
Abstract
The consequences of therapeutic hypothermia for neonatal hypoxic–ischemic encephalopathy are poorly understood. Adverse effects from suboptimal rewarming could diminish neuroprotection from hypothermia. Therefore, we tested whether rewarming is associated with apoptosis. Piglets underwent hypoxia–asphyxia followed by normothermic or hypothermic recovery at 2 hours. Hypothermic groups were divided into those with no rewarming, rewarming at 0.5 °C/hour, or rewarming at 4 °C/hour. Neurodegeneration at 29 hours was assessed by hematoxylin and eosin staining, TUNEL assay, and immunoblotting for cleaved caspase-3. Rewarmed piglets had more apoptosis in motor cortex than did those that remained hypothermic after hypoxia–asphyxia. Apoptosis in piriform cortex was greater in hypoxic–asphyxic, rewarmed piglets than in naive/sham piglets. Caspase-3 inhibitor suppressed apoptosis with rewarming. Rapidly rewarmed piglets had more caspase-3 cleavage in cerebral cortex than did piglets that remained hypothermic or piglets that were rewarmed slowly. We conclude that rewarming from therapeutic hypothermia can adversely affect the newborn brain by inducing apoptosis through caspase mechanisms.
INTRODUCTION
Neonatal hypoxic–ischemic encephalopathy (HIE) causes significant neurologic morbidity even with therapeutic hypothermia. 1 Indeed, the potential consequences of therapeutic hypothermia remain poorly understood despite its widespread clinical use.2–5 Although hypothermia is known to be neuroprotective in animal models,6,7 possible off-target actions of hypothermia, the effects of rewarming, and optimal rates of rewarming in developing brain require further evaluation.8,9
The mechanisms of therapeutic hypothermia are complex and may differentially affect the brain injury process during the acute, subacute, and delayed phases of HIE. In a neonatal piglet model of HIE, the brain regions that are selectively vulnerable to hypoxia–ischemia (HI) correspond to those in human term newborns. 10 HI causes cortical laminar necrosis in sensorimotor cortex, 11 and hypothermia decreases ischemic neuronal necrosis in this model. 12 In neonatal rodent models of HI, hypothermia attenuates apoptosis 13 and mitigates inflammation. 14 It is unclear whether hypothermia attenuates ischemic necrotic cellular degeneration or whether hypothermia delays or prevents cellular necrosis only to permit other forms of cell degeneration to emerge, as would be predicted by the concept of the cell-death continuum. 15 Alternatively, hypothermia might inhibit cell death in general and all its potential forms. Moreover, whether the rate of rewarming influences the efficacy of neuroprotection has not been fully studied in neonatal HI.
We evaluated these issues in a neonatal swine model of HIE and focused on two distinct regions of cerebral cortex: the neocortical motor cortex (six-layered region) and the allocortical piriform cortex (three-layered region). Differences in architecture, connectivity, and metabolism between these regions could result in differential vulnerability to HI. 16 In addition, vulnerability of the motor cortex to HI has been reported in human neonates. 17 We tested the hypotheses that (1) different cerebral cortical regions show differential vulnerability to HI and that hypothermia protects against ischemic vulnerability; (2) cortical injury after HI with hypothermia and rewarming is morphologically distinct from the ischemic necrosis that occurs after HI with normothermic recovery; (3) the rate of rewarming influences the severity and type of neurodegeneration; and (4) cortical injury after HI, hypothermia, and rewarming is caspase dependent.
MATERIALS AND METHODS
Animal Preparation
All procedures were approved by the Animal Care and Use Committee at Johns Hopkins University and complied with the United States Public Health Service Policy on the Humane Care and Use of Laboratory Animals and the Guide for the Care and Use of Laboratory Animals. Animal care was in accord with the National Institutes of Health Guidelines and ensured animal comfort. Male piglets (1 to 2.5 kg, 3 to 5 days old) underwent sham surgery or HI injury. Age-matched, male, naive piglets that did not receive anesthesia, surgery, or HI were used as an additional control group.
Anesthesia was induced with 5% isoflurane and 50%/50% nitrous oxide/oxygen. After intubation, the lungs were mechanically ventilated to maintain normocapnea. During insertion of catheters into the femoral artery and vein, anesthesia was maintained with 2% isoflurane and 70%/30% nitrous oxide/oxygen. Fentanyl was administered intravenously (20 μg/kg) after venous catheter placement. To determine which anesthetic regimen had the least potential neurotoxicity, we divided piglets without HI into two groups: one received 70%/30% nitrous oxide/oxygen+fentanyl (10 μg/kg to 20 μg/kg per hour, intravenously)+vecuronium (0.2 mg/kg per hour, intravenously), and the other received isoflurane 0.4%+70%/30% nitrous oxide/oxygen+fentanyl (10 μg/kg to 20 μg/kg per hour, intravenously)+vecuronium (0.2 mg/kg per hour, intravenously). 18 In piglets that were assigned to the no-isoflurane group, the isoflurane was discontinued after the femoral catheters were placed (Supplementary Figure 1). Placement of the femoral catheters took approximately 10 minutes. The anesthetic combination that caused the least apoptosis was identified and used in subsequent experiments of HI and temperature manipulation.
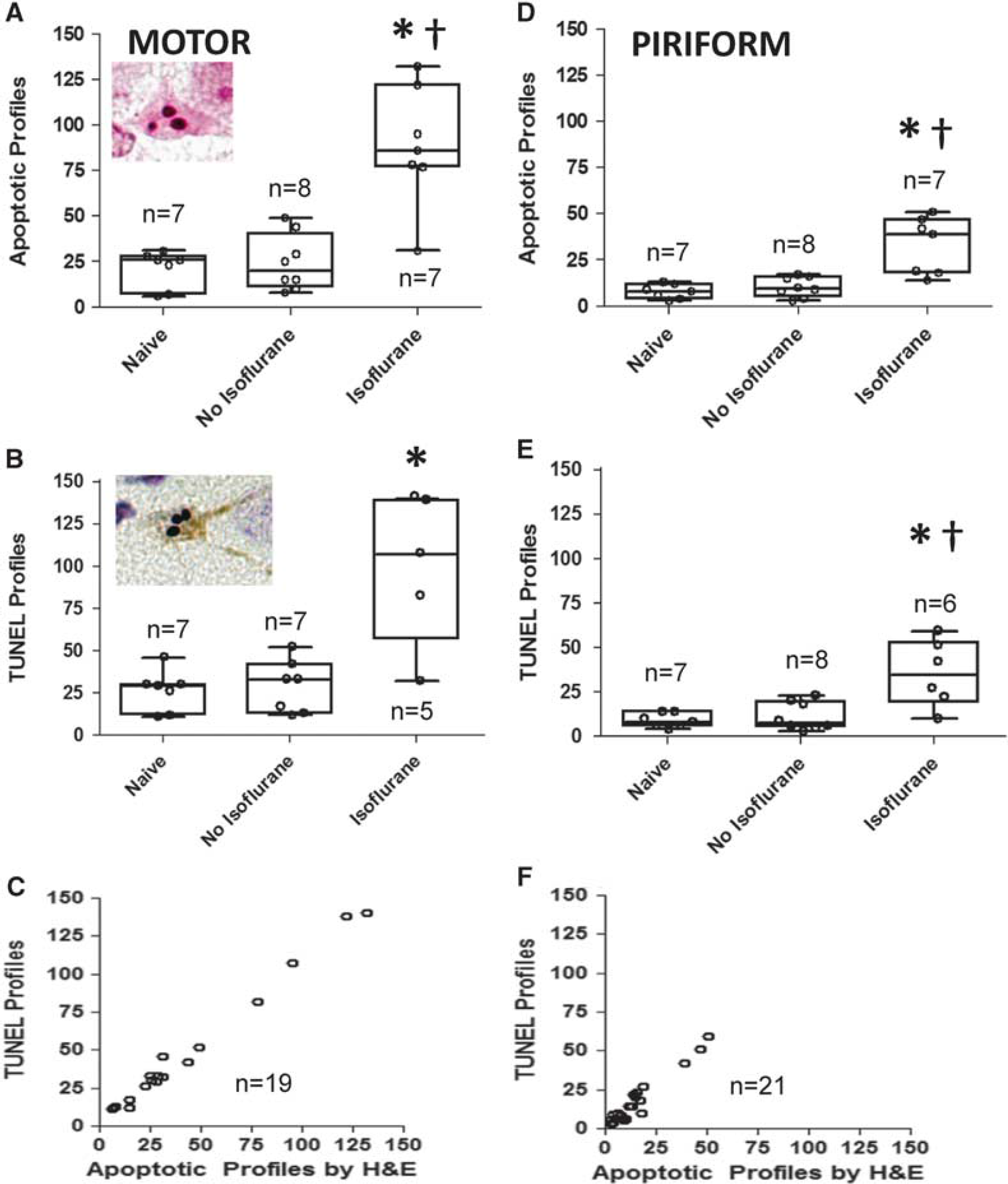
Identifying the anesthetic regimen with the least apoptosis in motor and piriform cortex. (
To prevent shivering during hypothermia and to ensure that all the groups received the same anesthetic regimen, we administered vecuronium (0.2 mg/kg per hour, intravenously) to all piglets. The piglets also received 5% dextrose in 0.45% saline at 10 ml/hour (intravenously). To ensure that the cerebral perfusion pressure exceeded the lower limit of autoregulation during anesthesia, we infused low-dose phenylephrine as needed to maintain mean arterial blood pressure (MAP) ≅ 45 mm Hg. 18
Hypoxic–Ischemic Injury
Injury severity in the piglet model of HI can be varied by modifying the duration of hypoxia. We have used 30 minutes6,10,12 to 45 minutes11,18,19 in previous studies with translational models of neuroprotection, including studies in cerebral blood flow autoregulation11,18,19 and hypothermia.6,12 In the current study, we used our more severe HI model, which utilizes 45 minutes of hypoxia, and added different rates of rewarming after a delayed onset of hypothermia.
