
Abstract
Induction of mild hypothermia improves neurologic outcome after global cerebral ischemia. This study measured levels of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in hippocampal tissue of rats after resuscitation from 8 minutes of normothermic, asphyxial cardiac arrest. After resuscitation, rats were maintained either at normal temperature (37°C) or cooled to mild hypothermia (33°C, beginning 60 minutes after resuscitation). After 12 or 24 hours, neurotrophin levels in hippocampus were measured by immunoblotting. Ischemia and reperfusion increased hippocampal levels of BDNF. Induction of hypothermia during reperfusion potentiated the increase in BDNF after 24 hours, but not after 12 hours. Levels of NGF were not increased by postresuscitation hypothermia. Hypothermia also increased tissue levels and tyrosine phosphorylation of TrkB, the receptor for BDNF. Increased BDNF levels were correlated with activation of the extracellularly regulated kinase (ERK), a downstream element in the signal transduction cascade induced by BDNF. In contrast to the many deleterious processes during ischemia and reperfusion that are inhibited by induced hypothermia, increasing BDNF levels is a potentially restorative process that is augmented. Increased activation of BDNF signaling is a possible mechanism by which mild hypothermia is able to reduce the neuronal damage typically occurring after cardiac arrest.
Hypothermia promotes neuronal recovery after both focal and global brain ischemia (Colbourne et al., 1999, 2000; Kawai et al., 2000; Coimbra and Wieloch, 1994). Recently, we reported that induction of mild hypothermia for 24 hours after resuscitation reduced the loss of CA1 hippocampal neurons after resuscitation from asphyxial cardiac arrest in rats (Hicks et al., 2000a). This histologic benefit was associated with an improvement in neurobehavioral recovery. In contrast to studies using briefer periods of hypothermia (Dietrich et al., 1993; Kuboyama et al., 1993; Xiao et al., 1998), the histologic and neurobehavioral improvement was identical between groups in which hypothermia was induced immediately and in which hypothermia was induced 1 hour after resuscitation. These data prompted speculation that prolonged (g12 hours) bouts of induced hypothermia may alter the subacute or evolving molecular events influencing ischemic neuronal death (Angelos et al., 2001).
Because most metabolic processes slow with hypothermia, induced resuscitative hypothermia may inhibit deleterious biochemical or cerebrovascular events during reperfusion (Dietrich et al., 1993). However, it also is possible that induced hypothermia increases restorative or neuronal survival-promoting processes. For example, one group of signal transduction pathways implicated in neuronal death and survival is the mitogen-activated protein kinase (MAPK) pathways. This class of kinases includes the p42/p44 MAPK, also known as the extracellularly regulated kinase ERK. In rat asphyxial cardiac arrest, a regimen of hypothermia that decreases hippocampal neuronal death increases ERK activation in the hippocampus (Hicks et al., 2000b). In certain systems, ERK activation is triggered by trophic factors and is associated with increased neuronal survival (Han and Holtzman, 2000). Thus, increased ERK signaling is one possible beneficial effect of hypothermia.
Neurotrophic factors also promote the development and survival of neurons in the central and peripheral nervous system. The neurotrophins consists of a family of structurally similar trophic factors including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3, and neurotrophin 4/5. In the brain, neurotrophins colocalize with their receptors, the tyrosine receptor kinases (Trk) (Kokaia et al., 1993). BDNF in particular binds to and is localized with the TrkB receptor (Kokaia et al., 1993; Squinto et al., 1991; Soppet et al., 1991; Klein et al., 1991). Brain-derived neurotrophic factor binding to TrkB receptors can activate the Raf-MEK-ERK signaling pathway (Yuen and Mobley, 1999), and can improve survival of neurons after various insults (Kiprianova et al., 1999; Schabitz et al., 1997). These observations prompted the hypothesis that hypothermia induced after resuscitation from cardiac arrest might increase activity of one or more neuronal trophic factors
Prior work has demonstrated that neurotrophin administration can improve brain recovery after ischemia (Kiprianova et al., 1999). Because hypothermia increases ERK activation, and ERK can be activated by neurotrophin receptors, increased neurotrophin activity is another possible mechanism for the beneficial effects of reduced brain temperature. Therefore, this study examined whether hypothermia during reperfusion altered neurotrophin levels and Trk receptor activation in the rat brain.
MATERIALS AND METHODS
Subjects and experimental design
Animal protocols were approved by the University of Pittsburgh Animal Care and Use Committee. Male Sprague-Dawley rats (Harlan Sprague-Dawley, Inc., Indianapolis, IN, U.S.A.) (N = 32) were housed individually with free access to food and water. The first 24 rats were randomly assigned to three treatment groups: sham (n = 4), postresuscitative normothermia (n = 10) and postresuscitative hypothermia (n = 10). Sham rats were not subjected to asphyxia and were kept in normothermic (brain temperature 37°C) conditions until they were killed. In the remaining rats, global cerebral ischemia was induced by 8 minutes of asphyxial cardiac arrest followed by resuscitation (Katz et al., 1995). For all rats, brain temperature was maintained at 37°C during asphyxia. Rats in the normothermia group were maintained at 37°C after asphyxia until killing 12 or 24 hours after reperfusion. Rats in the hypothermia group were initially maintained at 37°C after resuscitation and were then cooled to 33°C at 60 minutes after resuscitation. Hypothermic rats were maintained at 33°C until killing after 12 or 24 hours. This regimen of induced hypothermia reduces behavioral and histologic damage after this insult (Hicks et al., 2000a). The remaining eight rats underwent anesthesia and surgical preparation, but were not subjected to asphyxia but were maintained at normal temperature (37°C) (n = 4) or hypothermia (33°C) (n = 4) for 24 hours.
