanadate and vanadium complexes stimulate proliferation in mouse fibroblasts (Krady et al., 1997), they inhibit proliferation of NB41 neuroblastoma cells and C6 glioma cells (Faure et al., 1995). Inhibition of protein tyrosine phosphatases appears to underlie the biologic actions of vanadate species (Meier et al., 1998).
Vanadate species also activate the MAPK pathway (Zhao et al., 1996). Among the vanadate species, pervanadate, which is prepared from a combination of vanadate and H2 O2, is the most potent stimulant of both Akt and MAPK in situ (Zhao et al., 1996; Meier et al., 1998). However, the half-life of pervanadate is 1 to 2 hours in aqueous solution and its stability in biologic systems is questionable (Morinville et al., 1998). Moreover, H2 O2 itself is toxic for cells. Although prolonged incubation for 48 hours with 100 μmol/L orthovanadate has been reported to induce apoptosis of cultured neurons from rat dentate gyrus (Figiel et al., 1997), intraventricular administration of orthovanadate did not produce neurotoxic effects on hippocampal pyramidal neurons in the current study. In fact, the authors' observations are consistent with vanadate-induced inhibition of apoptosis induced by the following: TNFα in endothelial cells (Yang et al., 1996) and the mouse connective tissue cell line L-929 (Totpal et al., 1992); extracellular adenosine triphosphate in the murine mastocytoma cell line and a murine leukemia cell line (Bronte et al., 1996); and K252a in malignant glioma cells (Chin et al., 1999). Despite the weak activation by orthovanadate of MAPK in situ (Zhao et al., 1996) and Akt in the current study, orthovanadate exhibited a neuroprotective effect equivalent to that of IGF-1 when administrated into the ventricle. Interestingly, postadministration of orthovanadate also resulted in a neuroprotective effect 30 minutes and 1 hour after reperfusion as shown in Fig. 7.
In conclusion, the authors have shown that Akt and MAPK activation contributes to neuroprotection induced by treatment with orthovanadate. Activated Akt was predominantly located in pyramidal neurons, particularly in the nuclei and dendrites. The characteristic localization of the active Akt provides new insight regarding target molecules acting to protect neurons from apoptosis. These downstream targets of orthovanadate-induced Akt activation are now under investigation. The effects of vanadium compounds are of particular interest because these compounds have been tested as therapeutic agents for insulin-deficient and diabetic rats (Heyliger et al., 1985; Brichard et al., 1988; Dai et al., 1994; Blondel et al., 1990) and for genetically obese, hyperinsulinemic, and insulin-resistant rats and mice (Brichard et al., 1989, 1990; Meyerovitch et al., 1991). Furthermore, clinical studies of vanadyl sulfate or sodium metavanadate had been tried to improve insulin sensitivity of patients with noninsulin-dependent diabetes mellitus (Goldfine et al., 1995; Cohen et al., 1995; Brichard and Henquin, 1995). In the current study, the authors suggest that vanadate and vanadium compounds have pharmacologic effects on delayed neuronal death after brain ischemia.