
Abstract
Lysophospholipids (LPLs) are important intermediates in the synthesis and degradation of membrane phospholipids. Here we show that certain LPLs, particularly lysophosphatidylcholine and lysophosphatidylinositol, prevent neuronal death both in an in vivo model of transient global ischemia and in an in vitro model of excitotoxicity using primary cultures of cerebellar granule cells exposed to high extracellular concentrations of glutamate (20–40 μmol/L). The intravenous injection of lysophosphatidylcholine or lysophosphatidylinositol at a concentration of 200 nmol/kg induced a survival of CA1 pyramidal neurons as high as approximately 95%, even when the treatment was started 30 minutes after 15-minute global ischemia. In contrast, lysophosphatidic acid induced no protection. This work also provides evidence that a pretreatment with lysophosphatidylcholine or lysophosphatidylinositol (200 nmol/kg) injected as long as 3 days before a severe 6-minute ischemia provided a potent tolerance against neurodegeneration. Neuroprotection was also observed in in vitro experiments with LPLs. Taken together, in vivo and in vitro data suggest a potential therapeutic use of LPLs as antiischemic compounds. The potential role of 2P-domain K+ channels as targets of LPLs in this potent neuroprotective effect is discussed.
Cerebral ischemia is a major cause of death and long-term disability, annually affecting more than 700,000 people in the United States alone. During a stroke or other ischemic event in the brain, the cerebral blood flow is reduced and the resulting oxygen and glucose deprivation rapidly initiates a sequence of events leading to neuronal cell death. Neurons at risk die as a result of a neurotoxic biochemical cascade initiated by reduced energy stores, membrane depolarization, excessive excitatory amino acid release, accumulation of free fatty acids, and elevated intracellular calcium and neuronal hyperexcitability (Bazan, 1970; Choi and Rothman, 1990; Meldrum, 1995; Siesjö and Wieloch, 1985). While pharmacologic therapy to reduce ischemic damage is being pursued, neuroprotective compounds have failed in clinical trials despite promising preclinical data (Jain, 2000). Growing evidence indicates that the opening of potassium channels might provide a potent protective effect against cerebral ischemia/reperfusion injury. For example, activators of KATP channels have been described to prevent neuronal death induced by ischemic, epileptic, or excitotoxic injuries (Abele and Miller, 1990; Blondeau et al., 2000; Heurteaux et al., 1993; Lauritzen et al., 1997).
The recent discovery of a new family of mammalian two-pore (2P)-domain K+ channels (Lesage and Lazdunski, 2000) has suggested a new strategy for neuroprotection. These channels are present in the brain at synaptic and nonsynaptic sites (Hervieu et al., 2001; Lauritzen et al., 2000; Maingret et al., 2000a; Reyes et al., 2000; Talley et al., 2001). They have very peculiar functional properties, and many are background channels with an important role in the regulation of the resting potential (Lesage and Lazdunski, 2000; Patel and Honore, 2001; Patel et al., 2001). Some 2P-domain K+ channels (TREK-1, TREK-2, and TRAAK) are mechanosensitive channels. These same channels are also reversibly opened by polyunsaturated fatty acids (PUFAs). Since PUFAs are potent neuroprotectors in brain pathologies such as global ischemia and epileptic seizures (Lauritzen et al., 2000; Leaf et al., 1999) and induce ischemic and epileptic tolerance (Blondeau et al., 2001), it has been suggested (Blondeau et al., 2001; Lauritzen et al., 2000) that this effect is largely due to the opening of TREK-1, TREK-2, and TRAAK channels. Another interesting property of these particular TREK and TRAAK channels is that they are potently activated by lysophospholipids (LPLs) (Lesage et al., 2000; Maingret et al., 2000b). Lysophospholipids are produced in parallel with PUFAs by hydrolysis of membrane phospholipids by phospholipases A2 (Dennis, 1997; Valentin and Lambeau, 2000). Because PUFAs are potent neuroprotectors (Blondeau et al., 2001), the present work aims to investigate the potential protective effect of LPLs on neuronal death in the selectively vulnerable regions of rat hippocampus after transient global cerebral ischemia induced by four-vessel occlusion. The study also analyzes the effect of LPLs on the viability of cultured cerebellar granule cells treated with glutamate in neurotoxic concentrations in vitro.
MATERIALS AND METHODS
Animals
Animal experiments were approved by the local committee review board and were conducted according to policies on the care and use of laboratory animals of the Society of Neurosciences. Male Wistar rats weighing 250 to 300 g (Charles River Breeding Laboratories) were used. The animals, maintained on a 12/12 hours light/dark cycle, were given food and water ad libitum and were acclimated to the animal facility for at least 1 week prior to drug treatment or surgery. Anesthesia was induced with 2% halothane mixed with 30% oxygen and 70% nitrous oxide. Core temperature was monitored continuously with a thermometer (3-mm probe diameter; Harvard Apparatus, France) inserted into the rectum and maintained at physiologic temperatures using a thermostatically controlled heating blanket (Harvard Apparatus, France). Brain temperature was monitored with a temperature monitor (Licox pO2 Monitor, Germany) and, as previously reported (Cao et al., 2001), a 29-gauge thermocouple implanted 3.5-mm deep into the left caudate putamen (3.0 mm lateral and 1.40 mm occipital of the bregma) rather than in the hippocampus to avoid mechanical hippocampal injury. Core and brain temperatures were maintained at physiologic temperatures by a combination of the homeothermic blanket control unit and adjustment of a temperature regulated-heating lamp, which was placed over the head of the animal. The femoral artery was catheterized with polyethylene tubing (PE50) for continuous monitoring of mean arterial blood pressure (mm Hg) with a blood pressure transducer (Harvard Apparatus).
Global forebrain ischemia
Global cerebral ischemia lasting 6 or 15 minutes followed by reperfusion was induced by four-vessel occlusion in anesthetized rats as previously described (Pulsinelli and Brierley, 1979). Animals were placed in a Kopf stereotaxic frame and the vertebral arteries were irreversibly occluded by electrocoagulation at the junction of the C1 and C2 vertebrae. The common carotid arteries were then exposed and small-diameter silicon tubing was looped around the carotid arteries to facilitate subsequent occlusion. On the following day, the animals were ventilated with 1% halothane and atraumatic miniature aneurysm clips were attached to occlude both common carotid arteries. Anesthesia was then discontinued and carotid arteries were clamped for 6 or 15 minutes. Rats lost their righting reflex during ischemia. The EEG was monitored as previously described (Lauritzen et al., 2000) to ensure isoelectricity during the period of ischemia. Negative controls were sham-operated rats. Vehicle-injected rats treated 30 minutes prior to 15-minute ischemia or 3 days prior to 6-minute ischemia were used as positive controls.