Surgical procedures
Surgical procedures were identical to prior studies (Hicks et al., 2000a, b). At least 3 days before asphyxia, each rat was anesthetized with 1.5% halothane in oxygen, and a 4.5 mm × 20-gauge stainless steel cannula was stereotactically placed over the parietal cortex (AP-2.0 mm, ML +2.0 mm relative to bregma, according to the atlas of Paxinos and Watson [1997]), and secured with dental cement and skull screws. Battery-operated, wireless temperature probes (XH-FM-BP, MiniMitter, Sun River, OR, U.S.A.) that had been calibrated against a mercury thermometer were inserted into the guide cannula before asphyxia. The length and depth of the guide cannula was adjusted so that the tip of the temperature probe was adjacent to the cortex. The temperature at the tip of the brain probe was transmitted to an FM receiver and recorded every 3 seconds by a PC-compatible computer using commercial software (Vital View; MiniMitter), allowing continuous temperature monitoring. Temperature was regulated via software-driven relays connected to a 100-W heating lamp and a cooling fan (Colbourne et al., 1996; Hicks et al., 2000a).
Global cerebral ischemia was induced by asphyxial cardiac arrest as described previously (Katz et al., 1995; Katz et al., 1998). Anesthesia was induced with 3.0% halothane in oxygen, which was then titrated down to 0.8% halothane during the course of surgery. Rats were then orotracheally intubated with a 14-gauge intravenous catheter, and mechanically ventilated at a tidal volume of 9 mL/kg, 40 respirations/min and a positive end-expiratory pressure of 5 cm H2O (Harvard Rodent Ventilator; Harvard Apparatus, South Natick, MA, U.S.A.). The left femoral vein and artery were exposed via an incision and cannulated with polyethylene catheters (PE-50 tubing; Harvard Apparatus). The arterial catheter was used for continuous arterial blood pressure recording and blood gas analysis (I-STAT; Sensor Devices, Waukesha, WI, U.S.A.). Ventilation was adjusted to maintain eucapnia (Pco2= 32–45 mm Hg) and normal pH before asphyxia.
After reducing the fraction of inspired oxygen to 0.21 (room air) for 2 minutes, rats were chemically paralyzed with intravenous vecuronium (2 mg/kg), and the halothane was discontinued. Asphyxia was induced by disconnecting the ventilator at end-expiration for 8 minutes. Rats that were not paralyzed did not regain consciousness or move spontaneously during similar washout periods. Complete circulatory arrest defined by a decrease of central arterial blood pressure to equal central venous pressure occurred reliably within 180 seconds after onset of asphyxia. This timing results in 5 minutes of complete ischemia. After 8 minutes, the ventilator was reconnected, and ventilation was resumed with oxygen at a rate of 60 respirations/min. Intravenous epinephrine (0.005 mg/kg) and bicarbonate (1.0 mEq/kg) were administered, and external chest compressions were performed at a rate of 200 compressions/min. All rats had return of spontaneous circulation within 2 minutes. After stabilization for at least 60 minutes and confirmation of adequate spontaneous respirations, rats were extubated and weaned from oxygen to room air.
Hypothermia was induced using a cooling fan beginning at 50 minutes after reperfusion. The rats were misted with water during the induction of hypothermia, allowing the target temperature of 33°C to be reached within 10 to 15 minutes (at 60 minutes after resuscitation) (Fig. 1). In rats resuscitated from cardiac arrest, hypothermia could be maintained thereafter using just a cooling fan, and normothermia could be maintained using the computer-controlled heating lamp. Post—cardiac arrest rats exhibit spontaneous poikilothermia and will become spontaneously hypothermic if ambient temperature is not well controlled (Hickey et al., 2000). Resuscitated rats were comatose at the time that hypothermia was induced, and required no sedation or anesthesia to induce hypothermia. In contrast, the normothermic and hypothermic control rats were anesthetized with halothane at the beginning of their temperature control to reduce the potential stress and difficulty of inducing hypothermia in the uninjured awake rat as well as to control for any anesthetic effects. Sham rats not recovering from asphyxia required frequent misting of their fur with water to maintain the reduced cranial temperature. In contrast to rats subjected to cardiac arrest, sham rats were awake after anesthesia was discontinued. These rats did not tolerate indwelling catheters, and we did not attempt to perform chronic implants. Therefore, hemodynamic variables and blood gases are not available for sham rats.