We induced whole-body hypoxia by decreasing the inspired oxygen concentration to 10% for 45 minutes to reach an oxyhemoglobin saturation of 30% to 35% and then administered 21% inspired oxygen for 5 minutes. This brief reoxygenation period is required for cardiac resuscitation. We then occluded the endotracheal tube for 7 minutes to produce asphyxia. Piglets were resuscitated with 50% inspired oxygen, manual chest compressions, and epinephrine (100 μg/kg, intravenously).11,18 Piglets without return of spontaneous circulation (ROSC) within 3 minutes were excluded. The inspired oxygen was decreased to 30% after resuscitation. Sodium bicarbonate and calcium chloride were administered to correct metabolic acidosis and hypocalcemia. Sham-operated piglets received the same duration of anesthesia with 30% oxygen and normothermia, but without HI.
Temperature
Before inducing anesthesia, we randomized piglets to sham surgery or HI. Piglets destined to undergo HI were further randomized to four temperature groups: (1) normothermia (38.5 °C to 39.5 °C), (2) whole-body hypothermia (34.0 °C), (3) hypothermia+slow rewarming (0.5 °C/hour), or (4) hypothermia+rapid rewarming (4 °C/hour) until the goal sample size in each group was achieved. Normothermia was maintained with heating lamps and warming blankets. In groups destined to receive hypothermia, we delayed the induction of hypothermia by 2 hours to mimic clinical delays in inducing hypothermia.11,18,19 At 2 hours after ROSC, we induced whole-body hypothermia to a goal rectal temperature of 34 °C by using ice packs and cooling blankets.6,11,18,19 Rectal temperature decreases to 34 °C over ~30 minutes, and rectal temperature has been found to track brain temperature within 0.2 °C in this model. 6 Piglets in rewarming groups remained hypothermic for 18 hours. At 20 hours after ROSC, we initiated whole-body rewarming at 0.5 °C/hour or 4 °C/hour by turning off the cooling blanket, increasing the water temperature circulating through the warming blanket, and using whole-body warm packs until normothermia (38.5 °C) was reached. 18 All piglets were euthanized at 29 hours after ROSC or sham surgery (Supplementary Figure 2). Some piglets in the HI+rapid rewarming group received subdural artificial cerebrospinal fluid (aCSF) and were included in the experiments designed to test the efficacy of caspase-3 inhibitor in blocking apoptosis.
Representative images show histologic staining of tissue from the piglets that underwent the sham surgery or the hypoxia–ischemia injury (HI). Cortical layers 2 and 3 are shown in the motor gyrus. ‘I’ denotes layer 1. (
Caspase-3 Inhibitor
Piglets that received HI+hypothermia+rapid rewarming were randomized to receive either a subdural caspase-3 inhibitor or aCSF. To minimize the number of animals, piglets that received aCSF were also included in the HI+rapid rewarming group. During anesthesia with isoflurane 2%, nitrous oxide/oxygen 70%/30%, and fentanyl (20 μg/kg per hour, intravenously), PE10 catheters were threaded approximately 3 mm into the subdural space through small, cranial, bilateral burr holes. Catheter placement was verified by the return of CSF. Then the burr holes were sealed and the catheters secured. On completion of surgery, anesthesia was maintained by using the regimen identified to cause the least apoptosis. For example, if the anesthetic without isoflurane was identified as the regimen with the least apoptosis, then the isoflurane would be discontinued after placement of the subdural catheters. Placement of the subdural catheters took approximately 10 minutes. Therefore, piglets with subdural catheters would have approximately 10 minutes more of isoflurane exposure than those without subdural catheters had. In total, piglets that received subdural catheters received approximately 20 minutes of isoflurane (10 minutes for the placement of femoral catheters+10 minutes for placement of the subdural catheter), whereas piglets without subdural catheters received approximately 10 minutes of isoflurane. Then, piglets underwent HI and resuscitation. Beginning 15 minutes after ROSC and continuing every 30 minutes for the remainder of the experiment, we administered either 100 μg of the selective non-peptide caspase-3 inhibitor (50 μL; Tocris Bioscience, Bristol, UK; 2.5 mg/mL in 20% dimethyl sulfoxide (DMSO) and 80% aCSF) or an equivalent volume of 20% DMSO in aCSF into each subdural catheter bilaterally. This volume was substantially less than that required to increase the intracranial pressure in a previous piglet model of intracranial hypertension from our research group. 20 Because the optimal dosing for subdural caspase-3 inhibition in piglets is unknown, we used double the dose that has been found to protect against caspase-3-dependent apoptosis of cortical neurons in cell culture. 21 The induction of hypothermia was delayed by 2 hours, and rapid rewarming (4 °C/hour) was initiated at 20 hours after ROSC. Piglets were euthanized 29 hours after ROSC (Supplementary Figure 2).
Histologic Measurements
At 29 hours after HI or sham surgery, piglets were deeply anesthetized with 50 mg/kg pentobarbital and exsanguinated by transcardial perfusion with cold phosphate-buffered saline followed by 4% paraformaldehyde for brain fixation. The brains were removed, and forebrain slabs were embedded in paraffin and cut into 10-μm sections. Coronal sections were matched for anterior–posterior level as determined by anatomic regions.
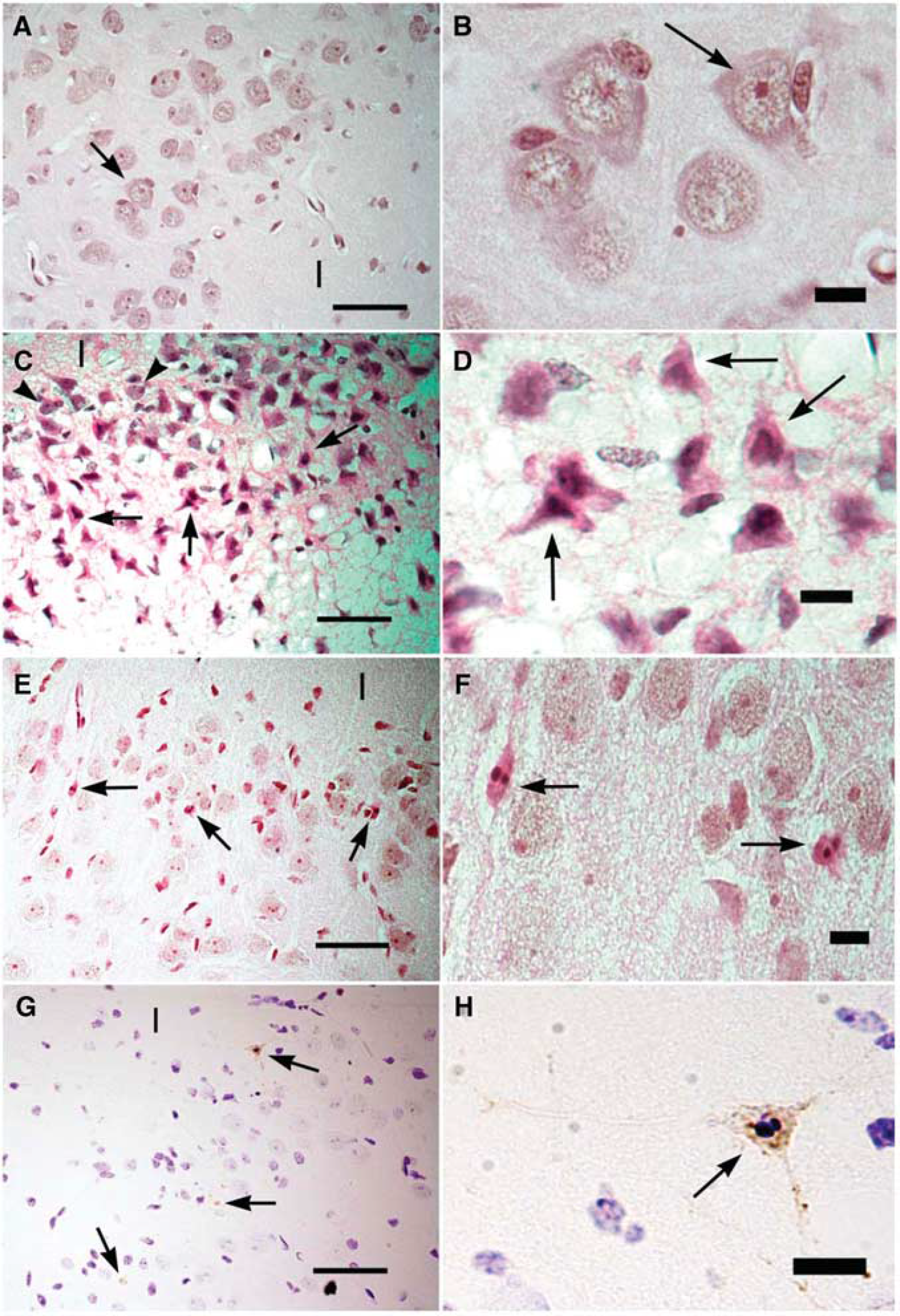
After staining the sections with hematoxylin and eosin (H&E), a single investigator (JKL), who was masked to the treatment group, counted ischemic and apoptotic profiles in cortical layers 2 and 3 of the entire motor gyrus at the hippocampal level on one slide (one level), and in layer 2 in the entire piriform cortex at an anterior striatal level on one slide (one level) in one hemisphere at ×400 magnification. Counter reliability was screened for accuracy by another investigator (LJM). Criteria for identifying a classic ischemic neuron included neuronal morphology with ischemic cytopathology, 12 which included eosinophilic cytoplasm, cytoplasmic vacuoles, condensed nucleus without a visible nucleolus or other nuclear organization, angular cell body and nucleus, and the absence of perinuclear pallor. Neurons with necrotic morphology (pyknotic nuclei and eosinophilic cytoplasm) 11 were included in the ischemic neuron counts. Criteria to classify a cell as an apoptotic profile included the identification of a few (< 4) apoptotic bodies, which appear as spherical or crescent-shaped chromatin clumps; intact cytoplasmic membrane; cytoplasmic condensation and shrinkage; and eosinophilic cytoplasm. 22 We excluded apoptotic profiles within or adjacent to blood vessels in an effort to avoid counting apoptotic endothelial and white blood cells.
