
Abstract
Delayed neuronal death in the hippocampal CA1 region after transient forebrain ischemia may share its underlying mechanism with neurodegeneration and other modes of neuronal death. The precise mechanism, however, remains unknown. In the postischemic hippocampus, conjugated ubiquitin accumulates and free ubiquitin is depleted, suggesting impaired proteasome function. The authors measured regional proteasome activity after transient forebrain ischemia in male Mongolian gerbils. At 30 minutes after ischemia, proteasome activity was 40% of normal in the frontal cortex and hippocampus. After 2 hours of reperfusion, it had returned to normal levels in the frontal cortex, CA3 region, and dentate gyrus, but remained low for up to 48 hours in the CA1 region. Thus, the 26S proteasome was globally impaired in the forebrain during transient ischemia and failed to recover only in the CA1 region after reperfusion. The authors also measured 20S and 26S proteasome activities directly after decapitation ischemia (at 5 and 20 minutes) by fractionating the extracts with glycerol gradient centrifugation. Without adenosine triphosphate (ATP), only 20S proteasome activity was detected in extracts from both the hippocampus and frontal cortex. When the extracts were incubated with ATP in an ATP-regenerating system, 26S proteasome activity recovered almost fully in the frontal cortex but only partially in the hippocampus. Thus, after transient forebrain ischemia, ATP-dependent reassociation of the 20S catalytic and PA700 regulatory subunits to form the active 26S proteasome is severely and specifically impaired in the hippocampus. The irreversible loss of proteasome function underlies the delayed neuronal death induced by transient forebrain ischemia in the hippocampal CA1 region.
Keywords
Many critical regulatory proteins that must be rapidly eliminated for normal growth and metabolism are degraded by the ubiquitin/proteasome pathway. This process involves two successive steps: covalent attachment of ubiquitin molecules to the target protein and degradation of the targeted protein by the 26S proteasome (Hershko and Ciechanover, 1998). The 26S proteasome, a eukaryotic ATP-dependent protease complex with a molecular mass of approximately 2,500 kd, is composed of a core proteinase (the 20S proteasome) and a pair of PA700 regulatory particles (the 19S complex) (Coux e al., 1996; Rechsteiner, 1998; Baumeister et al., 1998) PA700 attaches to both ends of the central 20S proteasome in opposite orientations to form the enzymatically active 26S proteasome (DeMartino and Slaughter, 1999) In vitro, these subunits associate and disassociate in an ATP-dependent manner (Tanahashi et al., 2000), forming the active complex under ATP-rich conditions and dissociating under ATP-poor conditions (Tanahashi e al., 2000). Therefore, ATP-depleting conditions, such as ischemia, would be expected to reduce proteasome activity.
Delayed neuronal death in the hippocampal CA1 region after transient forebrain ischemia and reperfusion i a unique type of cell death that progresses despite com plete recovery of metabolic parameters such as regional cerebral blood flow, glucose metabolism, and tissue ATP content (Kirino, 1982; Pulsinelli et al., 1982; Mies et al. 1990). Impairment of protein ubiquitination after transient forebrain ischemia had been pointed out in the hip pocampal CA1 region (Magnusson, 1989). We recently found that conjugated ubiquitin accumulates and free ubiquitin is depleted only in the hippocampal CA1 region, suggesting impairment of proteasome function (Morimoto et al., 1996; Ide et al., 1999). In the present study, we investigated the activity and molecular integrity of 20S and 26S proteasomes in the hippocampal CA1 region after transient forebrain ischemia in gerbils.
MATERIALS AND METHODS
Transient forebrain ischemia and sample preparation
Male Mongolian gerbils weighing 60 to 80 g were anesthetized with 3% halothane in 33% O2 and 67% N2. The bilateral common carotid arteries were exposed, and forebrain ischemia was induced by occluding these arteries with Sugita aneurysm clips. After 5 minutes, the clips were removed and halothane anesthesia was discontinued. During the operation, rectal and temporal muscle temperature were monitored and maintained at 37.5 ± 0.2°C with a heat lamp and warming blanket. Gerbils were killed by in situ freezing under anesthesia (n = 6 at each time point) and the brains were removed. Eight slices from 55-μm-thick frozen sections covering the hippocampus were prepared from each frozen brain. The hippocampal CA1 region, the CA3 and dentate gyrus, as well as the cerebral cortex were separately dissected under a microscope (Ide et al., 1999). These manipulations were performed in a cryotome at −20°C. Dissected brain tissues from 16 slices (from two brains) were used for each lysate preparation. We prepared three independent lysates at each time point.