Drug treatments
All chemicals were purchased from Avanti Polar Lipids and Sigma. Stock solutions were kept at −20°C and renewed weekly. Lysophosphatidylcholine (LPC; C14:0), 1-palmitoyl-LPC (C16:0), lysophosphatidic acid (LPA; C18:1), and LPI L-α-lysophosphatidylinositol) were complexed with an equal concentration of fatty acid-free bovine serum albumin dissolved in Ca2+-free Hanks balanced solution. Stock solutions of LPLs (10 μmol/L) were then diluted in NaCl 0.9% solution to reach final concentration. The pH of solutions was adjusted to 7. The LPLs were administered by the intraperitoneal route or injected as a bolus directly in the tail vein at different times before occlusion of carotid arteries or 30 minutes after the onset of ischemia. The range of concentrations used in that study was selected based on optimal neuroprotection results obtained from a previously obtained dose-response profile with PUFAs (Lauritzen et al., 2000).
Quantification of neuronal loss on cresyl violet-stained sections
Seven days after exposure to global ischemia, animals were deeply anesthetized and decapitated. Brains were quickly extracted and fresh frozen in isopentane at −40°C. Sections (10 μm) were cut on cryostat (Leica) and postfixed by immersion in 4% paraformaldehyde/10−2 mol/L phosphate-buffered saline (PBS) for 30 minutes. Slides were then dehydrated in ethanol baths (50%, 70%, and 100%), air dried, and stored at −70°C until further use. For each brain studied (n = 6 per time point and condition), two sections were placed on 3-aminopropylethoxysilane—coated slides, and 10 slides (randomly chosen) per rat were used in each stage of analysis. A neuropathologist who was blinded to the experimental conditions performed the histologic assessment using light microscopy. The neuronal density of hippocampal CA1 and CA3 subfields was determined by the method of Kirino (1982). Analysis of neuronal density was performed on coronal sections of the dorsal hippocampus stained with cresyl violet and corresponding to brain sections located between 3.14 and 4.16 mm posterior to bregma (Paxinos and Watson, 1986). The total linear length of CA1 and CA3 sectors was measured by means of a digitizer. The number of living neurons in the stratum pyramidale within CA1 and CA3 subfields was counted using a Leica Aristoplan photomicroscope at a magnification of 400×. The neuronal density of CA1 and CA3 sectors (i.e., the number of intact pyramidal cells per 1-mm linear length of the CA1/CA3 stratum pyramidale observed in each 10-μm section) was quantified. Thus, a mean value for each hippocampal CA1/CA3 substructure was obtained from 10 bilateral measurements on two sections per slide and 10 slides per rat, for the six animals in each of experimental groups. Measurements obtained from the 20 sections for a given animal were averaged to yield a single value of the CA1/CA3 neuronal density, which was then used in the statistical analysis. Results are reported as mean ± SD. The significance of differences between means was assessed by two-factor analysis of variance followed by the Tukey test for multiple comparisons, where P < 0.05 was considered statistically significant.
Isolation of hippocampal proteins and heat-shock protein 70 Western blotting
Rat hippocampi (n = 3 per experimental group) were homogenized in four volumes of lysis buffer (20 mmol/L Tris pH 7.5, 137 mmol/L NaCl, 2 mmol/L EDTA, 1% Triton ×100, 10% glycerol, and protease-inhibitor cocktail) on ice using a Dounce homogenizer. After being removed from ice and exposed to room temperature for 10 minutes, the homogenates were centrifuged at 12,000 g for 30 minutes at 4°C. The supernatant was stored at −70°C until further use. Protein concentrations were measured using conventional Bradford's method.
Expression of heat-shock protein 70 (HSP70) in hippocampi following LPI injection was determined using immunoblotting analysis. Aliquots containing 50 μg proteins were applied to 10% SDS PAGE gels and electrophoresed for 1 hour at 100 mA. Proteins were transferred onto nitrocellulose membrane (Hybond-C) in blotting buffer (156 mmol/L Tris, 1 mol/L glycine, PBS) for 90 minutes at 80 mA and blocked with 5% skim milk (Regilait) in PBS for 2 hours at room temperature. The blotted membrane was incubated with the mouse monoclonal HSP70 antibody (BD Transduction Laboratories, diluted 1/1000) for 2 hours at room temperature. After washing with 0.1% TWEEN/PBS (four times, 15 minutes each), peroxidase-conjugated goat antimouse IgG antibody was used as the second antibody (Jackson ImmunoResearch, diluted 1/15000) for 1 hour at room temperature.
Fluorography was performed on Kodak-X-OMAT AR film using Western blotting detection reagents (Pierce) following conventional enhanced-chemiluminescence technique. To control for sample loading, the membranes containing the original antibody were stripped and rehybridized with a α-tubulin antibody as internal control (data not shown). The specific bands obtained with both antibodies were quantified by densitometry using an imaging analysis system.
L-Lyso-3-phosphatidylcholine,1-[1–14C] palmitoyl transport across the blood—brain barrier
Intravenous L-Lyso-3-phosphatidylcholine,1-[1–14C] palmitoyl injection technique.
The transport of LPLs through the rat blood—brain barrier was determined by using radioactive of L-Lyso-3-phosphatidylcholine,1-[1–14C] palmitoyl and by measuring the uptake of this labeled compound into the brain after short in situ tail-vein perfusion. The in situ brain perfusion technique of Takasato et al. (1984) was used, except that the perfusion was in the tail vein, similar to the different treatments administrated in that study, and 1 mg/mL (14 μmol/L) fatty acid-free bovine serum albumin and no Ca2+ ions were included in bicarbonate-based salt solution (HBSS). The pH-7.4 medium containing 142.0 μmol/mL NaCl, 28.0 μmol/mL NaHCO3, 4.2 μmol/mL KH2PO4, 1.0 μmol/mL MgSO4, and 6 μmol/mL glucose was equilibrated with a gas mixture of 95% oxygen and 5% carbon dioxide at 37°C for 5 minutes. Radioactive L-Lyso-3-phosphatidylcholine,1-[1–14C] palmitoyl was dissolved in HBSS to give a 20 μmol/L concentration in the perfusate, with a specific radioactivity of 56 mCi/mmol (Amersham Pharmacia Biotech, France). Unlabeled L-Lyso-3-phosphatidylcholine,1-palmitoyl (final concentration, 1 mmol/L) was also added in the perfusion medium. The tail vein of rats anesthetized with a flow of O2/N2O gas mixture containing 1% halothane was cannulated with a 24-gauge catheter (Vitaflon Plus, Ohmeda, Becton Dickinson, France). At time zero, halothane was discontinued and the injection of the perfusate was begun at a constant rate of 83 μL/sec by a perfusion pump for 15 seconds and continued with HBSS solution for periods of up to 20 seconds. At the end of the perfusion, the brain was removed from the skull and rinsed in ice-cold saline. The meninges and blood vessels on the surface of the brain were carefully removed.