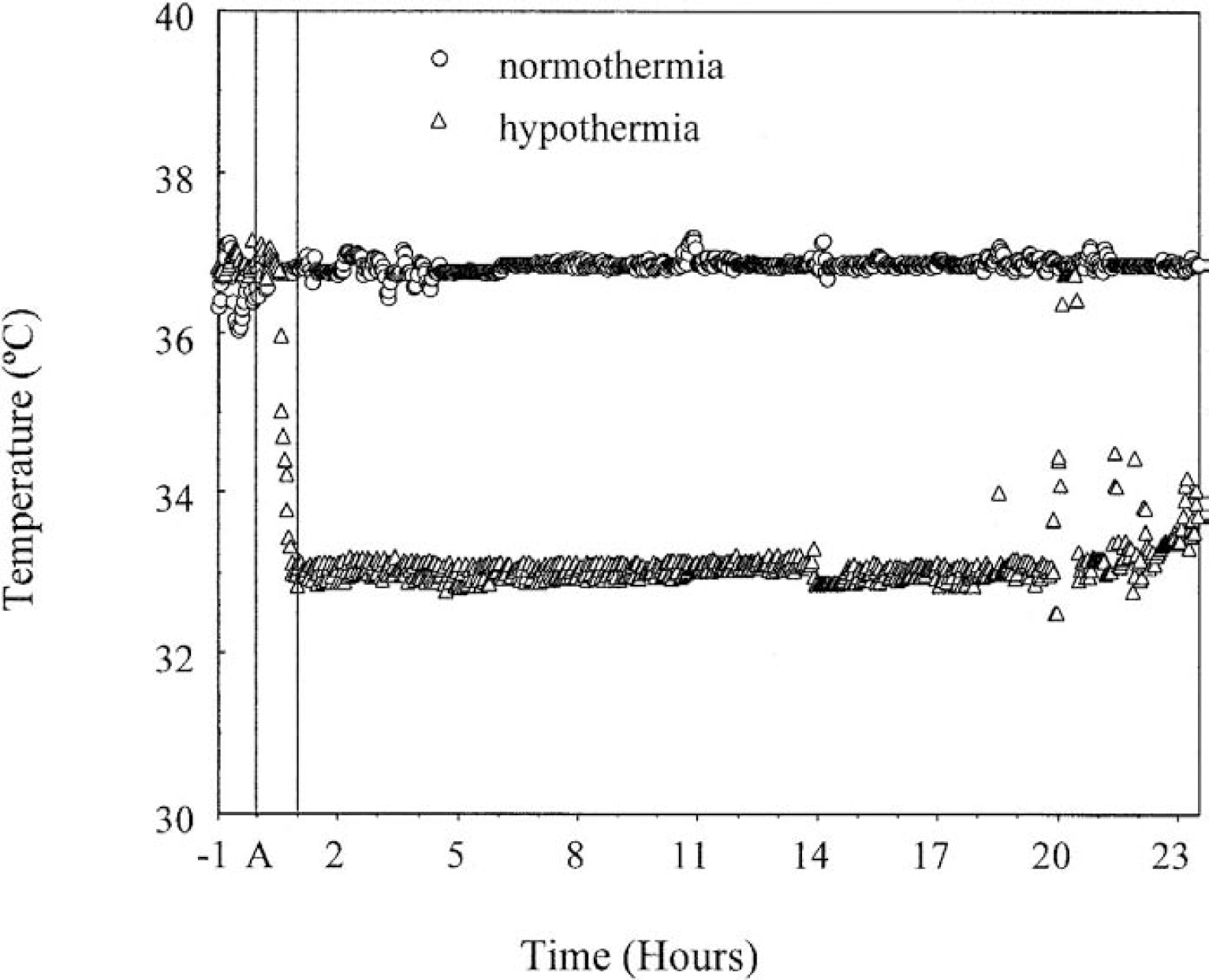
Example of temperature measurements from brain probes in separate rats subjected to normothermia or hypothermia regimens. Temperature was recorded every 3 seconds and controlled by heating lamps and cooling fans. This graph depicts average temperature for each minute. During the 1 hour before asphyxia
Immunoblotting
After 12 or 24 hours, rats were anesthetized with halothane and decapitated. Brains were dissected into chilled phosphate buffered saline (PBS) (0.01 mol/L, pH 7.4), for 1 minute. The hippocampus was dissected onto a cold metal stage and then frozen at −70°C. Frozen tissue was solubilized by sonication for 5 seconds on ice in 0.5 mL of buffer (50 mmol/L Tris-Cl, 100 mmol/L NaCl, 5 mmol/L EDTA, pH 7.4) containing detergent (1.0% Nonidet P-40; Sigma, St. Louis, MO, U.S.A.), protease inhibitors (100 mmol/L phenylmethylsulfonyl fluoride, 1 μg/mL aprotinin, 5 μg/mL leupeptin, 1 μg/mL pepstatin) and phosphatase inhibitors (1 mmol/L Na3VO4, 1 mmol/L NaF, and 1 μg/ml LR microcystin). Similar extractions have been used to quantify neurotrophins such as BDNF from tissue in prior studies (Nanda and Mack, 2000). After centrifugation of this lysate at 12,000 g for 15 minutes, the supernatant was collected and stored at −70°C. Protein concentration was determined by the Bradford method using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, U.S.A.).
Equal aliquots of protein (5 to 20 μg) were denatured by 5 minutes of boiling in sodium dodecyl sulfate sample buffer and separated by electrophoresis in 7.5% polyacrylamide gels containing sodium dodecyl sulfate. TrkB assays were performed in 15% gels. Protein was electrophoretically transferred to polyvinyl difluoride membranes (Immobilon-P; Millipore Corp., Bedford, MA, U.S.A.). Membranes were incubated for 1 hour with 5% dry milk in PBS with 0.5% Tween-20 (PBS-T) before overnight incubation at 4°C with primary antibody in 1:500 to 1,000 dilution. Primary antibodies included anti-BDNF (N-20), anti-TrkB (H-181), anti-TrkB(TK-) (C-13) (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), anti-NGF (Chemicon, Temecula, CA, U.S.A.), anti-p44/p42 MAP kinase, and anti-phospho-p44/42 MAP kinase (Thr202/Tyr204) (New England Biolabs, Beverly, MA, U.S.A.). Results for BDNF were confirmed with two other anti-BDNF antibodies (Sigma, St. Louis, MO, U.S.A.; Chemicon).
A horseradish peroxidase—conjugated species-specific anti-IgG secondary antibody with 5% dry milk in PBS-T was used in 1:5,000 dilution to label the primary antibody, and the resulting complexes were visualized by enhanced chemiluminescence using a commercial kit (Renaissance; New England Nuclear, Boston, MA, U.S.A.). The labeled membranes were exposed to x-ray film (X-OMAT; Kodak, Rochester, NY, U.S.A.) for 5 to 60 minutes. The resulting images were scanned and quantified using NIH Image software.
The molecular weights of the proteins to be quantified were determined by comparison with a lane loaded with protein molecular weight markers (Full Range Rainbow; Amersham Life Science, Little Chalfont, Buckinghamshire, U.K.). Equal loading of lanes was confirmed for immunoblots by stripping each membrane and reprobing with antibody against total p44/p42 MAPK or actin. In previous studies, total p42/p44 MAPK levels did not vary at these time points after cardiac arrest or hypothermia (Hicks et al., 2000b), and this antibody was used primarily to confirm equal lane loading. In all instances, total protein levels did not differ significantly between lanes, and uncorrected densitometry for the original antibody was used for data analysis. A linear relation between protein concentration and densitometry was confirmed within gels by loading different volumes of the same sample in separate lanes. Exposure times and concentrations were adjusted empirically to achieve a linear relation. All samples were run in duplicate, and results were confirmed by separate duplicate experiments.