Decapitation ischemia and sample preparation
Male Mongolian gerbils were decapitated under anesthesia. The resected heads were incubated at 37°C for 5 or 20 minutes and placed immediately in ice-cold phosphate-buffered saline (PBS). The brains were removed, and the hippocampus and cerebral cortex were dissected for the measurement of tissue ATP content and for analysis in a proteasome/ATP-regenerating assay. During these procedures, the tissue samples were kept in ice-cold PBS.
Assay of peptidase activity
Tissue samples were lysed in proteasome buffer (10 mmol/L Tris-HCl, pH 7.5, 1 mmol/L ethylenediaminetetraacetic acid [EDTA], 2 mmol/L ATP, 20% glycerol, 4 mmol/L dithiothreitol [DTT]), sonicated, and centrifuged at 13,000 g at 4°C for 10 minutes. The supernatants were assayed for their ability to degrade the activity of the synthetic peptide succinyl-
Sedimentation velocity analysis
The hippocampus and cerebral cortex removed after decapitation ischemia were homogenized in ice-cold lysis buffer (25 mmol/L Tris-HCl, pH7.5, 250 mmol/L sucrose, 1 mmol/L DTT) with a Potter-Elvenjen homogenizer. The homogenate was centrifuged at 8,000 g for 30 minutes, and the supernatant was incubated for 60 minutes at 37°C in 25 mmol/L Tris-HCl (pH7.5) containing 1 mmol/L DTT, 5 mmol/L MgCl2 and 2 mmol/L ATP with an ATP-regenerating system (10 mmol/L creatine phosphate, 10 μg/ml creatine kinase). Samples of freshly prepared or ATP-incubated extracts (1 mg of protein) were subjected to 10% to 40% glycerol density-gradient centrifugation in 25 mmol/L Tris-HCl buffer, pH7.5, containing 1 mmol/L DTT with or without 2 mmol/L ATP. After centrifugation at 83,000 g for 22 hours in a Hitachi SRP28SA1 rotor, the gradient was separated into 30 1-ml fractions.
Assay of 35S-ornithine decarboxylase degrading activity.
The degradation of recombinant 35S-labeled ornithine decarboxylase (ODC; 2,000 to 3,000 cpm) was assayed in the presence of ATP, an ATP-regenerating system, and antizyme (Murakami et al., 1992, 1999). 35S-ODC was produced by in vitro translation using rabbit reticulocyte lysate (Wako) containing rat ODC mRNA, 35S-labeled methionine, and 35S-labeled cysteine (DuPont NEN) and was purified by immunoaffinity chromatography before use. After incubation for 60 minutes at 37°C, trichloroacetic acid-soluble radioactivity in the reaction mixture was measured and expressed as a percentage of total 35S-ODC added.
RESULTS AND DISCUSSION
Proteasome activity after transient forebrain ischemia
As estimated by Suc-LLVY-MCA hydrolysis, 26S proteasome activity decreased by 60% in the frontal cortex, dentate gyrus, and hippocampus 30 minutes after transient forebrain ischemia. After 2 to 4 hours of reperfusion, 26S proteasome activity recovered to nearly normal levels except in the hippocampal CA1 region, where it remained low for up to 48 hours (Fig. 1). These findings are basically compatible with the previous report, which demonstrated that 26S proteasomal activity decreases transiently in the forebrain after 10 minutes of ischemia and recovers within 1 hour, and concluded that proteasome activity in the brain is not irreversibly impaired by transient ischemia (Kamikubo and Hayashi, 1996). In that study, however, the cerebral cortex and hippocampus were not analyzed separately. Because the hippocampus constitutes less than 10% of the volume of the forebrain, Kamikubo and Hayashi's findings mainly reflect proteasome activity in the cerebral cortex.