Lipid extraction.
Lipids were extracted from the brain with a mixture of chloroform, methanol, and water (50:25:25 by volume) and then two times with chloroform and methanol (2:1 volume:volume). The lipid extract was concentrated by evaporating the solvent under a stream of nitrogen and dissolved in 1 mL chloroform/methanol (2:1 volume:volume) to be partitioned according to the standard procedure of Folch et al. (1957). The washed organic phases were concentrated and the dissolved radioactive lipids were resolved by monodimensional TLC on activated silica gel 60-coated plates (20 × 20 cm, 0.20 mm thickness; Merck Eurolab, France), using a mixture of chloroform, methanol, and NH4OH (65:30:5 by volume) to develop the plates. Lipids, which were revealed by resublimed iodine, were identified using Rf values relative to authentic standards. The identified areas of silica gel were scraped off and eluted with chloroform and methanol (1:1 volume:volume). Aliquots of solution containing each individual or combined class of brain phospholipids were transferred to plastic vials, dried, dissolved in scintillation cocktail fluid (Ecolume, ICN), and measured for 14C radioactivity (Packard scintillation counter model 1600 CA).
Cerebellar granule cell culture
Cerebellar granule cells were prepared from 6- or 7-day-old OF1 mice (Iffa Credo, France) as previously described (Schousboe et al., 1989). Neurons were seeded on poly-L-lysine (50 μg/mL)-coated dishes at a density of 2.6 × 106 cells per 35-mm culture dish and cultured in Eagle basal medium (Sigma) supplemented with 10% fetal calf serum (Gibco), 25 mmol/L KCl, and 0.5% penicillin-streptomycin. To prevent growth of glial cells, cytosine arabinoside (10 μmol/L) was added to the cultures 24 hours following plating. Unless otherwise stated, experiments were performed using 8- or 9-day-old cultures.
In vitro model of excitotoxicity
Glutamate treatment was performed in magnesium-free glycine-supplemented PBS. Control cells were exposed to the same buffer without glutamate. Lysophospholipids (LPC [C14:0], LPA [C18:1], and LPI [L-α-lysophosphatidylinositol]; 10 μmol/L) were added as stated either 15 minutes before and during, or during and after the treatment with glutamate. Control cells were incubated with vehicle alone. Unless otherwise stated, cell viability was estimated 6 hours after treatment.
Assessment of cell viability
Neuronal injury was quantitatively assessed by the measurements of lactic dehydrogenase (LDH) in the medium (Koh and Choi, 1987). Lactic dehydrogenase activities, measured at OD340, were expressed as activity present in the total medium volume/total LDH activity (media + cells) and are presented as release of LDH as a percentage of that in glutamate-treated cells.
Cell death was determined using a fluorescent chromatin-fixing dye propidium iodide (PI) staining procedure. Cells were fixed with freshly prepared paraformaldehyde 4%, and left in 70% ethanol overnight. Cells were incubated in PBS containing PI (5 μg/mL) for 10 minutes. The nuclei of dead cells were highly fluorescent and condensed compared with living cells. Dead cells were scored by counting at least 600 cells in several randomized subfields for each sample from three different experiments. The percentage of LDH activities or of PI-pyknotic nuclei was used for statistical analysis by ANOVA followed by a Fischer PLSD for multiple comparisons (P < 0.05).
RESULTS
Lysophospholipids protect against glutamate-induced cell death in cerebellar granular cultures
First, we examined the effects of LPLs on glutamate-induced cell death in primary cultures of cerebellar granular neurons. Cell death was estimated by measuring cell leakage and nuclei condensation. When the granule cells were exposed to 20 μmol/L glutamate, no signs of cell death were observed in the first hours after the exposure, though cells died during the next several hours (Fig. 1A, left, and Fig. 1B). Cell leakage, measured by the release of LDH, remained relatively low (Fig. 1A, left), but the treatment led to the appearance of a high number of pyknotic nuclei, which may be explained by the presence of both necrotic and apoptotic neurons (Ankarcrona et al., 1995; Bachis et al., 2001; Lauritzen et al., 2000). We analyzed the effect of the lysophospholipid LPC on this glutamate-induced cell death. First, we wanted to establish the optimal concentration of LPC. Since it is well known that lysophospholipids from Avanti Polar Lipids are very pure and do not contain any contaminating lipids, as suspected in those obtained from Sigma (Liliom et al., 1998), we used LPC from Aventi Polar Lipids to establish the optimal concentration of the lysophospholipid to be used throughout all other in vitro experiments. Cells were exposed to 20 μmol/L glutamate in the presence of different concentrations of LPC (C14:0; 1, 5, 10, and 20 μmol/L) (Fig. 1A, left). Maximal protection was obtained at a concentration of 10 μmol/L, and the corresponding Sigma product, at an identical concentration, produced a similar protection (Fig. 1A, left). At a lower concentration (1 μmol/L), LPC was not able to prevent cell death, and a higher concentration (20 μmol/L) did not produce a protection better than that found with 10-μmol/L concentration. Since the LPC obtained from Sigma caused the same neuroprotective effect as LPC from Aventi, we used Sigma LPLs at a concentration of 10 μmol/L throughout all other experiments.