Immunoprecipitation
Phosphorylation of TrkB receptors was assessed by immunoprecipitation. Hippocampal extracts (100 μg) were incubated with 1 μg anti-TrkB (H-181; Santa Cruz Biotechnology) in PBS-T with protease inhibitors (1 μg/mL aprotinin, 5 μg/mL leupeptin, 1 μg/mL pepstatin) and phosphatase inhibitors (1 mmol/L Na3VO4, 1 mmol/L NaF) for 90 minutes. Immune complexes were precipitated by incubation with Protein A/G-agarose (Santa Cruz), followed by centrifugation at 12,000 g for 1 minute. After washing in the same buffer, precipitates were solubilized by boiling in sodium dodecyl sulfate sample buffer. The resulting samples were separated on 7.5% polyacrylamide gels containing sodium dodecyl sulfate, and blotted onto PVDF membranes. These membranes were probed with a 1:1,000 dilution of a horseradish peroxidase—linked antibody to phosphotyrosine (PY20; Santa Cruz) according to the protocols provided by the supplier, and the resulting labeling was visualized by enhanced chemiluminescence as mentioned earlier. Because the degree to which other proteins coprecipitate with TrkB is unknown, immunoprecipitation experiments were assumed to proceed with equal efficiency.
Statistics
Initial analyses were performed in 20 rats, and the findings were then replicated in a completely separate series of rats (N = 32, total as described herein). Because results of these series did not differ, and observed protein levels in shams from both series were identical, all data are presented in aggregate. Physiologic variables were compared between groups by repeated-measures analysis of variance with group as the between-subject factor and time as the within-subject factor. One- or two-factor analysis of variance was used to compare protein levels between groups at different time points. Data violating the sphericity assumptions for analysis of variance were examined using Huynh-Feldt and Greenhouse-Geisser corrections. Post hoc independent-sample t-tests were used to test for differences between hypothermia and normothermia groups at each time point, with a Bonferroni correction for multiple t-tests, and when necessary a degree of freedom correction for unequal variances. An alpha error rate of 0.05 was taken as the criterion for significance for all tests. All data are presented as mean ± SD.
RESULTS
A total of 24 rats were subjected to asphyxial cardiac arrest or sham treatment. An additional 8 rats underwent sham operation without asphyxia followed by normothermia or hypothermia for 24 hours. Initial hemodynamic variables, blood gas values, and serum chemistries did not differ between normothermic and hypothermic groups (Table 1). Asphyxia produced a transient metabolic acidosis at 10 minutes that resolved by 30 minutes (time: F3,18 = 6.64, P = 0.003). Blood gas values were not corrected for the temperature of the animal. Consequently, the hypothermia group exhibited an apparent elevation in pco2 levels (group × time: F3,18 = 5.56, P = 0.007) and a lower pH (group × time: F3,18 = 3.97, P = 0.025) relative to the normothermia group at 60 minutes (Table 1). Likewise, induction of hypothermia was associated with an increase in mean arterial blood pressure at this time point (group: F1,6 = 10.35, P = 0.018). Other hemodynamic and acid—base measures were not significantly different between normothermic and hypothermic groups.
Hemodynamic variables and blood gases measured in all rats subjected to asphyxia at baseline and 10, 30, and 60 minutes after resuscitation
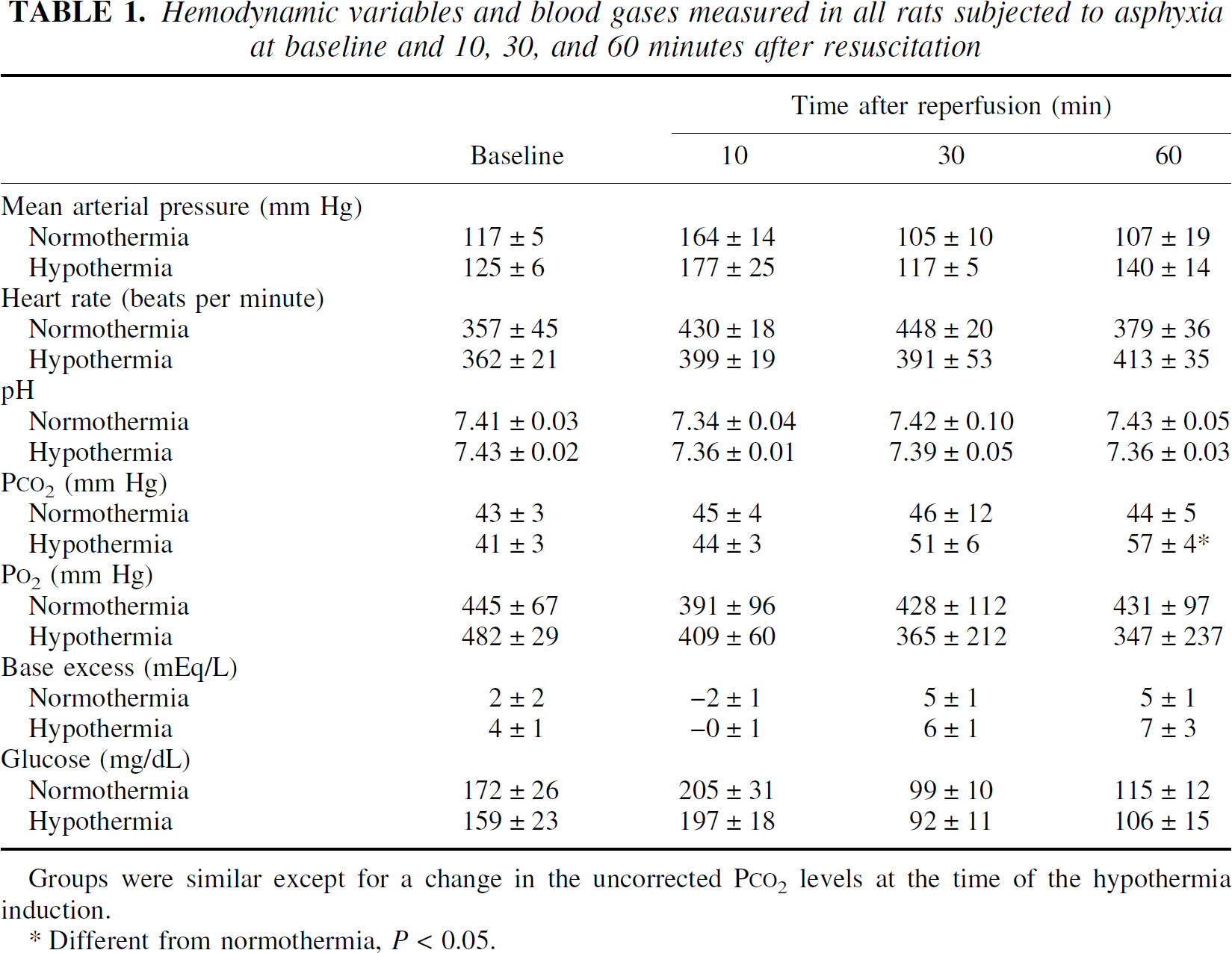
Groups were similar except for a change in the uncorrected P
Different from normothermia, P < 0.05.