Nuclei positive for terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) were identified with the In Situ Cell Death Detection Kit, POD (Roche Applied Science, Penzberg, Germany) in paraffin-embedded sections. Briefly, after endogenous peroxidase inactivation with 3% hydrogen peroxide and permeabilization with proteinase K for 30 minutes at 37 °C, tissue was blocked with 10% nonfat milk for 60 minutes. Terminal deoxynucleotidyl transferase was diluted 1:100 in distilled deionized water. The TUNEL cocktail was mixed by combining 50 μL of dilute terminal deoxynucleotidyl transferase with 450 μL of label solution. Each section was exposed to the TUNEL cocktail for 60 minutes in a humidified chamber. Converter POD was diluted 1:10 in distilled deionized water, and dilute POD was placed on each section for 30 minutes at 37 °C. The sections were then developed with 3,3′-diaminobenzidine substrate. A negative control was generated by omitting terminal deoxynucleotidyl transferase, and a positive control was generated by exposing the sections to DNase. One investigator (JKL), who was masked to treatment group, counted TUNEL+ cells in cortical layers 2 and 3 of the entire motor gyrus at the hippocampal level, and in layer 2 of the entire piriform cortex at the striatal level. We excluded TUNEL+ cells within or adjacent to blood vessels to avoid counting TUNEL+ white blood cells.
In the anatomic regions that showed differences in the number of apoptotic or TUNEL+ profiles by temperature treatment after HI, two investigators (JKL and BW), who were masked to the treatment group, quantified the number of morphologically normal (viable) neurons in the cortical layers described above with H&E stain at ×400. Criteria for identifying a viable neuron included a large cell body (usually 8 μm to 20 μm in diameter) with neuronal morphology (multipolar or triangular), an open non-condensed nucleus with chromatin strands and often a nucleolus, and no apoptotic or ischemic morphology.
Cleaved Caspase-3 Immunoblotting
Separate piglets were randomized to sham surgery with normothermia, HI+hypothermia, HI+slow rewarming, or HI+rapid rewarming. Naive piglets that did not receive anesthesia, surgery, or HI were prepared as an additional control group. Because we hypothesized that post-HI rewarming would increase caspase-3 cleavage relative to sustained hypothermia after HI, we did not include a normothermic HI group for cleaved caspase-3 immunoblotting. At 29 hours after HI or sham surgery, piglets were deeply anesthetized and killed by transcardial perfusion with cold phosphate-buffered saline to harvest the brain.
Caspase-3 cleavage was measured by immunoblotting for procaspase-3 fragments. Tissue samples were collected from fresh brain slabs by using micropunches. Frozen samples of sensorimotor and piriform cortex were treated with ice-cold 1 × RIPA buffer (Cell Signaling Technology; Danvers, MA, USA) and protease inhibitor cocktail (Invitrogen; Grand Island, NY, USA) with a weight-to-volume ratio of 0.1 g to 1 mL. The tissue was homogenized with a hand-held electric homogenizer. Homogenates were placed on ice to let the macroscopic tissue debris settle, homogenized again, and then sonicated for 60 seconds. Samples were centrifuged at 3,000 r.p.m. for 15 minutes at 4 °C. The supernatant was removed and the protein concentration determined with the Pierce BCA protein assay kit (Thermo Scientific; Rockford, IL, USA). Protein samples were denatured in loading buffer (4× LDS sample buffer, Invitrogen), separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 4% to 12% Tris– glycine gels, and transferred to nitrocellulose membranes. The membranes were blocked in 3% nonfat milk at room temperature and incubated overnight at 4 °C with the following primary antibodies: polyclonal goat anti-caspase-3 IgG (1:1,000, R&D Systems, Minneapolis, MD, USA) and monoclonal mouse anti-β-actin IgG (1:3,000, Santa Cruz Biotechnology, Dallas, TX, USA). Subsequently, the membranes were rinsed and incubated for 1 hour with the corresponding secondary antibodies (anti-goat IgG, Thermo Scientific; anti-mouse IgG, General Electric Healthcare Life Sciences, Pittsburgh, PA, USA) at room temperature. β-Actin was used as the protein loading control. After the membrane was washed three times, immunoreactivity was detected by enhanced chemiluminescence (Thermo Scientific) for the quantification of optical density. Immunoreactive band intensities were analyzed with ImageJ (National Institutes of Health, Bethesda, MD, USA). Optical density of the caspase-3 fragment was normalized to β-actin for analysis. In some experiments, recombinant active caspase-3 (Biovision, Milipitas, CA, USA) was loaded as a positive control. Protein from one piglet per group (naive, sham, HI+hypothermia, HI+slow rewarming, and HI+rapid rewarming) was run on a single gel in four separate experiments.
Sample Size Calculations
Because we had no a priori knowledge of the variability and magnitude of changes in apoptosis with hypothermia and rewarming in this model, we made a sample size estimate after completing three experiments in each of the HI+hypothermia and HI+rapid rewarming groups. Preliminary data of apoptotic profiles in piriform cortex indicated a difference in means of 9 between groups and a within-group standard deviation (s.d.) of 5. Thus, we calculated that a minimum sample size of 6 piglets/group would be necessary to reject the null hypothesis that the number of apoptotic profiles would differ by < 9 between piglets that remain hypothermic and those that undergo rapid rewarming at a power of 0.8 and alpha level of 0.05. We chose a sample size of 8 piglets/group to allow for some error in our initial estimate of variability.
After three experiments had been completed for groups that received HI+rapid rewarming and either aCSF or caspase-3 inhibitor, preliminary data indicated a difference in means of 15 apoptotic profiles in piriform cortex between groups and a within-group s.d. of 6. We calculated that a minimum sample size of 4 piglets/group would be necessary to reject the null hypothesis that the number of apoptotic profiles would differ by < 15 between piglets that received aCSF and those that received the caspase-3 inhibitor at a power of 0.8 and alpha of 0.05. A sample size of 6 was selected to permit some error in our initial estimate of variability.
Statistical Analysis
Analyses were conducted with SigmaPlot (v11.0, Systat Software, Chicago, IL, USA), and graphs were generated with GraphPad Prism (v5.03, GraphPad Software, La Jolla, CA, USA). Data are presented as means with s.d. or medians with interquartile ranges (IQR) as appropriate. Differences were considered significant at P < 0.05.
Differences in temperature at each time point before hypothermia were analyzed with one-way analysis of variance and the post hoc Holm–Sidak test or with the Kruskal–Wallis analysis of ranks and the post hoc Dunn's multiple comparisons test, as appropriate. MAP, pH, PaCO2, hemoglobin, hemoglobin oxygen saturation, sodium, potassium, ionized calcium, and glucose levels were compared among groups before and after HI with two-way repeated measures analysis of variance by the treatment group and time with the post hoc Holm–Sidak test when the F-value was significant. Differences in MAP, pH, PaCO2, and hemoglobin oxygen saturation during hypoxia and asphyxia were compared between the HI-injured groups by one-way analysis of variance or Kruskal–Wallis analysis of ranks, when appropriate.
We compared the numbers of apoptotic, TUNEL+, and ischemic profiles and number of viable neurons between the groups by using the Kruskal–Wallis analysis of ranks with post hoc Dunn's multiple comparison tests. The Mann–Whitney rank-sum test was used to compare apoptotic profiles, TUNEL+ profiles, and viable neurons between piglets that received the caspase-3 inhibitor and those that received aCSF and between piglets that did and did not receive phenylephrine. Among piglets that had apoptotic profiles assessed by both H&E and TUNEL, we used Spearman correlations to make pairwise comparisons between the number determined with each method in individual piglets. Western blot densities of the caspase-3 fragment normalized to β-actin were compared by Friedman two-way analysis of ranks in which the four independent gels were blocked as a between-subject factor. If treatment group was significant, post hoc comparisons were made with the Student–Newman–Keuls multiple comparison test.
RESULTS
Identifying the Least Neurotoxic Anesthetic Regimen in Developing Brain
Given the recent concerns about anesthesia neurotoxicity in the pediatric brain, 23 we addressed this issue in piglets. After the sham surgery, nine piglets were anesthetized with nitrous oxide+fentanyl+vecuronium, and seven were anesthetized with isoflurane+nitrous oxide+fentanyl+vecuronium. Seven age-matched, naive piglets that did not receive anesthesia or surgery were prepared as a control group. One piglet that did not receive isoflurane died during anesthesia. Thus, we analyzed the data for 22 piglets to assess the effects of 29 hours of anesthesia on cortical apoptosis.
Examples of apoptotic profiles by H&E and TUNEL staining are shown in Figure 1. As assessed by H&E staining, isoflurane-anesthetized piglets (n = 7) had more apoptotic profiles in the motor cortex than did naive piglets (n = 7) and piglets that were anesthetized without isoflurane (n = 8; P = 0.002). Post hoc multiple comparisons revealed that isoflurane-anesthetized piglets had more apoptotic profiles than either group individually (both P < 0.05; Figure 1A). The number of TUNEL+ profiles in the motor cortex was also greater in piglets that received isoflurane (n = 5) than in naive piglets (n = 7) and those anesthetized without isoflurane (n = 7; P = 0.016). In post hoc multiple comparisons testing, piglets that received isoflurane had more TUNEL+ profiles than did naive piglets (P < 0.05; Figure 1B). The degree of apoptosis was similar in naive piglets and those that received nitrous oxide+fentanyl+vecuronium (P > 0.05 by both H&E and TUNEL). TUNEL+ profiles in motor cortex at the hippocampal level could not be counted in one piglet that was anesthetized without isoflurane because of damage to the mounted tissue and in two piglets that received isoflurane because paraffin-embedded tissue at the appropriate anatomic level was unavailable. In the motor cortex, H&E-stained apoptotic profiles and TUNEL+ profiles had good correlation (r = 0.96; P < 0.0001; n = 19; Figure 1C).