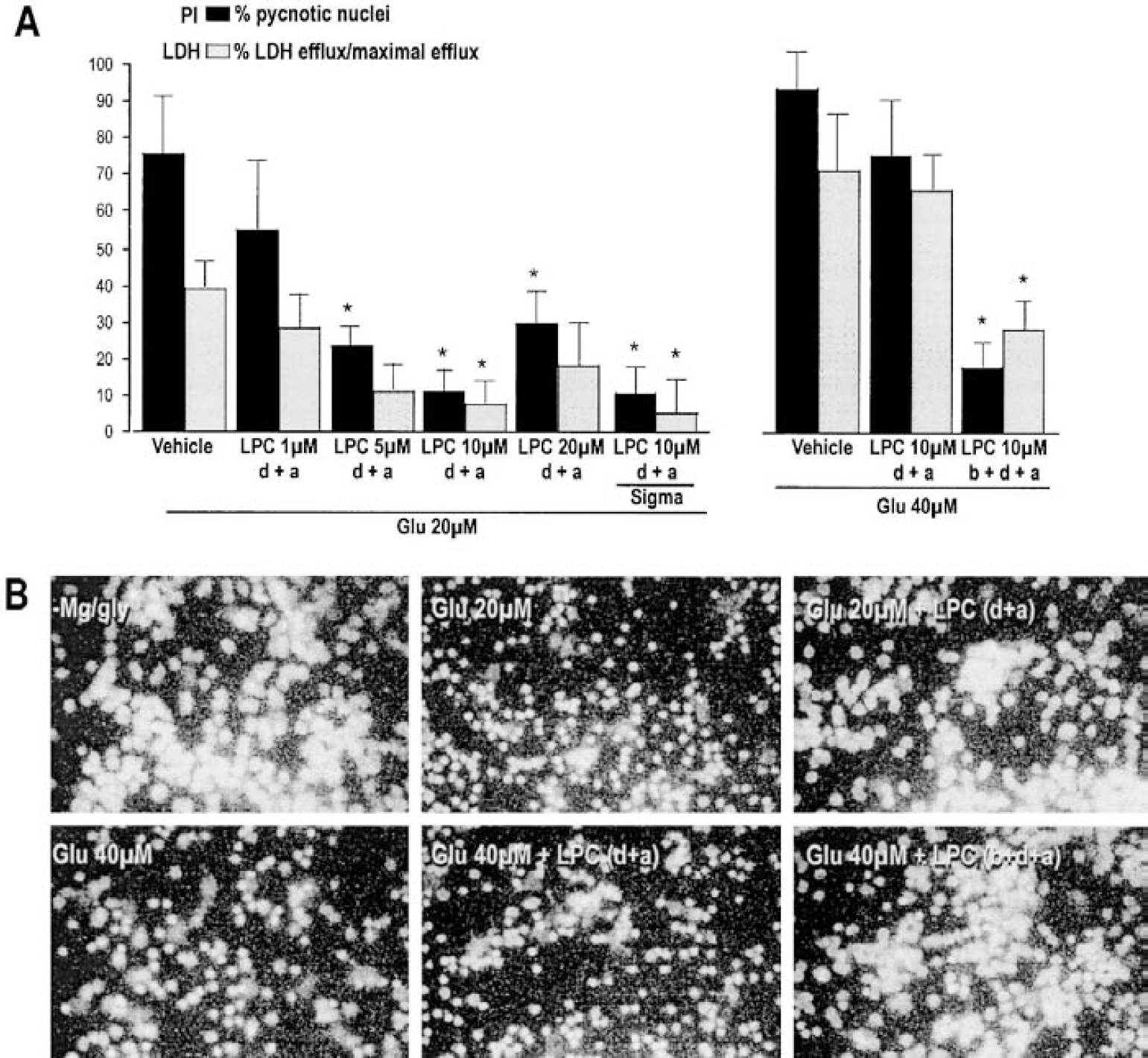
Lysophosphatidylcholine (LPC)-induced neuroprotection in glutamate-treated granular cell cultures.
The cells were then exposed to a higher concentration of 40 μmol/L glutamate for 15 minutes, which led to cell swelling and cell death in the first hours after treatment (Fig. 1A, right, and Fig. 1B). At 6 hours after treatment, the LDH activity in 40 μmol/L glutamate-treated cells was almost twice the activity of 20 μmol/L glutamate-treated cells, but the numbers of pyknotic nuclei were almost identical in the two cases (Figs. 1A and 1B). In 40 μmol/L glutamate-treated cells, the presence of LPC (10 μmol/L) only during and after the glutamate exposure did not prevent cell death (Fig. 1A, right, and Fig. 1B). However, if cells were preincubated with LPC (10 μmol/L) for 15 minutes before the toxic insult, the appearance of pyknotic nuclei and LDH activity were significantly reduced (Fig. 1A, right, and Fig. 1B).
Next, we analyzed the effect of other LPLs on excitotoxicity. We used LPI, another lysophospholipid that activates 2P-domain K+ channels, and LPA, which has no action on these K+ channels (Maingret et al., 2000b). Neither LPI nor LPA (10 μmol/L) affected cell viability in normal conditions. When cells were exposed to glutamate (20 μmol/L), LPI completely protected against cell death, whereas LPA had no beneficial effects (Fig. 2). These findings were confirmed using both PI staining and LDH-efflux measurements.