Brain-derived neurotrophic factor
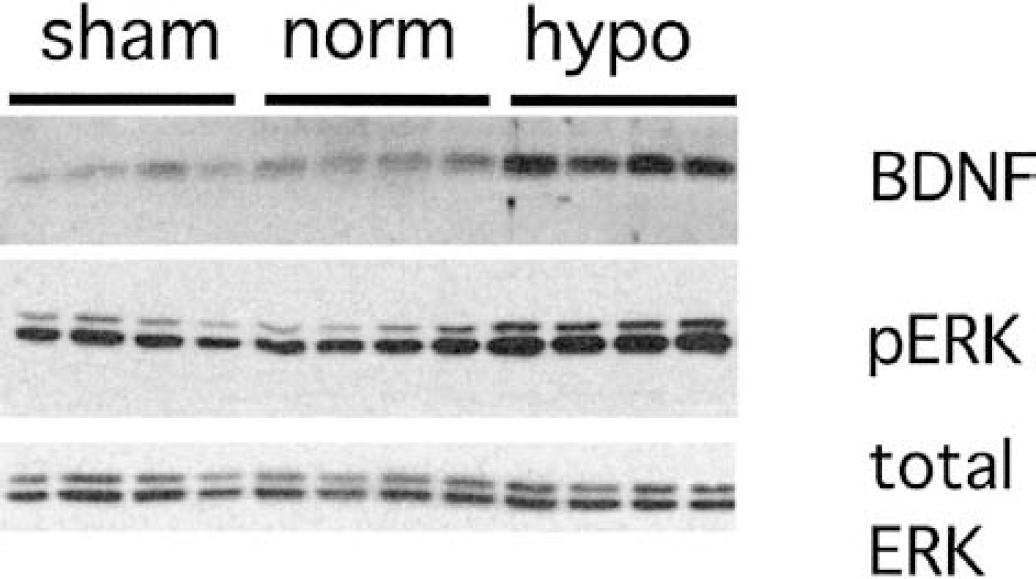
Immunoblotting detected a protein in tissue extracts that was approximately 15 kd in weight and corresponded to BDNF (Fig. 2). Separate experiments using recombinant human BDNF as a positive control identified the same band (data not shown). The antibodies obtained from all vendors labeled several bands at higher molecular weights in tissue extracts and were not suitable for immunohistochemistry.
Immunoblots of BDNF, phosphorylated ERK (P-ERK), and total ERK in rat hippocampal extracts 24 hours after resuscitation from asphyxial cardiac arrest. Each lane contains protein from a separate rat. Samples from rats that had no asphyxia (sham) are shown along with samples from rats subjected to asphyxial cardiac arrest and reperfusion at 37°C (norm) or reperfusion at 33°C (hypo). Hypothermia during reperfusion increased BDNF and activated (phosphorylated) ERK levels. Total protein concentration as reflected by total ERK did not change.
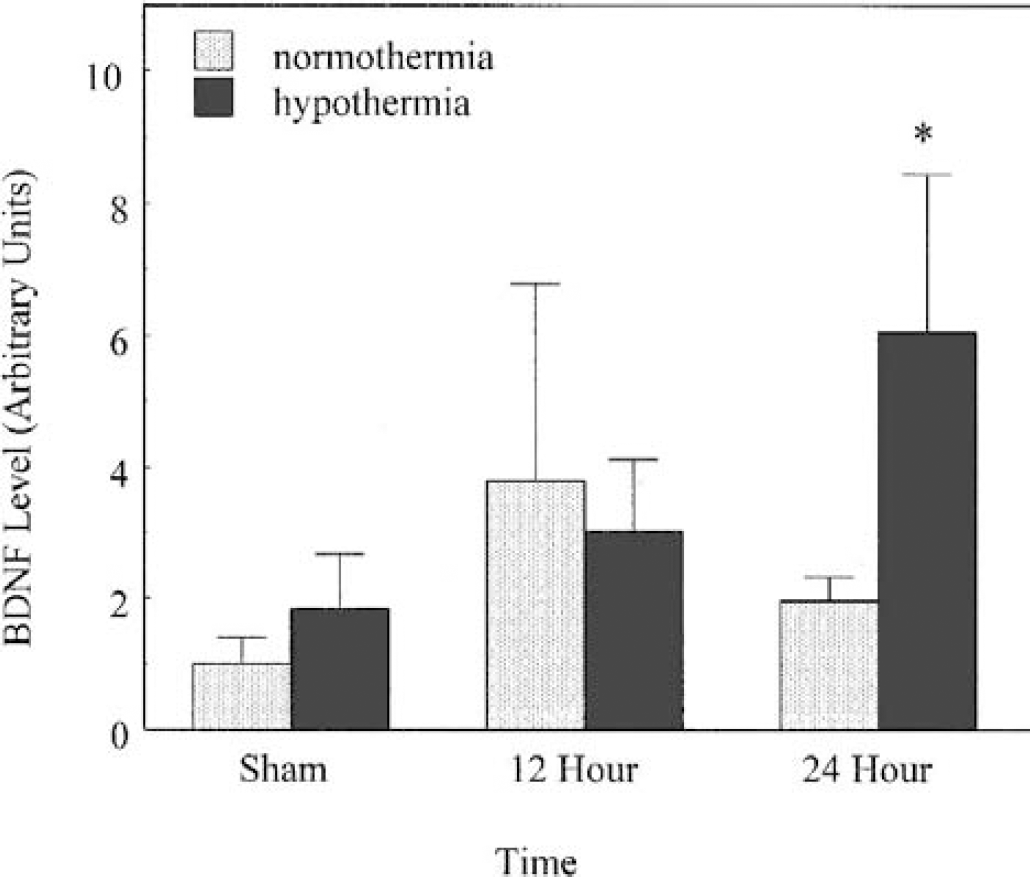
There were significant effects of time (F2,27 = 6.47, P = 0.006) and of temperature group (F1, 27 = 11.70 P = 0.002) on BDNF levels. The main effect of time reflects the fact that BDNF levels increased after ischemia in both normothermia and hypothermia-treated rats relative to sham-operated rats (Fig. 3). The main effect of temperature group reflects the fact that BDNF levels after 24 hours of reperfusion were significantly greater in the hypothermia rats than that in the normothermia rats (t12 = 4.42, P = 0.004). Induction of hypothermia in sham rats did not increase BDNF levels significantly relative to normothermia sham rats (t6 = 1.78, NS). The 95% confidence interval for the observed difference between mean BDNF levels for hypothermic and normothermic sham rats was (−44%, +200%). Likewise, there was no difference between BDNF levels after 12 hours of reperfusion in normothermia and hypothermia rats (t4 = 0.4, NS). This time-dependent effect of hypothermic reperfusion was reflected by a significant interaction between time and temperature group on BDNF levels (F2,27 = 5.48, P = 0.012).