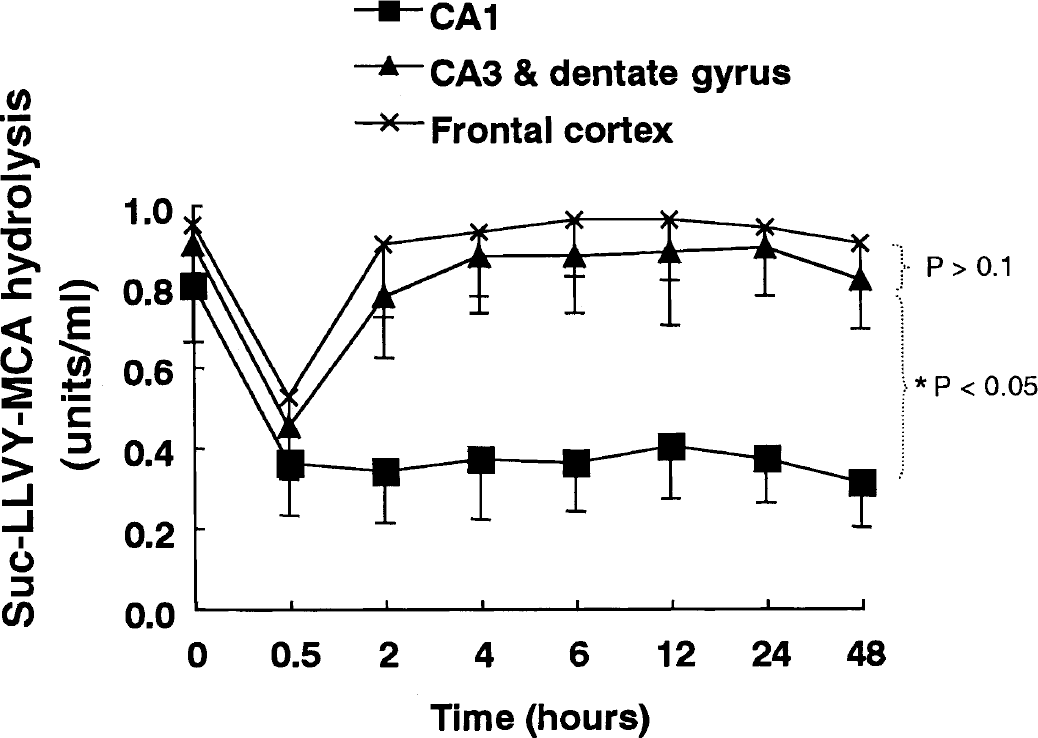
Proteasome activity after transient forebrain ischemia. Six gerbils were killed by in situ freezing at each time point after 5 minutes of forebrain ischemia. The frozen brains were sliced into 55-μm-thick blocks with a cryotome. Under the operating microscope, the CA1 region, the CA3/dentate gyrus, and the frontal cerebral cortex were separately cut from each block in a freezing box. Dissected brain tissues from 2 animals were lysed for one analysis of proteasome activity. Proteasome activity of the cytoplasmic extracts was measured as described in Materials and Methods. Proteasome activity decreased to about half of normal levels 30 minutes after transient ischemia in all three brain regions. Proteasome activity recovered to a normal level after 2 hours of reperfusion in the CA3/dentate gyrus and the frontal cortex, but failed to recover for up to 48 hours in the hippocampal CA1 region. The analysis was repeated three times independently using 6 animals altogether at each time point. Bars designate standard deviation. The proteasome activity curves for the frontal cerebral cortex and the CA3/dentate gyrus differed significantly from that for the CA1 region (P < 0.05, as determined by comparing the curves using two-way ANOVA with a Bonferroni post hoc test for multiple comparisons) but did not differ significantly from each other (P > 0.1).