In the piriform cortex, the number of apoptotic profiles by H&E stain differed among isoflurane-anesthetized piglets (n = 7), piglets anesthetized without isoflurane (n = 8), and naive piglets (n = 7; P = 0.002). Post hoc multiple comparisons showed that isoflurane-anesthetized piglets had more apoptotic profiles than did piglets in the other two groups (both P < 0.05; Figure 1D). Isoflurane-anesthetized piglets (n = 6) also had more TUNEL+ profiles than did naive piglets (n = 7) and piglets anesthetized without isoflurane (n = 8; P = 0.011). Post hoc comparisons confirmed that isoflurane-anesthetized piglets had more TUNEL+ profiles than piglets in the other two groups (both P < 0.05; Figure 1E). The piglets anesthetized without isoflurane and naive piglets had similar numbers of apoptotic and TUNEL+ profiles (P > 0.05 for both). One piglet in the isoflurane-anesthetized group did not have TUNEL+ profiles counted because the TUNEL assay did not stain all of the motor cortex. Quantification of apoptotic and TUNEL+ profiles had good correlation (r = 0.87; P < 0.0001; n = 21; Figure 1F). On the basis of these data, we anesthetized all the piglets in subsequent experiments with nitrous oxide+fentanyl +vecuronium (without isoflurane) to test the effects of temperature on neurodegeneration after HI.
Mortality for Hypoxia–Ischemia and Temperature Experiments
Fifty-eight piglets underwent HI, and 13 piglets received sham surgery for histologic examination of apoptosis or cleaved caspase-3 immunoblotting. To reduce the number of animals, we used piglets that were anesthetized without isoflurane in the experiment described above (Figure 1) as the sham group when we tested the effects of temperature on apoptosis after HI. Eleven naive piglets were used as additional controls. Four piglets could not be resuscitated from HI (including three piglets destined for histologic examination of apoptosis and one piglet for cleaved caspase-3 immunoblotting), resulting in a 93% resuscitation rate. Six HI-injured piglets (including five piglets for histologic examination of apoptosis and one piglet for cleaved caspase-3 immunoblotting) and one sham-operated piglet (destined for histologic examination of apoptosis) died during anesthesia. With the HI+rapid rewarming protocol, six piglets received subdural aCSF and six piglets received subdural caspase-3 inhibitor. To minimize the number of animals, we combined the six piglets that received subdural aCSF with two piglets that received HI+rapid rewarming without subdural catheters to achieve eight piglets in the rapid rewarming group. Seventy-one piglets were used in the final analysis.
Physiology
Table 1 lists the temperature, pH, PaCO2, hemoglobin oxygen saturation, MAP, hemoglobin, sodium, and glucose levels of piglets that underwent histologic analysis. All the HI groups had similar levels of arterial O2 saturation during the HI insult. Piglets exhibited severe acidosis and hypercapnea during asphyxia, but the levels returned to normal after ROSC. Rectal temperature was similar among the HI groups before initiation of hypothermia, although temperature in the shams was slightly higher at 1 hour of recovery (Table 1). Temperature was maintained near 34 °C during hypothermia, and rewarming did not result in an overshoot of the goal temperature. The piglets became more acidotic with longer periods of anesthesia, but no differences in pH were detected between the treatment groups at any time point.
Physiologic variables of piglets for histologic quantification of apoptotic profiles
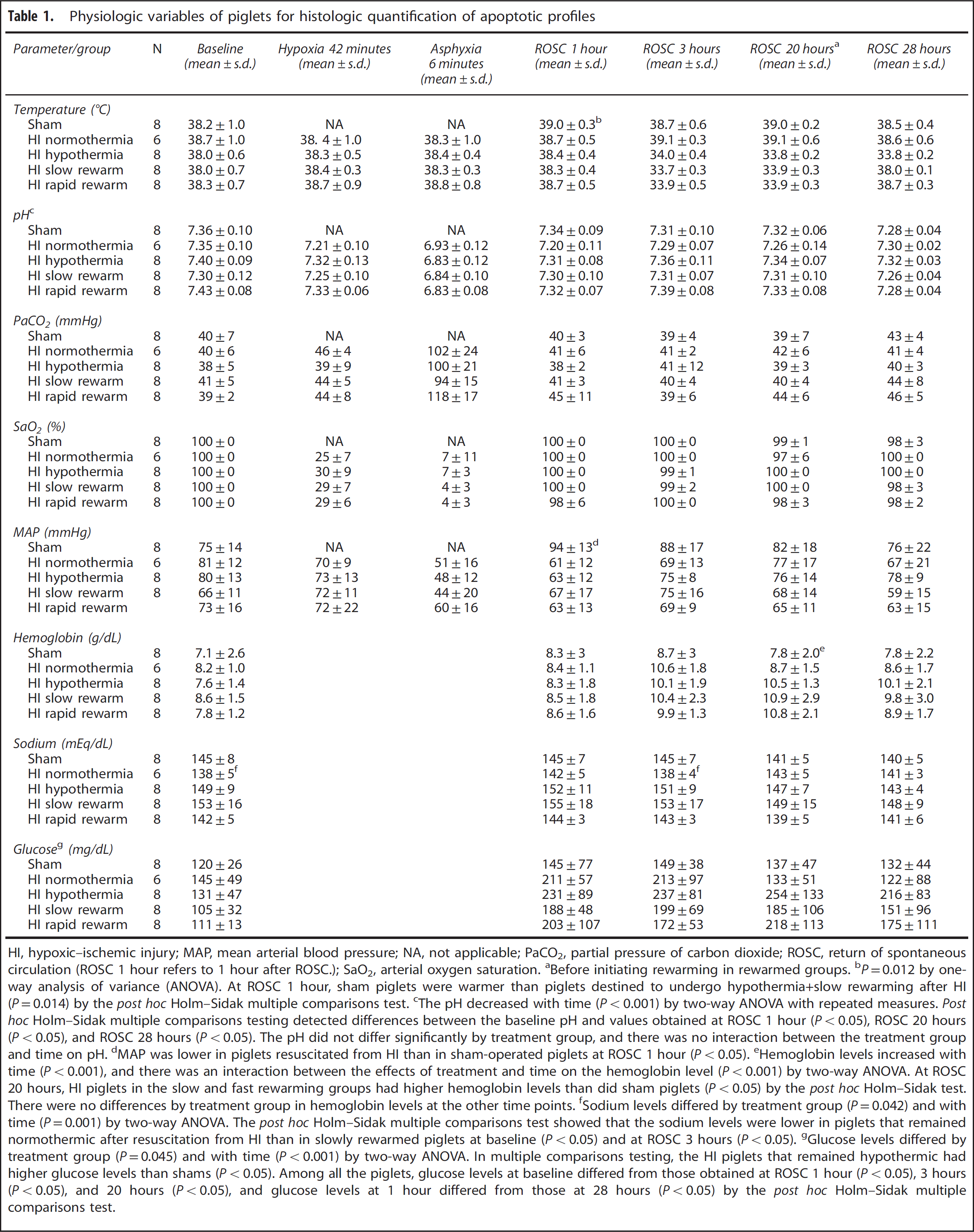
HI, hypoxic–ischemic injury; MAP, mean arterial blood pressure; NA, not applicable; PaCO2, partial pressure of carbon dioxide; ROSC, return of spontaneous circulation (ROSC 1 hour refers to 1 hour after ROSC.); SaO2, arterial oxygen saturation.
Before initiating rewarming in rewarmed groups.
P=0.012 by one-way analysis of variance (ANOVA). At ROSC 1 hour, sham piglets were warmer than piglets destined to undergo hypothermia+slow rewarming after HI (P=0.014) by the post hoc Holm–Sidak multiple comparisons test.
The pH decreased with time (P<0.001) by two-way ANOVA with repeated measures. Post hoc Holm–Sidak multiple comparisons testing detected differences between the baseline pH and values obtained at ROSC 1 hour (P<0.05), ROSC 20 hours (P<0.05), and ROSC 28 hours (P<0.05). The pH did not differ significantly by treatment group, and there was no interaction between the treatment group and time on pH.
MAP was lower in piglets resuscitated from HI than in sham-operated piglets at ROSC 1 hour (P<0.05).
Hemoglobin levels increased with time (P<0.001), and there was an interaction between the effects of treatment and time on the hemoglobin level (P<0.001) by two-way ANOVA. At ROSC 20 hours, HI piglets in the slow and fast rewarming groups had higher hemoglobin levels than did sham piglets (P<0.05) by the post hoc Holm–Sidak test. There were no differences by treatment group in hemoglobin levels at the other time points.
Sodium levels differed by treatment group (P=0.042) and with time (P=0.001) by two-way ANOVA. The post hoc Holm–Sidak multiple comparisons test showed that the sodium levels were lower in piglets that remained normothermic after resuscitation from HI than in slowly rewarmed piglets at baseline (P<0.05) and at ROSC 3 hours (P<0.05).
Glucose levels differed by treatment group (P=0.045) and with time (P<0.001) by two-way ANOVA. In multiple comparisons testing, the HI piglets that remained hypothermic had higher glucose levels than shams (P<0.05). Among all the piglets, glucose levels at baseline differed from those obtained at ROSC 1 hour (P<0.05), 3 hours (P<0.05), and 20 hours (P<0.05), and glucose levels at 1 hour differed from those at 28 hours (P<0.05) by the post hoc Holm–Sidak multiple comparisons test.