Hypothermia increased BDNF immunoreactivity after 24 hours of reperfusion. Sham rats were subjected to anesthesia, surgery, and 24 hours of temperature regulation, but no asphyxia. After 12 hours of reperfusion, BDNF levels were increased in both groups relative to shams. (*Significantly different from normothermia group, P h 0.05).
Nerve growth factor
Immunoblotting detected proteins at approximately 15 kd corresponding to NGF (Fig. 4). Levels of NGF did not rise in normothermia or hypothermia-treated rats as compared with sham-operated animals (F2,11 =1.542, P = 0.265).
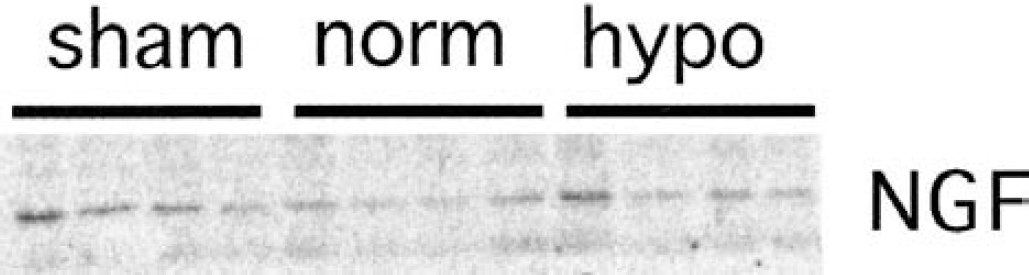
Immunoblot of NGF in rat hippocampus 24 hours after resuscitation. Ischemia and reperfusion did not alter NGF levels in any group. Abbreviations are as in Fig. 2.
ERK activation
The phosphorylated p42/p44 MAPK antibody detected activated ERK-1 (44 kd) and ERK-2 (42 kd) (Fig. 2). Levels of active ERK-2 were increased after ischemia. Less intense labeling of active ERK-1 was noted, although this kinase also increased after ischemia. Hippocampal ERK activation was greater in rats subjected to hypothermia than in rats subjected to normothermia (Fig. 2) (F2,11 = 20.44, P h 0.001).
After being probed for a neurotrophin or active ERK, membranes were stripped and reprobed for total ERK using the phosphorylation-insensitive p42/p44 MAPK antibody. In all conditions, total ERK concentration did not differ between samples (Fig. 2) confirming equal protein loading in all lanes.
TrkB receptors
Immunoblotting detected proteins corresponding to the 145-kd TrkB receptor. In addition, the TrkB(TK-) antibody detected a 95-kd protein corresponding to the truncated TrkB receptor. Levels of TrkB receptors increased in the hypothermia group (F2,11 = 21.9, P h 0.001) (Fig. 5). TrkB(TK-) receptor levels were also increased by hypothermia (F2,11 = 4.34, P = 0.048) (Fig. 5). Moreover, tyrosine phosphorylation of the TrkB receptor as assessed by immunoprecipitation was increased in the ischemia and hypothermia groups, although the measured phosphorylation was highly variable (F2,11 = 1.58, P = 0.259) (Fig. 6).
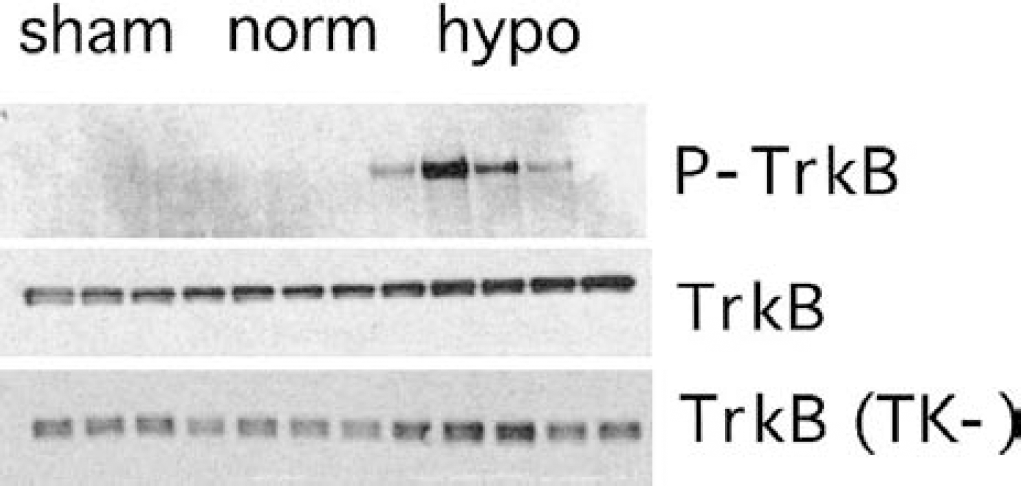
Immunoblots of TrkB receptor in hippocampus after 24 hours of reperfusion. Tyrosine phosphorylated TrkB (P-TrkB) was detected by immunoprecipitation with anti-TrkB and immunoblotting with antiphosphotyrosine. P-TrkB levels were elevated in several of the rats with hypothermic reperfusion. Total TrkB receptor and tyrosine kinase-deficient TrkB receptor (TrkB(TK-)) were detected in tissue extract by immunoblotting with their respective antibodies. Other abbreviations are as in Fig. 2.