In contrast to Kamikubo and Hayashi (1996), we measured proteasome activity in the hippocampal CA1 region and in the frontal cortex over time. First, we used the degradation of Suc-LLVY-MCA as a measure of 26S proteasome activity. The Suc-LLVY-MCA-degrading activity in tissue extracts is due to the 26S proteasome, because its removal by immunoprecipitation with anti-proteasome antibodies results in almost complete loss of activity (Tanahashi et al., 2000). In addition, although proteasomes exist in two forms (20S and 26S) in cells, the contribution of the 20S proteasome is quite low because of its functional latency. In the CA1 region, 26S proteasome activity decreased and remained at a low level for up to 48 hours after transient forebrain ischemia, indicating irreversible impairment because morphologic changes of cell death become prominent after 48 hours. These data are consistent with the depletion of free ubiquitin and the accumulation of multi-ubiquitinated proteins, even at 48 hours after transient ischemia, observed in our previous study (Ide et al., 1999).
Molecular dynamics of proteasomal subunit assembly
Next, we investigated the molecular mechanism underlying the impairment of the 26S proteasome in the CA1 region. Because the 26S proteasome is reversibly assembled from its constituents (i.e., the 20S proteasome and PA700) in an ATP-dependent fashion (Tanahashi et al., 2000), we hypothesized that the assembly process is selectively impaired in the CA1 region. After 5 minutes of decapitation ischemia, the tissue ATP content decreased almost to zero (data not shown), as is compatible with the previous report (Ljunggren et al., 1974). Then, we estimated molecular dynamics of subunit assembly of the 20S and 26S proteasomes by velocity sedimentation analysis. Cytoplasmic extracts from the frontal cortex (Fig. 2A) and hippocampal CA1 region (Fig. 2C) of brains subjected to 5-minute decapitation ischemia were fractionated by glycerol density-gradient centrifugation. Low peptidase activity was found in slowly sedimenting fractions corresponding to the sedimentation position of purified 20S proteasome. Addition of 0.05% sodium dodecyl sulfate (SDS), a potent artificial