The HI protocol significantly affected the MAP of piglets (P = 0.012), and there was an interaction between the effects of HI and time on MAP as time progressed from resuscitation (P = 0.004) by two-way analysis of variance. More specifically, piglets resuscitated from HI had lower MAP than did sham-operated piglets at 1 hour after ROSC (ROSC 1 hour; P < 0.05). There were no statistically significant differences in MAP between sham and HI-injured groups after ROSC 1 hour (Table 1).
Sodium levels differed by treatment group (P = 0.042) and with time (P = 0.001). In multiple comparisons testing, piglets that remained normothermic after resuscitation from HI had lower sodium levels than did slowly rewarmed piglets at baseline (P < 0.05) and ROSC 3 hour (P < 0.05). Glucose levels also differed by treatment group (P = 0.045) and with time (P < 0.001). In multiple comparisons testing, glucose levels were higher in the HI piglets that remained hypothermic than in the shams (P < 0.05). Among all the piglets, glucose levels at baseline differed from those obtained at ROSC 1 hour (P < 0.05), 3 hours (P < 0.05), and 20 hours (P < 0.05), and between those measured at ROSC 1 hour and 28 hours (P < 0.05; Table 1). Potassium and ionized calcium levels were similar between the groups at each time point (data not shown).
Ischemic Neurons
Figure 2 shows examples of H&E-stained normal and ischemic neurons. Neurons with classic apoptotic morphology were not observed in the piglets exposed to HI+normothermia, and the HI injury was morphologically consistent with ischemic necrosis. The ischemic neuron counts in motor cortex of sham (n = 8; median: 0; IQR: 0, 0) and naive (n = 7; median: 0; IQR: 0, 0) groups were similar (P =0.96 by Mann–Whitney rank-sum tests). Likewise, the number of ischemic neurons in the piriform cortex of sham (n = 8; median: 0; IQR: 0, 0) and naive (n = 7; median: 0; IQR: 0, 0) groups was similar (P = 0.69 by Mann–Whitney rank-sum tests). In the subsequent statistical comparisons, the naive and sham groups were combined to increase statistical power.
In the motor cortex, the numbers of ischemic neurons were not statistically different among the sham/naive (n = 15), HI+normothermia (n = 6), HI+hypothermia (n = 8), HI+rapid rewarming (n = 8), HI+slow rewarming (n = 8), and HI+rapid rewarming +caspase inhibitor groups (n = 6; P = 0.15). Ischemic neurons were observed in select piglets with HI+normothermia (Figures 2 and 3A). Piglets that remained hypothermic, received slow or rapid rewarming, or rapid rewarming with the caspase inhibitor after HI did not show ischemic cytopathology. Piriform cortex was resistant to HI-related accumulation of ischemic neurons, and there were no differences in the number of ischemic neurons within piriform cortex among sham/naive (n = 15), HI+normothermia (n = 6), HI+hypothermia (n = 8), HI+rapid rewarming (n =8), HI +slow rewarming (n = 8), and HI+rapid rewarming+caspase inhibitor groups (n = 6; P = 0.54; Figure 3B).
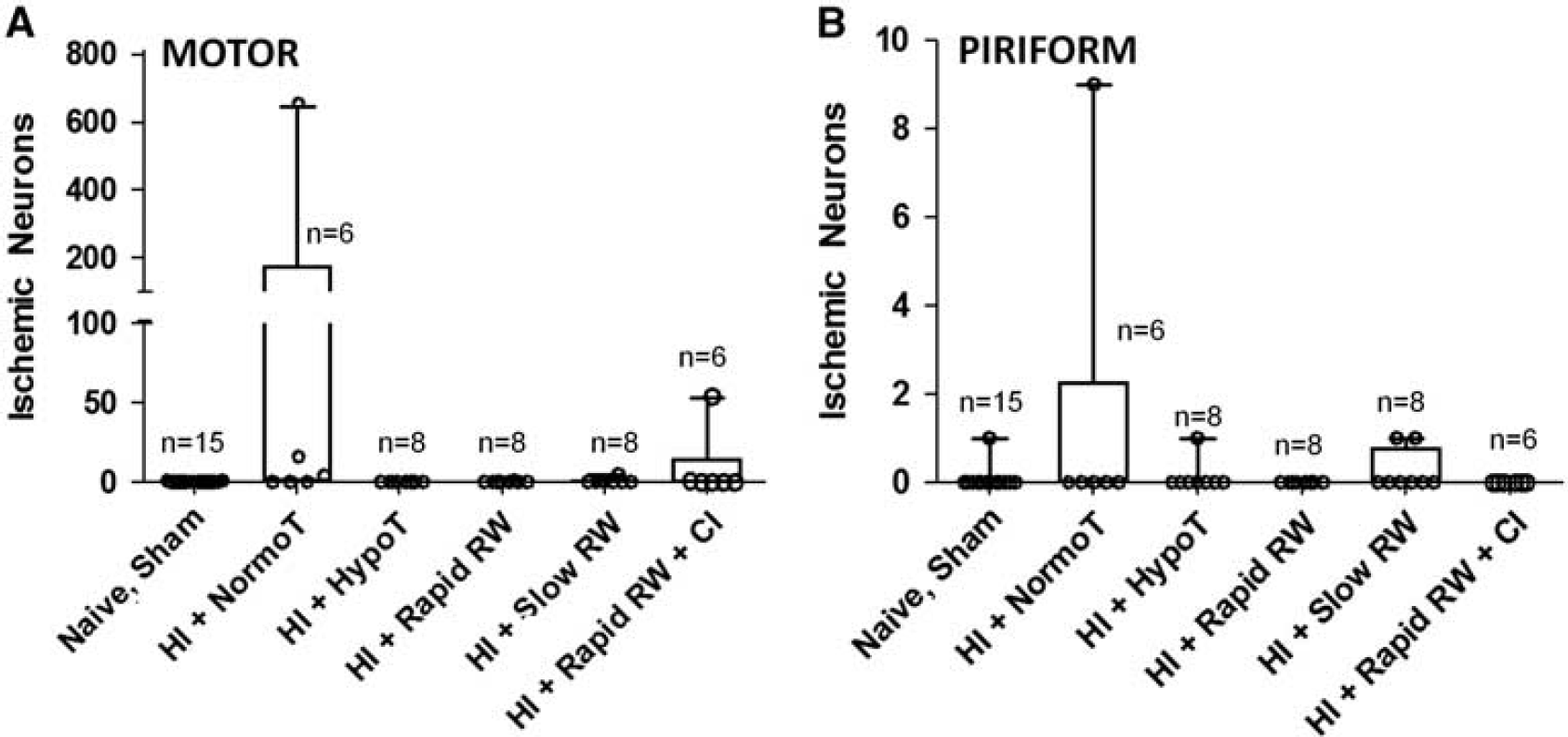
Ischemic neurons in the motor and piriform cortex. The number of ischemic neurons was similar among piglets in the sham/naive, hypoxia–ischemia injury (HI)+normothermia (HI+NormoT), HI+hypothermia (HI+HypoT), HI+rapid rewarming (HI+Rapid RW), HI+slow rewarming (HI+Slow RW), and HI+rapid rewarming+caspase inhibitor (CI) groups in both the motor cortex (P = 0.15,
Apoptosis in Motor Cortex with Rewarming Apoptotic profiles were uncommon in sham-operated piglets (anesthetized without isoflurane; Figures 1, 2A and 2B). Piglets from the naive and sham groups had similar numbers of apoptotic and TUNEL+ cells in the motor and piriform cortex (P ≥ 0.40 by Mann–Whitney rank-sum tests; Figure 1). In the subsequent statistical comparisons, the naive and sham groups were combined to increase statistical power.
On the basis of H&E staining, the number of apoptotic profiles in the motor cortex differed among the naive/sham (n = 15), HI+normothermia (n = 6), HI+hypothermia (n = 8), HI+rapid rewarming (n = 8), and HI+slow rewarming groups (n = 8; P < 0.001). In post hoc multiple comparisons, rapidly rewarmed piglets had more apoptotic profiles than did piglets that remained hypothermic (P < 0.05) or normothermic (P < 0.05) after HI. HI-injured, rapidly rewarmed piglets also had more apoptosis than naive/shams (P < 0.05). Moreover, piglets that were rewarmed slowly after HI had more apoptosis than did naive/sham piglets (P < 0.05) or piglets that remained hypothermic after HI (P < 0.05). The number of apoptotic profiles in the rapidly and slowly rewarmed groups did not differ significantly (P40.05; Figure 4A).