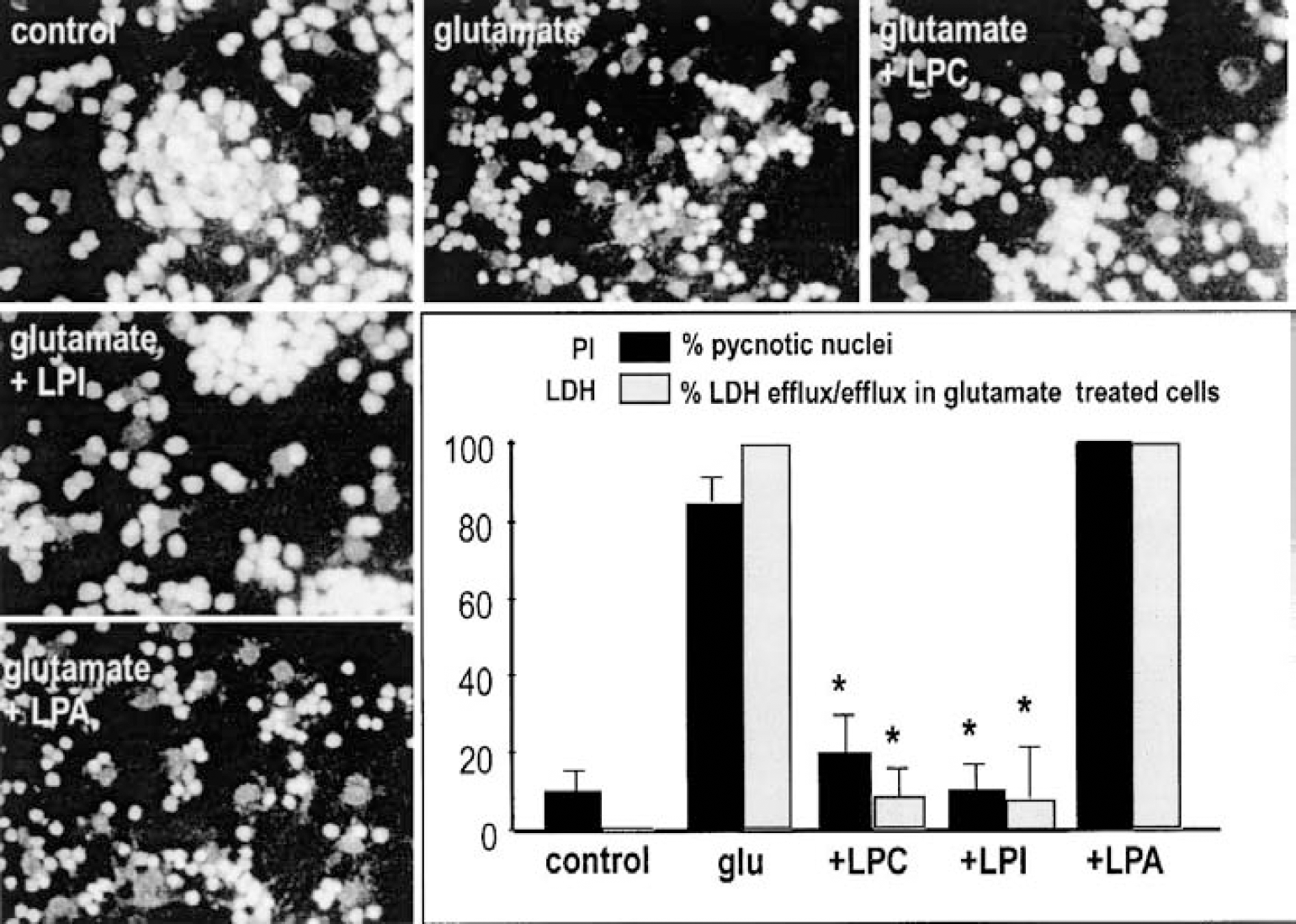
Effects of distinct lysophospholipids (LPLs) on glutamate-induced cell death. Granular cells were exposed to glutamate (20 μmol/L for 15 minutes) in the absence or presence of distinct LPLs: lysophosphatidylcholine (LPC; C14:0), and lysophosphatidylinositol (LPI) and lysophosphatidic acid (LPA; Sigma, each at 10 μmol/L). Cell death was estimated 6 hours after treatment using propidium iodide (PI) staining and lactic dehydrogenase (LDH)-efflux measurements. Viability is expressed as percentage of pyknotic nuclei for PI staining, and as percentage of LDH efflux of the efflux in glutamate-treated cells for LDH-efflux measurements. Photomicrographs correspond to representative experiments of PI staining.
Physiologic parameters before, during, and after transient global forebrain ischemia
Mean arterial blood pressure and core and brain temperatures measured at 15 minutes before the induction of ischemia, 5 minutes after the induction of ischemia, and at the end of 15-minute ischemia are given in Table 1. Before the induction of ischemia, mean arterial blood pressure was maintained close to 76 to 82 mm Hg, reduced to 30 to 33 mm Hg during ischemia, and progressively returned to preischemic baseline values during reperfusion. There were no significant differences among the groups (Table 1) The average core and brain temperatures were maintained at 36.9°C to 37.5°C before ischemia. During the ischemic interval, brain and core temperatures were continually maintained within the physiologic range, and the temperature stability of the animals in the different groups was preserved throughout the reperfusion period. Table 1 shows no significant differences among the groups (P < 0.05)
Mean arterial blood pressure and core and brain temperatures measured at 15 minutes before, and 5 minutes after, induction of ischemia, and after 15 minutes’ ischemia
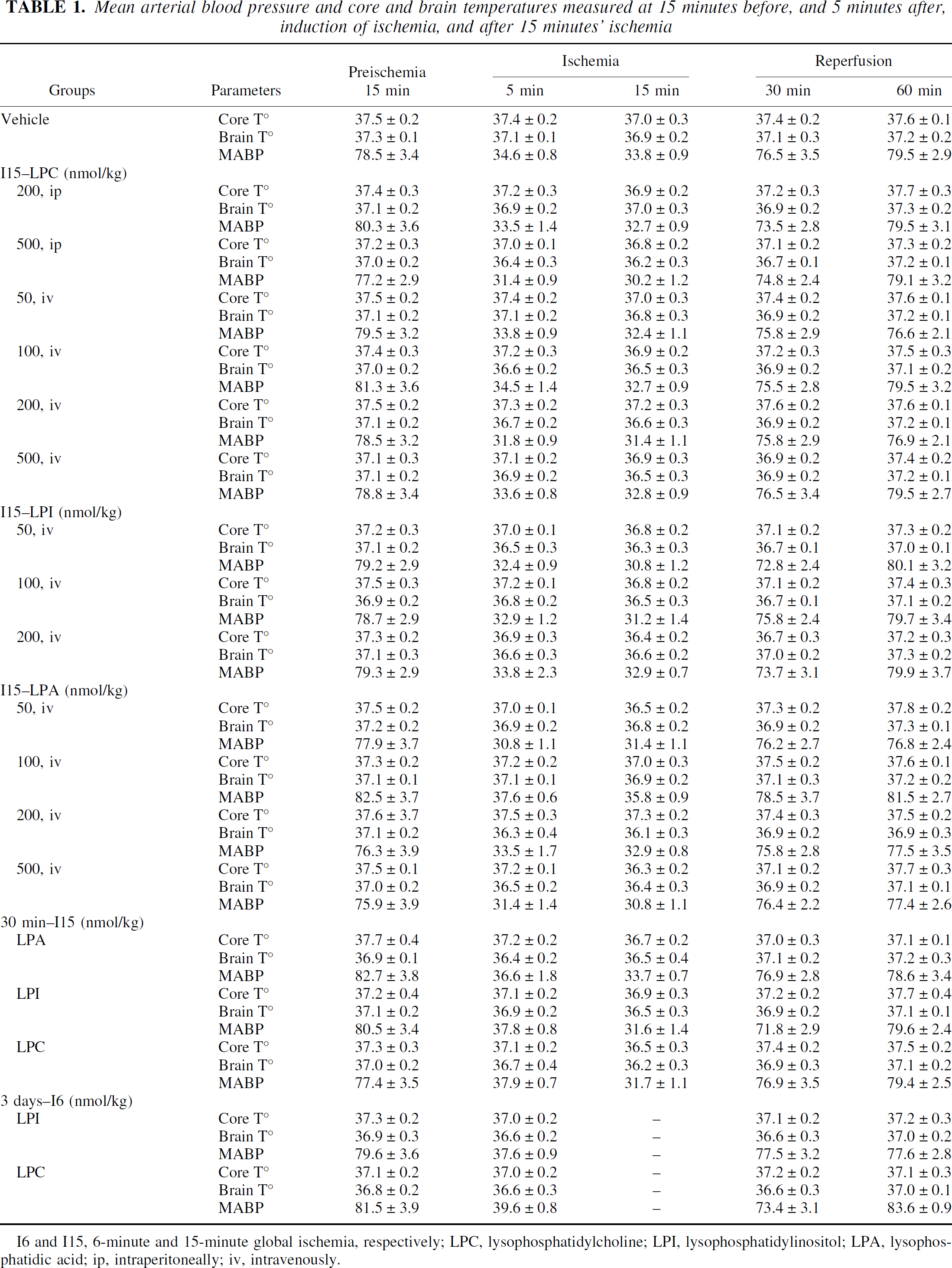
I6 and I15, 6-minute and 15-minute global ischemia, respectively; LPC, lysophosphatidylcholine; LPI, lysophosphatidylinositol; PA, lysophosphatidic acid; ip, intraperitoneally; iv, intravenously.