activator of the latent 20S proteasome, caused marked activation of the enzyme in those fractions, as previously reported (Tanahashi et al., 2000). However, when the cortical extract was incubated with ATP and an ATP-regenerating system, the enzyme activity in the fraction corresponding to the 26S proteasome increased markedly, as reflected in a large peak, and SDS-augmented activity in the 20S proteasome was greatly reduced (Fig. 2B). The sedimentation profiles were similar to those of extracts from freshly prepared intact cortex (data not shown).
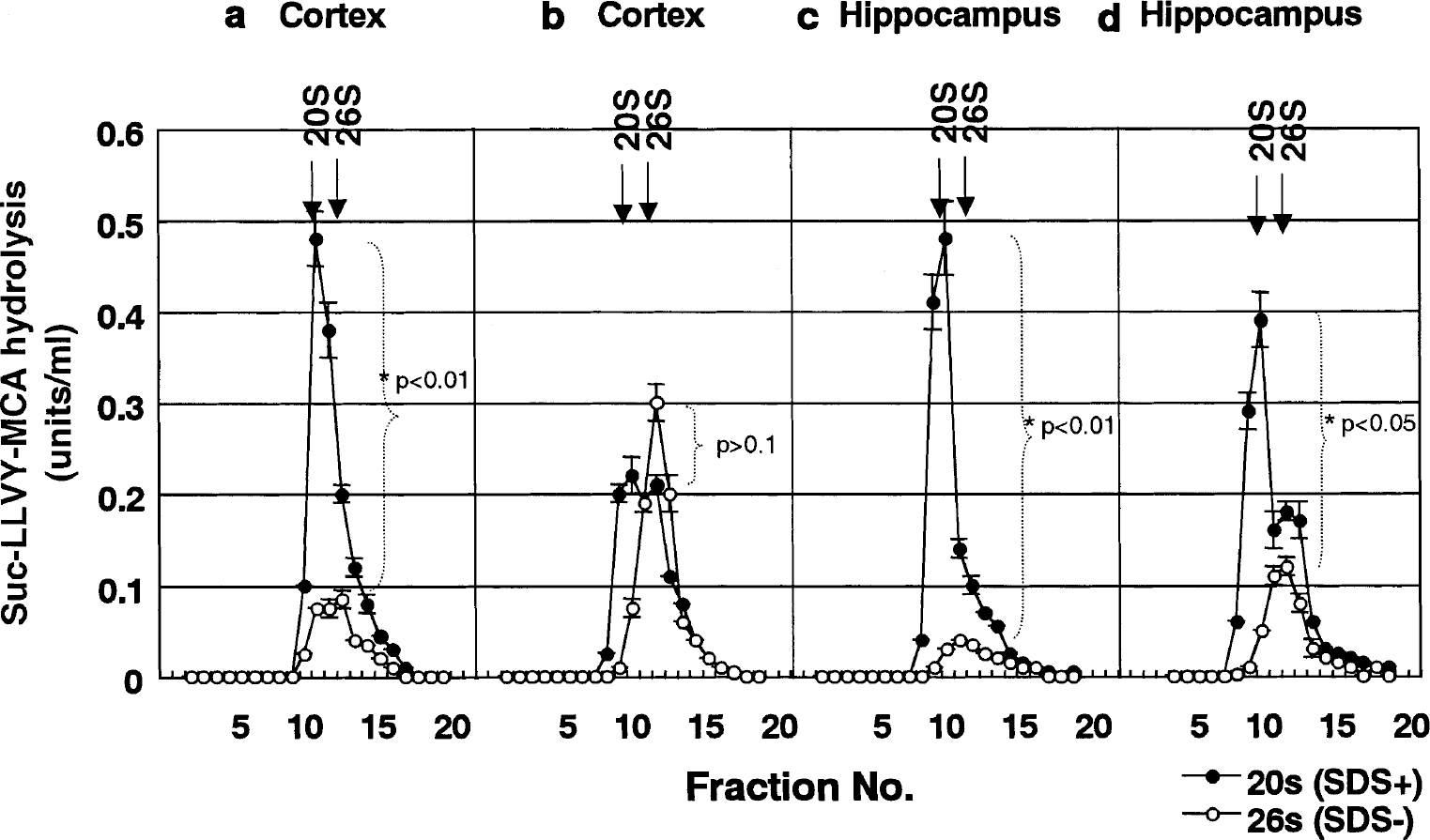
Effects of 5 minutes of decapitation ischemia and ATP on the integrity of the 26S proteasome analyzed by density-gradient centrifugation. The decapitated head was soaked in ice-cold phosphate-buffered saline (PBS) after ischemia, and the frontal cortex (A and B) and the whole hippocampus (C and D) were dissected in ice-cold PBS. The dissected brain tissues were immediately lysed on ice. Samples (1.0 mg protein) of these extracts were preincubated for 60 minutes at 37°C with 2 mmol/L ATP and an ATP-regenerating system (10 mmol/L creatine phosphate, 5 mmol/L MgCl2, and 10 μg/ml creatine kinase). The freshly prepared extracts (A and C) and ATP-regenerated extracts (B and D) were then fractionated by glycerol density-gradient centrifugation (10% to 40% glycerol), as described in Materials and Methods. After fractionation, aliquots (20 μl) of individual fractions were used for assay of Suc-LLVY-AMC hydrolysis with (+) or without (−) 0.05% sodium dodecyl sulfate. Arrows indicate the elution positions of purified 20S and 26S proteasomes. 20S proteasome activity is significantly dominant before ATP regeneration (A and C) both in the cortex and the hippocampus (P < 0.01, as determined by comparing the curves using two-way ANOVA with a Bonferroni post hoc test for multiple comparisons). Whereas the dominance disappeared in the cortex (B) (P > 0.1), it is maintained in the hippocampus (D) (P < 0.05) after ATP regeneration. The whole experiment was repeated five times independently. Bars designate standard deviation. Similar data were obtained after 20 minutes of decapitation ischemia (data not shown).