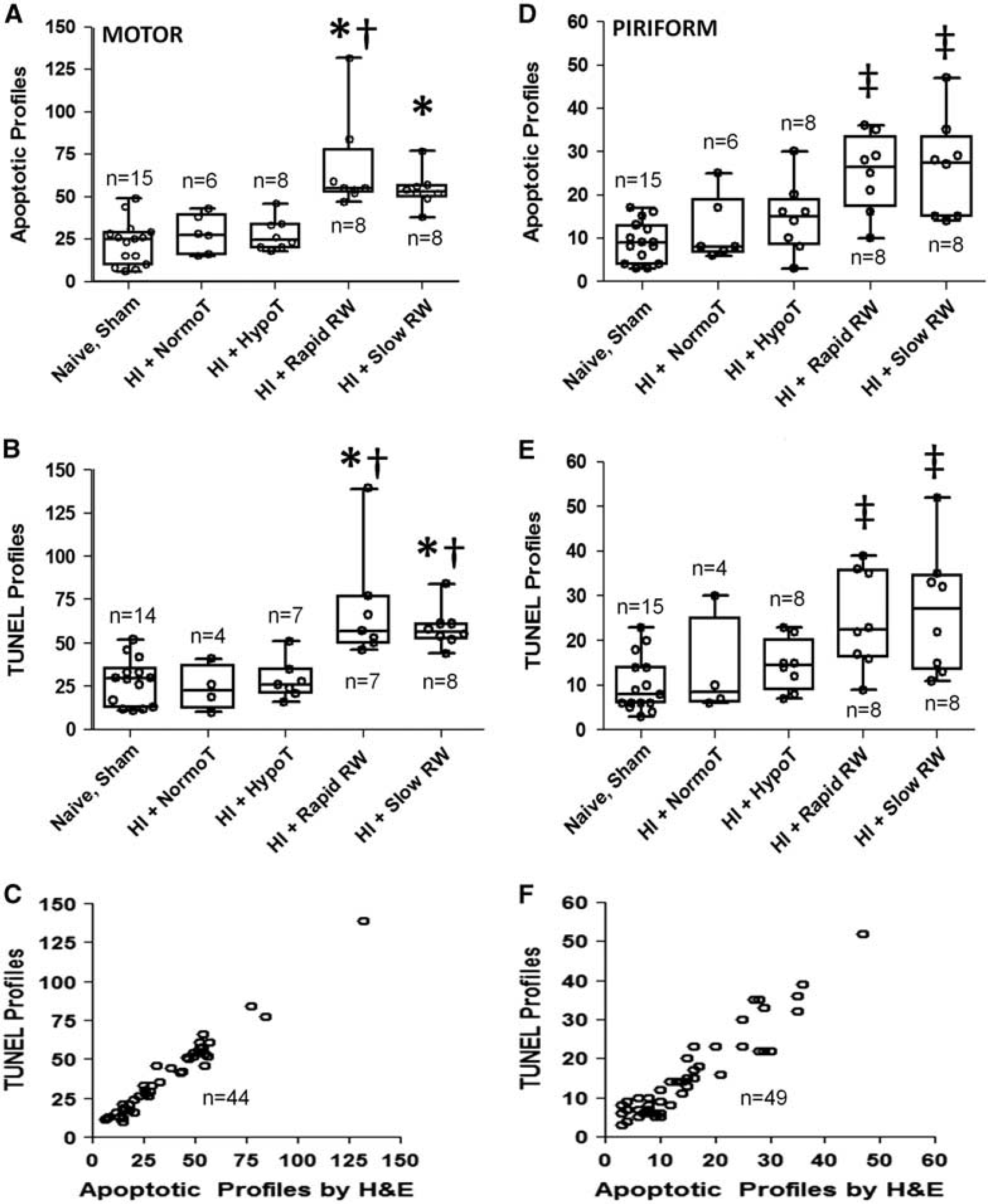
Apoptosis in motor (
We also observed differences in the number of TUNEL+ profiles in the motor cortex between naive/sham (n = 14), HI+normothermia (n = 4), HI+hypothermia (n = 7), HI+rapid rewarming (n = 7), and HI+slow rewarming groups (n = 8; P < 0.001). Both the slowly rewarmed (P < 0.05) and rapidly rewarmed (P < 0.05) piglets had more TUNEL+ cells after HI than did piglets that remained hypothermic. In addition, normothermic piglets had fewer TUNEL+ profiles after HI than did the slowly rewarmed (P < 0.05) or rapidly rewarmed (P < 0.05) piglets. The number of TUNEL+ cells did not differ significantly by rewarming rate (P40.05; Figure 4B). We were unable to quantify TUNEL+ profiles in one piglet each of the sham, HI+normothermia, HI+hypothermia, and HI+rapid rewarming groups because of tissue damage on the slide. One HI +normothermia piglet did not have paraffin-embedded tissue at the appropriate anatomic level available for TUNEL staining. The classification of cells as apoptotic by H&E stain and TUNEL had good correlation (n = 44; r = 0.964; P < 0.0001; Figure 4C).
The number of viable neurons in the motor cortex of each treatment group was similar. The median viable neuron counts were 5,096 (IQR: 4,530 to 6,430; n = 15) for naive/sham, 4,899(3,902 to 5,419; n = 6) for HI+normothermia, 6,008 (5,117 to 7,131; n = 8) for HI+hypothermia, 4,728 (4,253 to 6,595; n = 8) for HI+rapid rewarming, and 4,901 (4,274 to 6,679; n =8) for HI+slow rewarming piglets (P = 0.363).
Apoptosis in Piriform Cortex with Rewarming
On the basis of H&E staining, the number of apoptotic profiles in the piriform cortex differed among naive/sham (n =15), HI +normothermia (n = 6), HI+hypothermia (n =8), HI+slow rewarming (n = 8), and HI+rapid rewarming groups (n = 8; P < 0.001). Both rapidly rewarmed (P < 0.05) and slowly rewarmed (P < 0.05) HI-injured piglets exhibited more apoptosis than naive/sham piglets (Figure 4D). However, the difference in apoptotic profile counts between the rewarmed piglets and piglets that remained hypothermic or normothermic after HI did not reach statistical significance in post hoc multiple comparisons testing, possibly because the magnitude of the rewarming effect in the piriform cortex (Figure 4D) was smaller than that observed in the motor cortex (Figure 4A).
The number of TUNEL+ profiles also differed among naive/sham (n = 15), HI normothermia (n =4), HI+hypothermia (n = 8), HI+slow rewarming (n = 8), and HI+rapid rewarming groups (n = 8; P = 0.002). Compared with that of naive/sham piglets, both rapidly rewarmed (P < 0.05) and slowly rewarmed HI-injured piglets (P < 0.05) had more TUNEL+ cells in post hoc multiple comparisons (Figure 4E). Two HI+normothermia piglets did not have tissue at the striatal level available for TUNEL staining. The classification of cells as apoptotic and TUNEL+ had good correlation (n = 49; r = 0.893; P < 0.0001; Figure 4F).
Effects of Caspase-3 Inhibitor
Hematoxylin and eosin staining of motor cortex showed that among the piglets subjected to HI+rapid rewarming, those that received the subdural caspase-3 inhibitor (n = 6) had fewer apoptotic profiles than those that received aCSF (n = 6; P = 0.002; Figure 5A). The number of TUNEL+ profiles was also lower in piglets that received the caspase-3 inhibitor (n = 4) than in those that received aCSF (n = 5; P = 0.016; Figure 5B). We were unable to quantify TUNEL+ profiles for two piglets in the caspase-3 inhibitor group and one in the aCSF group because of damage to the mounted tissue specimens. The correlation was good for the classification of cells as apoptotic by H&E stain and TUNEL+ in motor cortex (n = 9; r = 0.937; P = 0.0007). Viable neuron counts in the motor cortex were similar in piglets that received the caspase-3 inhibitor (median: 5,274; IQR: 4,661 to 5,476; n = 6) and those that received aCSF (median: 4,615; IQR: 4,130 to 5,579; n = 6; P = 0.240).
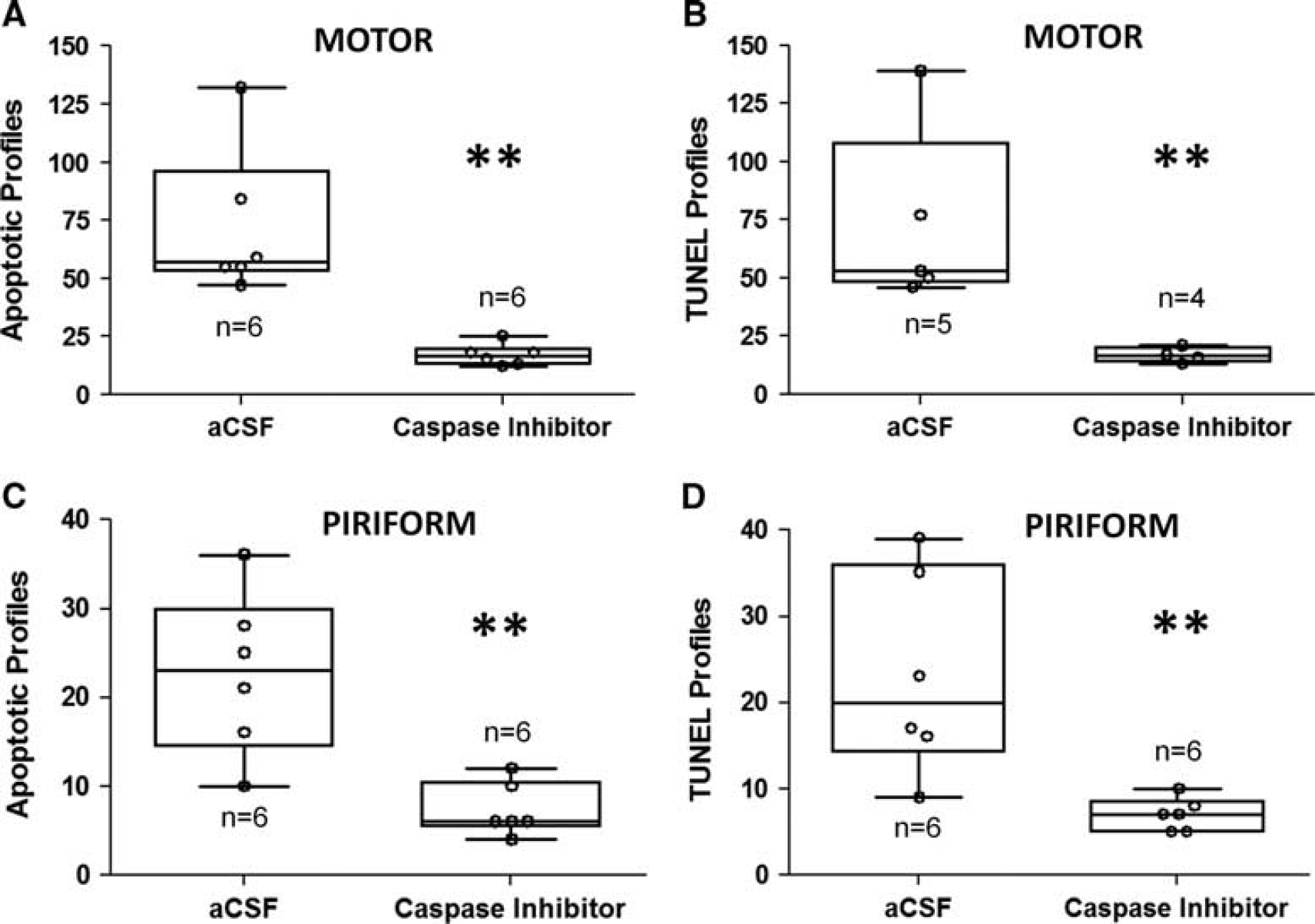
The piglets underwent hypoxia–ischemia followed by hypothermia and rapid rewarming. In comparison to artificial CSF (aCSF), subdural administration of caspase-3 inhibitor decreased the number of (
The piglets that underwent HI+rapid rewarming and received the caspase-3 inhibitor (n = 6) also had less apoptosis in the piriform cortex than those that received aCSF (n = 6), as assessed by H&E (P = 0.004; Figure 5C) and TUNEL (P = 0.004; Figure 5D). The classification of cells as apoptotic by H&E stain correlated well with TUNEL classification (n = 12; r =0.834; P < 0.001).