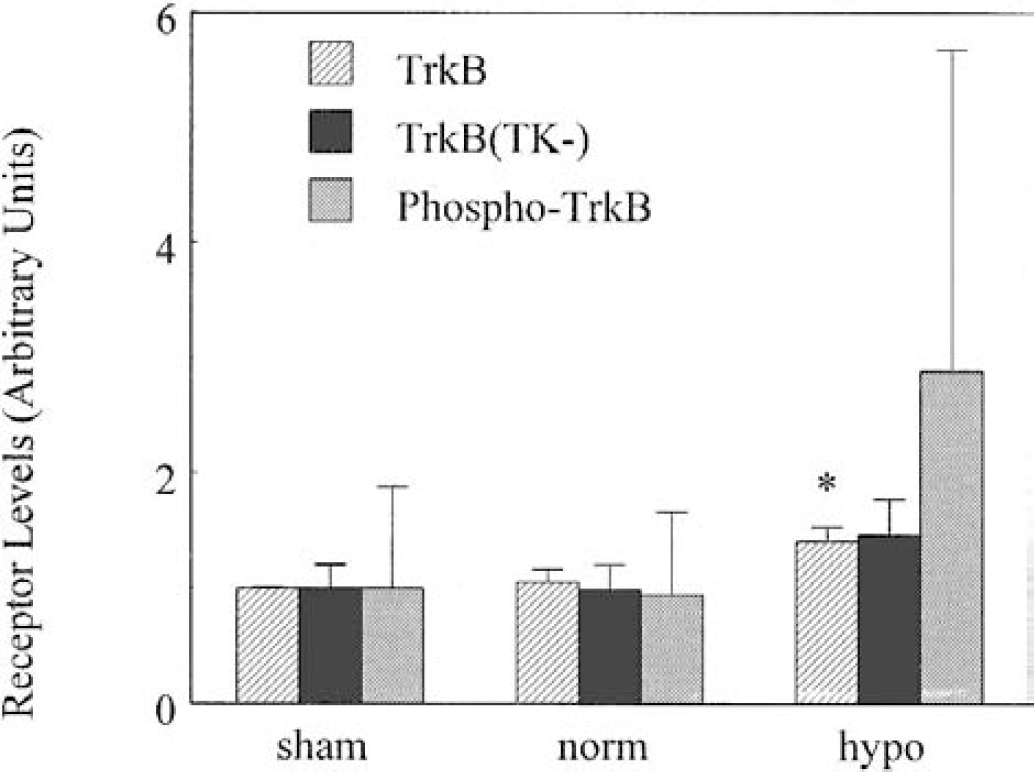
The total levels of TrkB and the tyrosine kinase deficient TrkB(TK-) receptor were increased at 24 hours after ischemia and reperfusion in the hypothermia group. Tyrosine-phosphorylated (active) TrkB was also increased after hypothermia reperfusion. However, the increases in TrkB(TK-) and phospho-TrkB were not statistically significant. (*Significantly different from normothermia group, P h 0.05).
DISCUSSION
These data demonstrate that induced hypothermia increases activity of at least one neurotrophic factor (BDNF) at later time points after reperfusion from cardiac arrest. This effect is specific because NGF levels were not affected. The fact that more than 12 hours were required to see significant differences between the levels of BDNF in normothermic and hypothermic rats supports the idea that this effect is triggered by prolonged but not by brief hypothermia. Furthermore, hypothermia potentiates both phosphorylation of the BDNF receptor, TrkB, and activation of ERK, an intracellular signaling pathway that has been associated with activation of the BDNF receptor. These data suggest that increased trophic factor release is a potential restorative process triggered in the brain after ischemic injury, and that enhancement of this response is a potential mechanism for the beneficial effects of prolonged hypothermia during reperfusion.
Prior studies indicated that ischemia alters the expression of NGF and BDNF in brain. In particular, brain ischemia increases BDNF mRNA (Merlio et al., 1993), immunoreactivity (Yamasaki et al., 1998; Lee et al., 1998), and TrkB levels (Merlio et al., 1993). Furthermore, increased BDNF levels have been measured in the cerebrospinal fluid of children who had asphyxia (Korhonen et al., 1998). Hypothermia induced during cerebral ischemia enhances the increase in BDNF mRNA levels in ischemia-resistant areas of the brain (Boris-Moller et al., 1998). Taken together with the present data, these observations suggest that hypothermia induced after reperfusion increases BDNF activity, a potential neuron survival-promoting pathway.
The timing of postischemic hypothermia influences its efficacy. Induction of hypothermia during ischemia is optimal (Dietrich et al., 1993; Kuboyama et al., 1993; Tadler et al., 1998), but not practically achievable in most clinical settings (Bernard et al., 1997; Zeiner et al., 2000; Callaway et al., 2002). Brief periods of hypothermia (less than 3 hours) must be instituted immediately after ischemia to have any effect, and this method does not block later damaging events (Kuboyama et al., 1993; Dietrich et al., 1993). In contrast, prolonged hypothermia during reperfusion after forebrain ischemia is extremely effective in preventing neuronal damage, even if instituted several hours after ischemia (Coimbra and Wieloch, 1994; Colbourne et al., 1999; Hicks et al., 2000a) and appears to be long lasting (Colbourne et al., 1999).