In contrast, when extracts from the hippocampus were subjected to the same ATP-regenerating treatment, the SDS-insensitive Suc-LLVY-MCA-degrading activity of the 26S proteasome recovered only partially, as reflected in a small peak, whereas the SDS-augmented activity of the 20S proteasome remained significantly high (Fig. 2D). This is in marked contrast to the profiles of both proteasomes from cortical extracts (Fig. 2B) or fresh hippocampal extracts (data not shown). Similar results were obtained after 20 minutes of decapitation ischemia (data not shown). These findings indicate that ATP-dependent regeneration of the 26S proteasome is specifically impaired in the hippocampus.
To evaluate 26S proteasome activity more accurately, we used ODC, a natural substrate of the 26S proteasome that does not require ubiquitination and is employed for quantitative and sensitive measurement of ATP-dependent proteolytic activity in vitro (Murakami et al., 1992). Antizyme, an ODC-inhibitory protein, which is needed for the process, and ODC are available as recombinant proteins (Murakami et al., 1999). Using this assay system, we remeasured 26S proteasome activity in all fractions used for the experiments shown in Fig. 2. In the presence of antizyme, no obvious ATP-dependent degradation of 35S-ODC was detected in sedimentation fractions derived from fresh samples of the cortex or hippocampus (Fig. 3A and 3C). Thus, 5 minutes of decapitation ischemia induced almost complete dissociation of the 26S proteasome complex into its constituents, the latent 20S proteasome and PA700. However, after ATP regeneration, the 35S-ODC-degrading activity in the hippocampal extracts was significantly low, nearly half that of cortical extracts (Fig. 3B and 3D; P < 0.05). These findings confirmed that reassembly of the 26S proteasome is impaired in the hippocampus (Fig. 4).
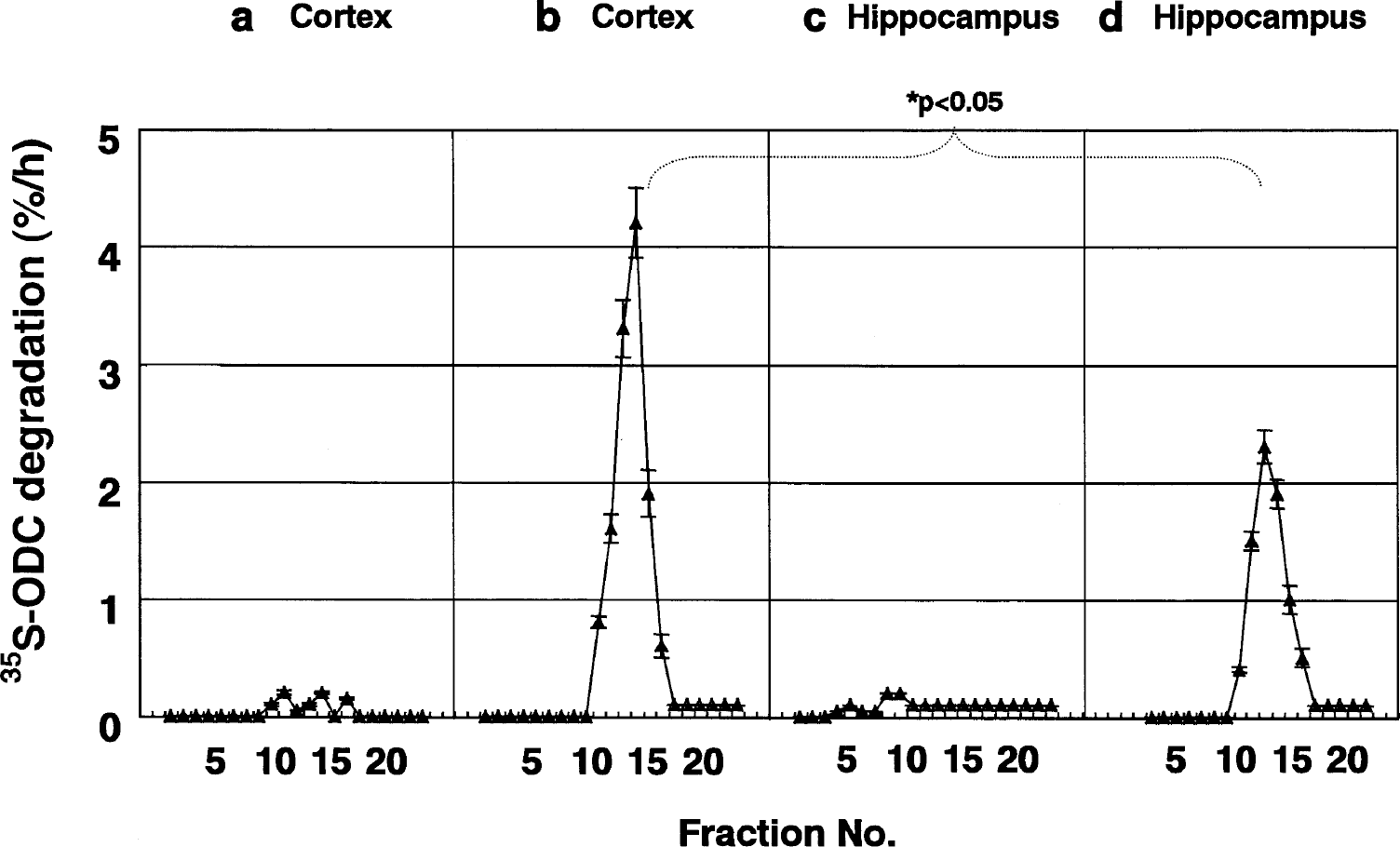