Western Blots for Cleaved Caspase-3
Western blot analysis revealed differences in the levels of cleaved caspase-3 in the sensorimotor cortex among naive (n = 4), sham (n =4), HI+hypothermia (n = 4), HI+rapid rewarming (n =4), and HI+slow rewarming (n =4) groups (P = 0.013). In post hoc multiple comparisons, piglets that were rewarmed rapidly after HI had more cleaved caspase-3 than piglets that were rewarmed slowly after HI (P < 0.05), piglets that remained hypothermic after HI (P < 0.05), sham-operated piglets (P < 0.05), and naive piglets (P < 0.05; Figures 6A and 6C).
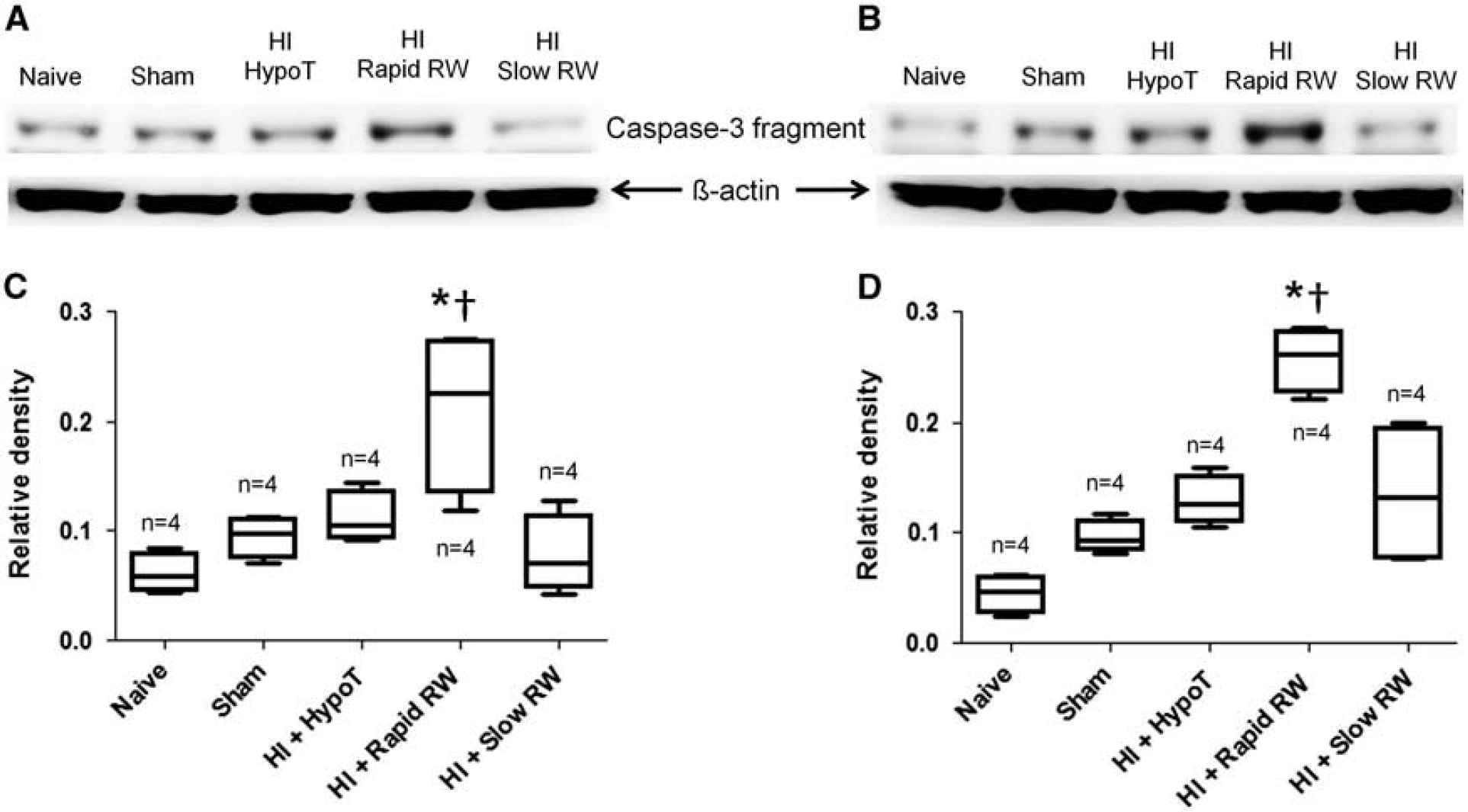
Protein expression of cleaved caspase-3. Western blots for cleaved caspase-3 in naïve, sham, hypoxia–ischemia injured+hypothermia (HI+HypoT), HI+rapid rewarming (HI+rapid RW), and HI+slow rewarming (HI+slow RW) groups in the sensorimotor cortex (
Cleaved caspase-3 levels in the piriform cortex also differed by treatment group (n = 4; P = 0.009). Post hoc multiple comparisons showed more caspase-3 cleavage after HI in rapidly rewarmed piglets than in slowly rewarmed piglets (P < 0.05) or piglets that remained hypothermic (P < 0.05; Figures 6B and 6D). Rapidly rewarmed piglets also had greater cleaved caspase-3 levels than did sham (P < 0.05) and naive (P < 0.05) piglets. Naive piglets had less caspase-3 cleavage than did piglets in the HI+hypothermia (P < 0.05), HI+slow rewarming (P < 0.05), and sham (P < 0.05) groups.
Effects of the Subdural Catheter, Dimethyl Sulfoxide, or Phenylephrine
In the HI+rapid rewarming group, six piglets received subdural aCSF in DMSO and two did not. The median number of apoptotic profiles by H&E stain among piglets that had subdural catheters with aCSF in DMSO was 57 (IQR: 55, 78) in the motor cortex and 23 (17, 27) in the piriform cortex. Piglets without catheters (and therefore without aCSF in DMSO) had median apoptotic profile counts of 53 in the motor cortex and 32 in the piriform cortex. The median number of TUNEL+ profiles among piglets with catheters and aCSF in DMSO was 53 (50, 77) in the motor cortex and 20 (16, 32) in the piriform cortex. Piglets without catheters, aCSF, or DMSO had median TUNEL+ profile counts of 62 in motor cortex and 29 in the piriform cortex.
Six piglets received phenylephrine during anesthesia, including one in the HI+rapid rewarming, one in the HI+hypothermia, two in the HI+slow rewarming, and two in the HI+normothermia groups. Piglets that received phenylephrine (n = 6) and those that did not (n = 24) had similar apoptotic profile counts in the motor cortex (P = 0.24) and piriform cortex (P = 0.19). Piglets that did and did not receive phenylephrine also exhibited similar numbers of TUNEL+profiles in the motor cortex (n =6 and 20, respectively; P = 0.61) and the piriform cortex (n = 5 and 23, respectively; P = 0.51). Among piglets that received subdural catheters, one piglet in the caspase-3 inhibitor group and no piglet in the aCSF group received phenylephrine.
DISCUSSION
This study demonstrated several findings relevant to neonatal anesthesia neurotoxicity and the treatment of HI brain injuries. First, in a piglet model, we identified and used an anesthetic regimen that produced minimal apoptotic effects. Second, rewarming from hypothermia after HI was associated with apoptosis in the motor and piriform cortex. Increased apoptosis with rewarming was confirmed by the TUNEL assay and was prevented by a subdural caspase-3 inhibitor. Third, western blotting showed that caspase-3 cleavage was greater with rapid rewarming than with slow rewarming or sustained hypothermia after HI. These results suggest that rewarming from hypothermia after HI may promote apoptosis through pathways involving caspases. Some piglets displayed ischemic neurons in the motor cortex after HI and normothermic recovery, although differences in ischemic neuron counts among experimental groups were not statistically significant. This finding likely reflects variability in the acute neuropathology early after resuscitation. The piriform cortex was resistant to ischemic injury in all piglets.
The anesthetic regimen of nitrous oxide+fentanyl+vecuronium caused less apoptosis than the same regimen combined with isoflurane and was therefore selected for our HI experiments. The comparable levels of apoptosis observed among naive, unanesthetized piglets and piglets anesthetized without isoflurane indicate that the anesthesia did not increase apoptosis above normal neurodevelopmental levels. These results support a previous report that isoflurane, but not fentanyl, increases apoptosis in piglets. 24 It is important to note, however, that we tested the effects of anesthesia on uninjured animals. Anesthetics may have a different effect on post-hypoxic neurons that are compensating or that have already entered the cell-death continuum. Hypoxic pretreatment has been reported to increase anesthesia-related neuroapoptosis, 25 whereas the administration of nitrous oxide after ischemic brain injury may be neuroprotective. 26 A progressive acidosis developed with prolonged anesthesia in all the groups. The combination of nitrous oxide and fentanyl for short periods is known to produce general anesthesia with hemodynamic stability and minimal change in cerebral blood flow, oxygen consumption, and oxygen supply in experimental models of the developing brain. 27 However, the physiologic effects of prolonged anesthesia for 29 hours are not well studied. The acidosis may have been because of a mild respiratory acidosis observed in some piglets and/or to dehydration. Blood pressure remained above the lower limit of autoregulation 11 throughout the experiments.