Pretreatment with lysophosphatidylcholine or lysophosphatidylinositol, but not with lysophosphatidic acid, prevents hippocampal neuronal cell death induced by transient global forebrain ischemia
We analyzed the potential neuroprotective effect of different LPLs injected shortly before a severe ischemic insult. Intravenous injection of 200 nmol/kg LPC or LPI30 minutes before the onset of ischemia almost completely inhibited neuronal loss, and 97% and 86% of CA1 pyramidal cells were respectively preserved (Fig. 3A). In contrast, intravenous administration of LPA failed to protect the brain. When animals were treated with 200 nmol/kg LPA 30 minutes before ischemia, neuronal counts in CA1 and CA3 pyramidal layers were not significantly different from counts obtained after ischemia (vehicle-injected group) alone; 79% of CA1 neuronal loss was observed in LPA-treated rats (Fig. 3A).
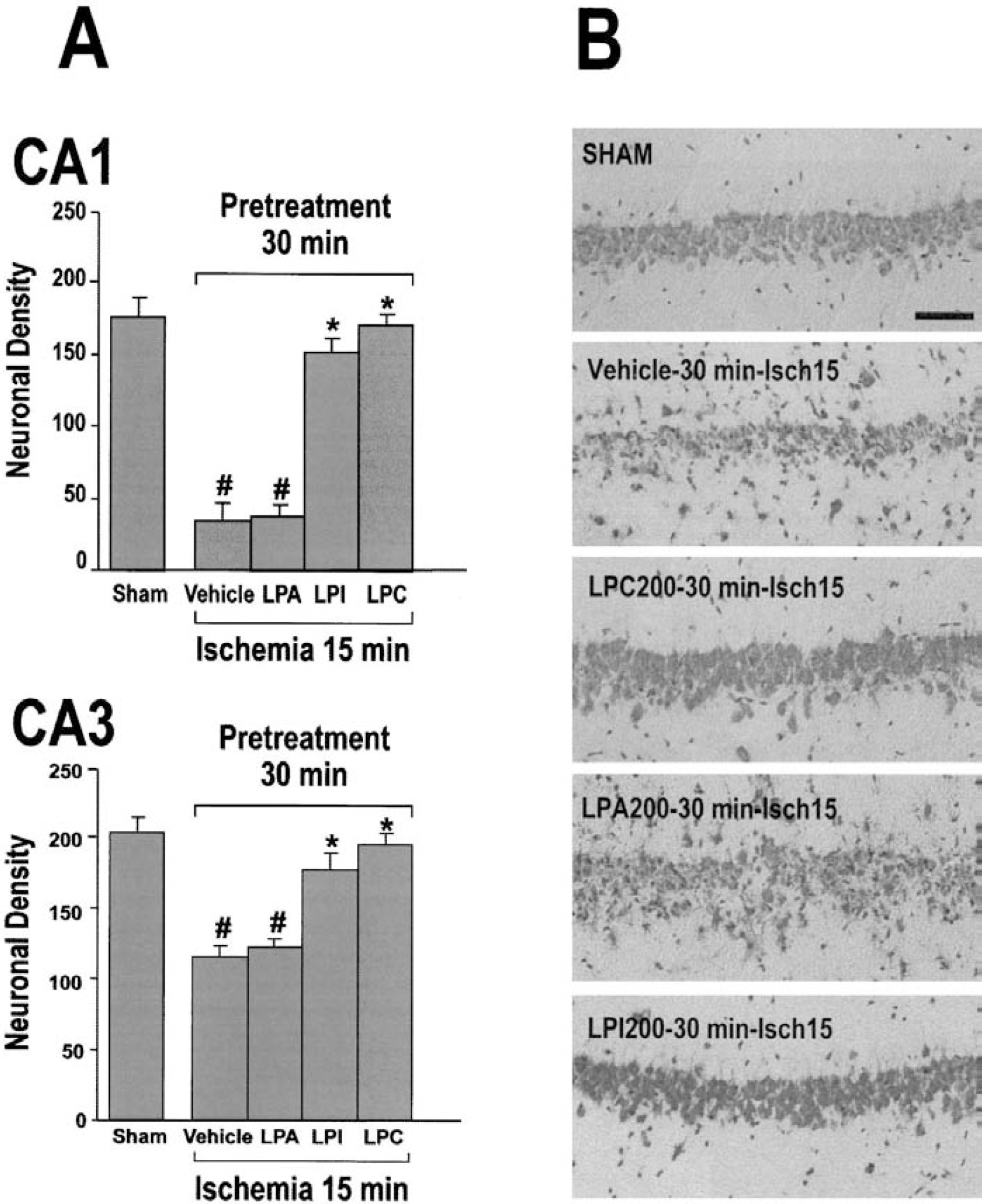
Effects of different lysophospholipids (LPLs; lysophosphatidylcholine [LPC], lysophosphatidylinositol [LPI], and lysophosphatidic acid [LPA]), injected 30 minutes before 15-minute ischemia, on neuronal survival.