Sedimentation velocity analysis of the 26S proteasome monitored by the degradation of 35S-labeled ornithine decarboxylase (ODC). The same fractions obtained by glycerol density-gradient centrifugation shown in Fig. 2 were used for the assay of ATP- and antizyme-dependent degradation of 35S-ODC as described in Materials and Methods. Arrows indicate the elution positions of purified 20S and 26S proteasomes. 35S-ODC-degrading activity (26S proteasome activity) is almost null in the cortex and hippocampus before ATP regeneration (
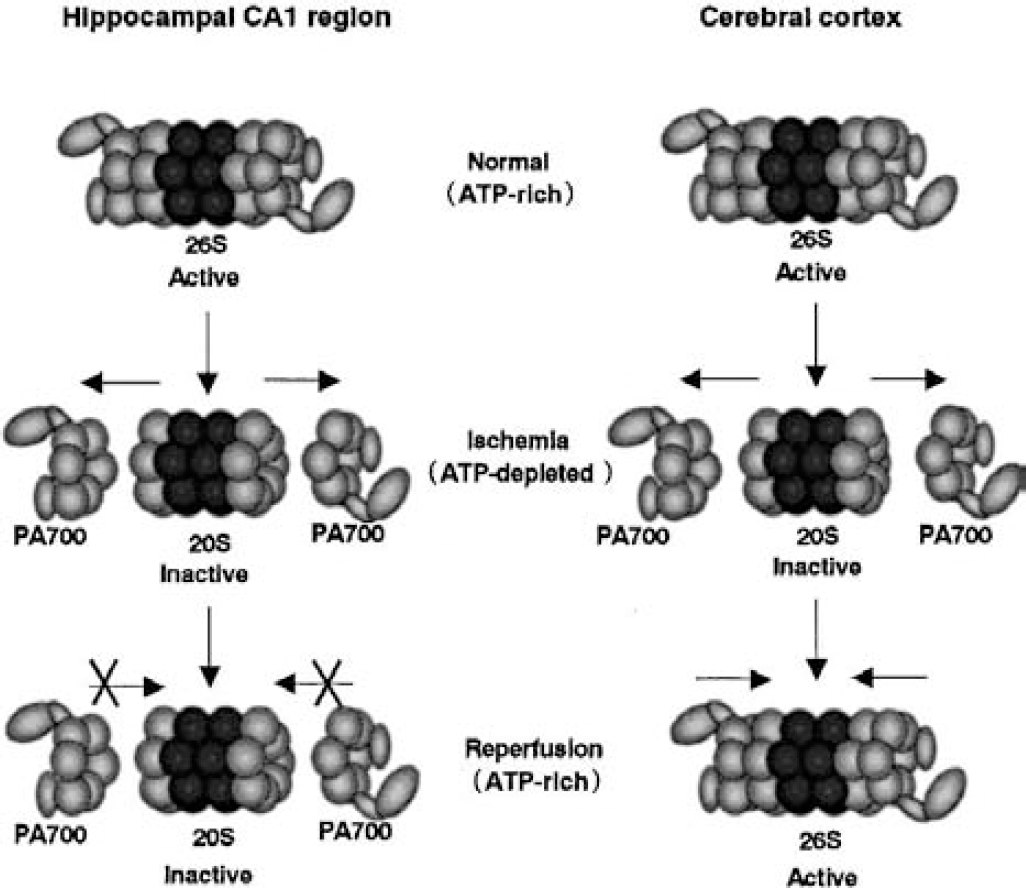
Schematic model of 26S proteasome reassembly failure in the hippocampus after ischemia. Under normal (ATP-rich) conditions, a large amount of the 26S proteasome (26S) exists in both the cerebral cortex and the hippocampus. Under ischemic (ATP-poor) conditions, the 26S complex dissociates into its constituents, the 20S proteasome (20S) and PA700. After reperfusion (ATP recovered), 20S and PA700 reassemble to form the 26S proteasome in the cerebral cortex. In the hippocampus, however, the reassembly process is impaired.
For these assays, we used the total hippocampus because the ATP-regenerating system functions reliably only when fresh samples are used, and dissecting the CA1 region is difficult without freezing. Therefore, some 26S proteasome recovery in the hippocampus was observed after ATP addition because the total hippocampus contains CA3 and dentate gyrus, where proteasomal recovery was complete.
Next, we assessed the expression levels of the constituents of the 26S proteasome, such as the 20S proteasome (Tanahashi et al., 1998), PA700 (S5a, p45, and p40.5) (Tanahashi et al., 1998, 2000), and PA28, another proteasomal activator consisting of a and β subunits (Tanahashi et al., 1997), by Western blotting. This analysis showed no change in expression levels in the CA1 region or the frontal cortex (data not shown) after 5 minutes of decapitation ischemia. These findings indicated that the decrease in 26S proteasome activity after ischemia is due to dissociation of the active 26S proteasome into its constituents, not to changes in the expression levels of those constituents, and that the reassembly process is impaired in the CA1 region (Fig. 4).