Neuronal cell death manifests along a continuum between ischemic necrosis and apoptosis. 15 After neonatal HI, dying neurons can display necrotic or apoptotic morphology depending on the location and recovery time.28–31 Apoptotic cell signaling is prominent in neonatal HI, 32 but impaired protein synthesis and mitochondrial dysfunction can interrupt apoptosis and lead to necrosis. 22 In our HI piglet model, neurons in sensorimotor cortex appear necrotic rather than apoptotic with 2 days of normothermic recovery. 11 Although hypothermia can suppress the load of oxidative stress 12 that is thought to contribute to regulated and unregulated forms of necrosis, it may preserve organelle function necessary for executing caspase-dependent apoptosis once rewarming occurs. Thus, we theorized that rewarming would permit the cleavage of caspase-3 and the appearance of apoptosis in motor cortex, where necrosis ordinarily predominates. We also examined the piriform cortex where necrosis is not normally predominant after HI but where others have reported apoptosis in piglet brain. 33
All the piglets recovered for 29 hours after HI, and the rewarmed piglets received 18 hours of hypothermia. Hypothermia for as little as 3 hours is neuroprotective in this model, 12 and ischemic cortical neuron cytopathology is evident by 2 days of normothermic recovery after HI. 11 Therefore, we selected a recovery period of 29 hours to explore the evolution of cell death at approximately 1 day after HI. We delayed hypothermia by 2 hours to mimic the potential clinical delay in initiating hypothermia. Clinically, patients may be passively warmed during resuscitation, and transport to a tertiary care hospital may delay the induction of hypothermia. In animal models, delaying hypothermia by 3 to 8 hours reduces neuroprotection.34,35 In our study, piglets that received hypothermia with or without rewarming did not have neuronal ischemic injury despite the 2-hour delay in initiating hypothermia.
Ischemic neurons were evident in the neocortex of some piglets with HI+normothermia, although we found no statistical differences in the ischemic neuron counts across experimental groups. The variability seen in the amount of neuronal injury among HI normothermic piglets likely reflects variation in the acute neuropathology early after resuscitation. We counted ischemic neurons 29 hours after resuscitation, but this time might have been too early to detect robust injury. In our piglet model of HI, neuronal ischemic necrosis is abundant in the cortex after 2 days of normothermic recovery. 11 The motor cortex was more vulnerable to ischemia than the piriform cortex in some individual piglets. The motor cortex is a neocortical (six-layered) region, whereas the piriform cortex is an allocortical (three-layered) region. Thus, differential vulnerability could rely on the differences in architecture, connectivity, and metabolism. 16 Neocortical neurons are more vulnerable than allocortical neurons to oxidative stress, 36 a fact that might explain the motor cortex vulnerability to HI observed in some piglets. In contrast, although both neocortical and allocortical neurons are sensitive to protein-folding stress, allocortical neurons have enhanced sensitivity, 36 possibly contributing to the hypothermia–rewarming cytotoxicity. In addition, our sample size calculations were based on apoptotic profile counts rather than ischemic neuron counts.
Our finding that rewarming is associated with cortical apoptosis suggests that rewarming could negate some of the neuroprotective effects of hypothermia. Apoptosis in both the motor and piriform cortex were increased in rewarmed piglets. Many apoptotic cells were neurons based on morphology, but others were smaller and likely glia. Since HI affects neurons 11 and glia, 37 the effects of rewarming on multiple cell types could indicate a global impact that might influence cortical function. This possibility must be tested in future experiments. Whether the occurrence of apoptosis with rewarming requires ischemic stress or is a manifestation of rewarming and independent of HI remains unclear. The findings in piriform cortex indicate that rewarming can augment apoptosis in a region largely devoid of ischemic injury and thus independent of the ischemic–necrotic milieu. Perhaps hypothermia suspends normal developmental apoptosis, and rewarming permits its continuation in a backlog of neurons that were in different phases of apoptosis at the onset of hypothermia.
It is also important to note that these experiments represent a short period after HI. Experiments with longer duration of recovery are necessary to delineate the effects of rewarming on cell death. The observed apoptosis may or may not continue after rewarming, and ischemic cell death might re-emerge. The variability in the ischemic neurons at 29 hours of recovery in a model known to produce cortical neuron injury at 2 days of recovery 11 emphasizes the importance of examining later time points. Therefore, the cumulative, time-dependent effects of rewarming after HI and hypothermia cannot be determined in this study.
Rewarming has been reported to induce cellular stress and activate pro-apoptotic pathways in cell cultures. 38 Apoptosis also occurred after rewarming from hypothermic cardiopulmonary bypass in swine. 39 Although our histologic quantification of apoptotic cells did not show a difference by rewarming rate, caspase-3 cleavage assessed by western blotting was greater in piglets that were rewarmed rapidly after HI than in piglets that were rewarmed slowly after HI. However, these data have some caveats. The western blots included not just neuronal and glial proteins, but also proteins from vascular and blood-borne cells. We excluded apoptotic endothelial and blood cells in our histologic counts, but we did not quantify these cells; therefore comparisons between the treatment groups cannot be made. Although the detected increase in cleaved caspase-3 serves as biochemical confirmation of the observed histologic cortical apoptosis, the results would also include apoptosis among nonneuronal and non-glial cells. Furthermore, we did not conduct immunohistochemistry for cleaved caspase-3. We tested multiple caspase-3 antibodies in piglet tissue by western blotting and compared the bands with a recombinant caspase-3 positive control (Biovision). All the antibodies that we tested revealed bands for both procaspase and cleaved caspase fragments and were therefore not appropriate for immunohistochemistry in piglets (unpublished results).
The slow rewarming rate of 0.5°C/hour reflects clinical practice for neonatal HIE. 5 We reasoned that a fast rewarming rate of 4°C/hour would have a stronger effect on apoptosis and caspase-3. Indeed, rapid rewarming was associated with greater caspase-3 cleavage than slow rewarming was, although the exact cell type affected cannot be determined in a tissue homogenate-based assay. It should also be noted that rewarming started at 20 hours of recovery in both the groups and that tissue was harvested at 29 hours. Thus, the rapidly rewarmed piglets experienced 8 hours more of post-rewarming normothermia and had more time to accumulate cleaved caspase-3 than the slowly rewarmed piglets.
Although apoptosis in the motor cortex was elevated in piglets that were rewarmed after HI compared with that in piglets that remained normothermic or hypothermic after HI, viable neuron counts in the motor cortex did not differ among the treatment groups. This lack of difference may have been because of the relatively short recovery time of 29 hours after HI. In this model, the viable neuron count decreases after 2 days of recovery from HI. 11 Alternatively, some apoptotic cells are probably not neurons. The overall effect of rewarming on cortical cell apoptosis may be small relative to the total density of neurons. To determine the full impact of rewarming on cortical injury, additional experiments with longer recovery after HI and rewarming are needed.
This study had several limitations. Piglets remained under anesthesia for 29 hours, which does not occur clinically. However, the piglets must remain anesthetized to prevent shivering during hypothermia and to ensure comfort. Nevertheless, the numbers of apoptotic and TUNEL+ profiles were similar between sham-operated piglets (that did not receive isoflurane) and unanesthetized naive piglets. All the animals received the same anesthetic regimen to maintain comparability across experimental groups. We did not decrease the anesthesia during hypothermia to account for the expected decrease in minimum alveolar concentration for nitrous oxide or slowed metabolism of fentanyl or vecuronium. The age range of piglets in this study (3 to 5 days) includes a period of rapid brain growth that might introduce more neuropathologic variability than that observed in studies that use piglets < 24 hours old. 40 The 18 to 27 hours of hypothermia that we used is shorter than the clinical treatment of 72 hours 5 but longer than the 3 to 8 hours used in many rodent studies.41,42 Although the 20-hour start for rewarming is later than the 3- to 8-hour start typically used in rodent studies,41,42 it is considerably earlier than the 72-hour start time used clinically. 2 Prolonging hypothermia beyond 1 day may suppress apoptosis, as has been reported in older pigs. 43 For the sake of focus, we did not examine ischemic neurons or apoptosis in regions other than the piriform or motor cortex. Our focus was on cerebral cortex rather than subcortical areas because clinically adverse effects of rewarming might manifest as subtle cognitive or intellectual disabilities. We could not assess the effects of the subdural catheter or DMSO because too few piglets in the HI+rapid rewarming group did not have a catheter with aCSF in DMSO. DMSO may have some neuroprotective properties after ischemia, 44 but we could not examine whether DMSO affected cortical apoptosis after HI and rewarming in this study. Some piglets were hypernatremic, and hypertonicity can promote apoptosis in certain conditions. 45 Nonetheless, mean sodium levels were within normal clinical limits. Glucose control was poor, and severe hyperglycemia may promote apoptosis. 46 However, no differences in glucose were detected between rewarmed piglets and the other treatment groups. Finally, we did not study female piglets.
CONCLUSION
Despite the increasing use of therapeutic hypothermia to treat neonatal hypoxic brain injuries, little research exists on the effects of rewarming. In a neonatal swine model of HI injury, rewarming after overnight hypothermia increased apoptosis involving caspase mechanisms. Rapid rewarming may induce more biochemical caspase-3 cleavage than slow rewarming. Although it is not clear which specific types of cells in cerebral cortex are intrinsically vulnerable to rewarming, neurons are involved. This study identifies a neuropathologic consequence of therapeutic hypothermia that warrants further examination of the cell-type vulnerability, cellular mechanisms, and long-term neurologic ramifications.
DISCLOSURE/CONFLICT OF INTEREST
JKL has received research support from Covidien for clinical research.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.