Figure 3B shows the most representative effects of LPL pretreatment on morphologic changes observed in the CA1 pyramidal cell layer following severe ischemia. It confirms the quantitative analysis of neuronal density for the different experimental conditions. Similar to the vehicle-injected group, LPA-treated rats exhibited severe damage of pyramidal neurons indicated by pyknotic nuclei and an irregular shape. In contrast, LPC-and LPI-treated animals displayed many healthy CA1 neurons indicated by their round soma and distinct nuclei (Fig. 3B).
Posttreatment with lysophosphatidylcholine or lysophosphatidylinositol, but not with lysophosphatidic acid, prevents hippocampal neuronal cell death induced by transient global forebrain ischemia
The efficiency of posttreatment with LPLs against ischemia-induced neuronal damage has been tested in relation to the type of injection (intraperitoneal vs. intravenous) and the dose used. Figures 4 and 5 summarize the scores of quantified neuronal damage in hippocampal cell layers of ischemic, LPC-treated (Fig. 4), LPI-treated, and LPA-treated rats (Fig. 5). Figure 6 presents the most representative histopathological examples obtained for the corresponding experimental conditions. As previously observed (Lauritzen et al., 2000; Schmidt-Kastner and Freund, 1991), complete forebrain ischemia for 15 minutes induced significant neuronal cell death in the CA1 subfield and, to a lesser extent, in the CA3 pyramidal layer. Compared with the sham-operated group, 85% and 43% of CA1 and CA3 pyramidal cell populations showed signs of neuronal damage in the ischemic group, respectively (Figs. 4 and 6). Intraperitoneal injection of 200-nmol/kg or 500-nmol/kg LPC 30 minutes after the end of global ischemia did not induce neuronal protection. The neuronal density in CA1 and CA3 pyramidal layers of both LPC-treated groups was not significantly different from that of the ischemic vehicle-injected group (which showed an approximate 80% neuronal loss in the CA1 field;Figs. 4 and 6). When animals were given LPC intravenously 30 minutes after the end of ischemia, there was a dose-dependent increase in neuronal survival that reached maximal levels (94% of sham-operated CA1 controls) at 200 nmol/kg. At higher dose (500 nmol/kg), neuronal survival declined (Figs. 4 and 6). Compared with the sham-operated group, intraperitoneal or intravenous injection of 500-nmol/kg LPC alone did not change neuronal counts (Figs. 4 and 6). Treatment with doses of LPI ranging from 50 to 200 nmol/kg given intravenously after the onset of ischemia induced a potent neuronal protection. The three doses tested (50, 100, and 200 nmol/kg) limited the CA1 and CA3 cell destruction to approximately 7% and 11% of neurons (Figs. 5 and 6). In contrast, intravenous injections of different doses of LPA (50–500 nmol/kg) had no beneficial effect on neuronal cell death (Fig. 6). Neuronal counts in CA1 and CA3 pyramidal layers were not significantly different from counts obtained after ischemia (vehicle-injected group). Approximately 82% and 40% of neuronal loss in CA1 and CA3 was observed, respectively (Fig. 5). For control, a single injection of 500-nmol/kg LPA did not affect the neuronal counts. The average neuronal density in pyramidal cell layers of LPC-and LPI-treated rats was not significantly different from that of the sham-operated group (Figs. 5 and 6).
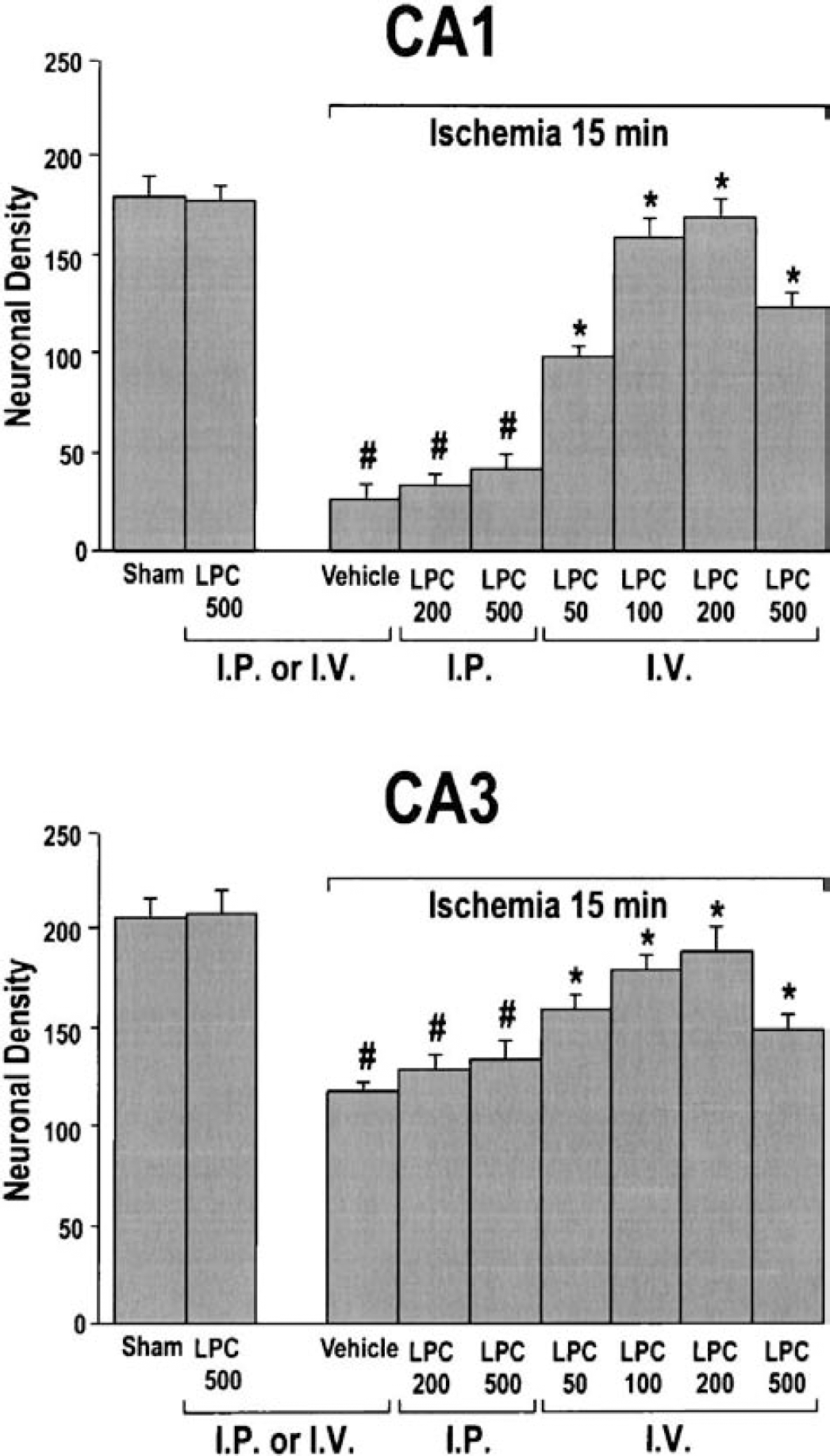
Quantitative analysis of CA1 and CA3 cell survival obtained with different concentrations of lysophosphatidylcholine (LPC) injected 30 minutes after 15-minute global ischemia. Histograms represent the neuronal density assessed in cresyl violet-stained sections per 1-mm linear length of hippocampal CA1 and CA3 pyramidal layers in the different experimental groups subjected to 15 minutes global ischemia. Data represent the mean values ± SEM from counts of two brain sections per slide and 10 slides per rat (n = 6 per group). LPC50, LPC100, LPC200 and LPC500 correspond to LPC (C14:0) administration at a dose ranging from 50 to 500 nmol/kg injected intraperitoneally or intravenously 30 minutes after 15-minute global ischemia. Sham corresponds to sham-operated rats injected with vehicle and vehicle to rats subjected to 15-minute global ischemia and injected with vehicle 30 minutes after the end of the ischemic period. Differences were considered statistically significant when P < 0.05 (Tukey test). *Significantly different from vehicle-injected ischemic animals, #significantly different from sham-operated animals.