The molecular mechanism whereby ATP-dependent reassembly of the 26S proteasome is impaired exclusively in hippocampal CA1 region remains unknown. Recently, it was reported that assembly of the 26S proteasome involves the phosphorylation of certain proteasome subunits (Satoh et al., 2001), but the exact mechanism for the ATP-dependent process of the assembly remains elusive. We speculate that essential factors required to reassemble the subunits are lacking or that factors that impair reassembly are located in the CA1 region. Further investigation is required to clarify this point.
Several studies have demonstrated that proteasome dysfunction induces apoptosis in various types of cells, including neurons (Drexler, 1997; Lopes et al., 1997; Qiu et al., 2000; Canu et al., 2000; Keller and Markesbery, 2000; McNaught and Jenner, 2001). Proapoptotic proteins targeted by the ubiquitin/proteasome system include, among others, p53, cytochrome c, bid, and bax (Maki et al., 1996; Breitschopf et al., 2000; Pearce and Sherman, 1997; Li and Dou, 2000). Although the mechanism by which proteasomal dysfunction induces apoptosis remains controversial, we assume that these proapoptotic proteins can induce apoptosis if ubiquitin/proteasome-dependent degradation of these proteins is impaired. Thus, prolonged impairment of proteasome function after transient ischemia in CA1 neurons probably leads to their delayed death. Further study is needed to clarify the direct relation between the prolonged impairment of proteasome function and delayed death in hippocampal CA1 neurons.
Footnotes
Acknowledgements
The authors thank Mr. Stephen Ordway for editorial assistance.