The fact that the therapeutic window for initiating postischemic hypothermia is dependent on the duration of hypothermia effects suggests that different mechanisms mediate the effects of brief and prolonged hypothermia. For example, induced hypothermia inhibits damaging free radical production (Dietrich et al., 1993) and excitatory amino acid release (Ooboshi et al., 2000). However, oxidative stress and excitotoxic amino acid release occur during early reperfusion after cerebral ischemia (Globus et al., 1991; Katz et al., 1998), and this effect cannot explain the beneficial effects of hypothermia induced hours after reperfusion. It is possible that prolonged hypothermia augments restorative processes rather than just slowing down harmful events. For example, hypothermia selectively potentiates the late activation of the extracellular signal-regulated kinase (ERK or p42/p44 MAPK) (Hicks et al., 2000b). ERK activation is associated with neuronal survival in several models (Han and Holzman, 2000), although the mechanism whereby ischemia and hypothermia influence this intracellular signaling pathway is unknown.
Exogenous BDNF can promote neuronal survival after ischemia. For example, intracerebroventricular infusion of BDNF can reduce neuronal injury after both focal and global ischemia (Schabitz et al., 1997; Kiprianova et al., 1999). Conversely, inhibition of BDNF activity by continuous intraventricular infusion of antibody-TrkB receptor conjugates worsens recovery from forebrain ischemia (Larsson et al., 1999). Thus, the increased activation of BDNF by postischemic hypothermia is consistent with the increased neuronal survival observed with this intervention.
Increased BDNF levels may be related to the prolonged increase in ERK activity after ischemia as well as its augmentation by hypothermia (Hicks et al., 2000b). BDNF exerts its effects via cell membrane—bound TrkB receptors (Squinto et al., 1991; Soppet et al., 1991; Klein et al., 1991), which are linked to intracellular MAP kinase cascades (Hetman et al., 1999). On BDNF binding, Trk receptors dimerize and become phosphorylated at tyrosine residues on their cytoplasmic domain (Jing et al., 1992).
This study did not attempt to define the anatomic distribution of BDNF within the hippocampus. However, prior studies indicate that the anatomic distribution of BDNF and TrkB immunoreactivity in the hippocampus parallels the distribution of active ERK after ischemia. In particular, TrkB immunoreactivity is localized to the dendritic fields of the pyramidal neurons (Lee et al., 1998), where ERK activation is most apparent after ischemia (Hicks et al., 2000b). Although BDNF immunoreactivity has been noted in both neuronal bodies and dendritic fields, immunohistochemistry cannot identify its site of release (Dugich-Djordjevic et al., 1995; Iritani et al., 2000).
In the present study, NGF and BDNF were differentially affected by hypothermia. Consistent with this difference, the intracellular processing of NGF and BDNF differs in cultured hippocampal neurons (Mowla et al., 1999). In particular, BDNF is localized into vesicles usually involved in regulated secretion, whereas NGF is processed in the Golgi apparatus along a pathway usually resulting in constitutive secretion (Farhadi et al., 2000). Consistent with the present results, induction of hypothermia after brain trauma does not increase and may decrease NGF levels (Goss et al., 1995). Although trauma can increase BDNF mRNA in brain (Pringle et al., 1996), the effects of hypothermia after brain trauma on BDNF are not known. This differential regulation suggests distinct physiologic roles for these neurotrophins in response to ischemic brain injury.
The present study, along with prior studies using the same model (Hicks et al., 2000a, b), demonstrates a correlation between BDNF levels, ERK activation, and improvement in neurologic outcome. However, these studies do not establish a causal connection. The contribution of BDNF to the effects of hypothermia could be examined by blocking BDNF synthesis or action at its receptor, while observing both functional recovery and activation of intracellular MAPK. Infusion of antibodies or antisense oligonucleotides are possible approaches to this question. Conversely, future studies could also examine whether infusion of BDNF is sufficient to increase ERK activation.
It is possible that improved neurologic recovery and increased BDNF levels are both secondary to other effects of hypothermia such as increased blood pressure. Although the blood pressure was elevated in the hypothermic rats, the absolute magnitude of the increase was small (Table 1). Furthermore, we have conducted experiments on separate rats subjected to a similar regimen in which arterial catheters were left in place for prolonged monitoring (Hicks et al. 2000a). In those studies, the initial elevation of blood pressure observed in hypothermic rats at 60 minutes was not evident at 12 or 24 hours. Those data suggest that the difference in blood pressure was not sustained during prolonged hypothermia. Because brief hypothermia is less effective for reducing brain injury, and BDNF levels continued to increase during 24 hours (Fig. 2), we doubt that this change in systemic hemodynamics is related to the differences in outcome between groups. A contribution of other variables that were not measured cannot be excluded.
Clinically, a better understanding of the mechanisms promoting neuronal recovery after global cerebral ischemia could lead to advances in reducing neurologic damage for those patients surviving the cardiopulmonary crisis after cardiac arrest, drowning, or severe shock (Callaway, 1997). Currently, no routine therapy is directed at improving neurologic recovery after resuscitation, although induction of prolonged hypothermia after resuscitation from cardiac arrest or other ischemia is feasible for humans using current technology (Bernard et al., 1997; Zeiner et al., 2000). Mild hypothermia induced during reperfusion reduces the magnitude of brain damage after ischemia in animal models (Coimbra and Wiloch, 1994; Colbourne et al., 1999; Hicks et al., 2000a), and possibly in humans (Bernard et al., 1997; Zeiner et al., 2000). Induced hypothermia is also effective at reducing neuronal damage in a variety of other circumstances, including traumatic head injury (Marion et al., 1997).
In summary, induction of hypothermia augments the activity of the neurotrophin BDNF after global ischemia. This effect is specific because NGF is not affected. Increased intracellular ERK signaling is associated with this increase in BDNF levels. Even if other processes contribute to the beneficial effects of induced hypothermia, these data are significant in that they illustrate how a known restorative process can be specifically stimulated by a nonspecific physical manipulation.