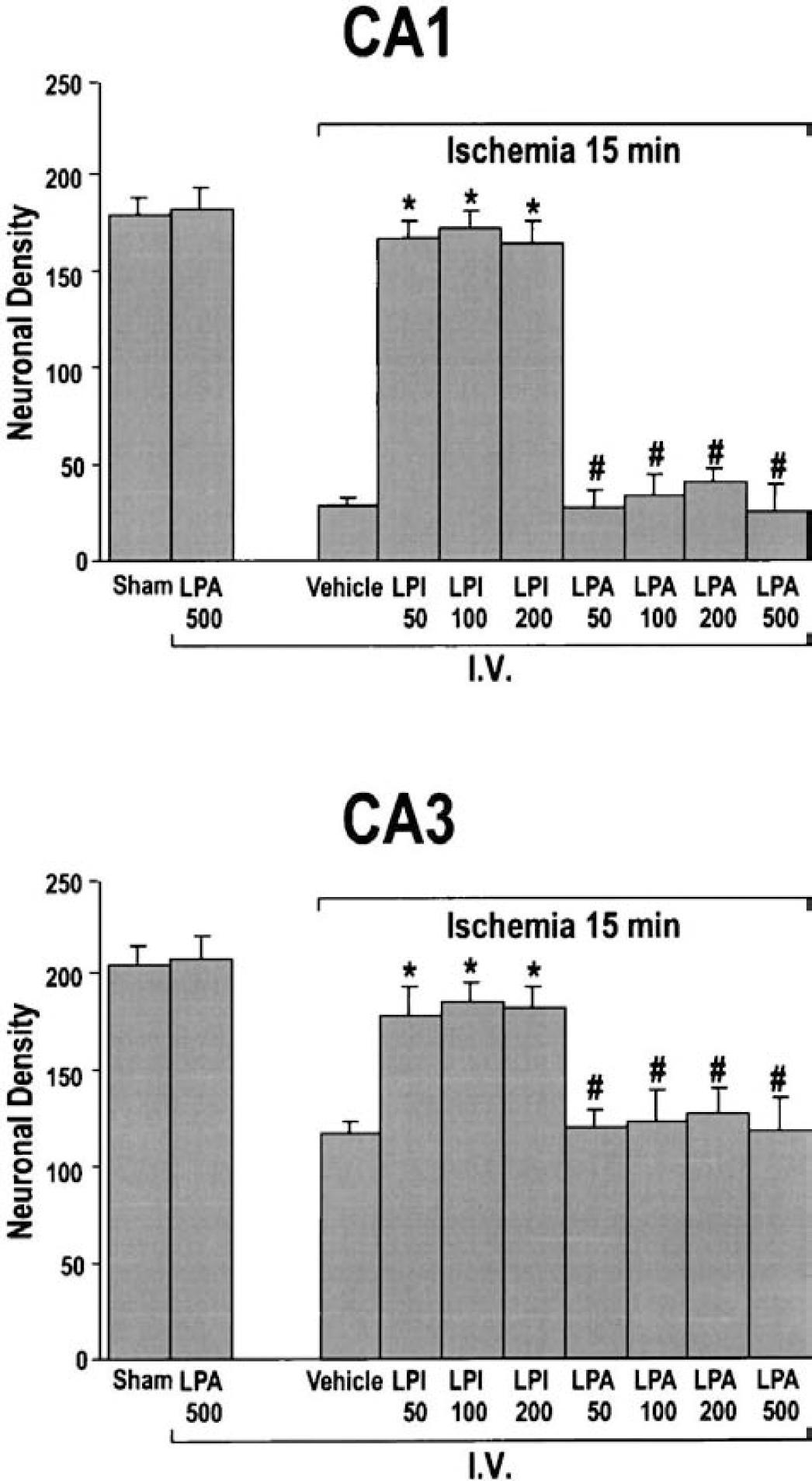
Quantitative analysis of CA1 and CA3 cell survival obtained with different lysophospholipids (LPLs; lysophosphatidylinositol [LPI] and lysophosphatidic acid [LPA]) injected 30 minutes after the end of the ischemic period. Histograms represent the neuronal density assessed in cresyl violet-stained sections per 1-mm linear length of hippocampal CA1 and CA3 pyramidal layers in the different experimental groups submitted to 15-minute global ischemia. Data represent the mean values ± SEM from counts of two brain sections per slide and 10 slides per rat (n = 6 per group). LPI50, LPI100, and LPI200 correspond to LPI administration at a dose ranging from 50 to 200 nmol/kg. LPA50, LPA100, LPA200, and LPA500 correspond to LPA administration at a dose ranging from 50 to 500 nmol/kg. All LPLs were intravenously injected. Sham corresponds to sham-operated rats intravenously injected with vehicle and vehicle to rats subjected to 15-minute global ischemia and intravenously injected with vehicle 30 minutes after the end of the ischemic period. Differences were considered statistically significant when P < 0.05 (Tukey test). *Significantly different from ischemic animals, #significantly different from sham-operated animals.
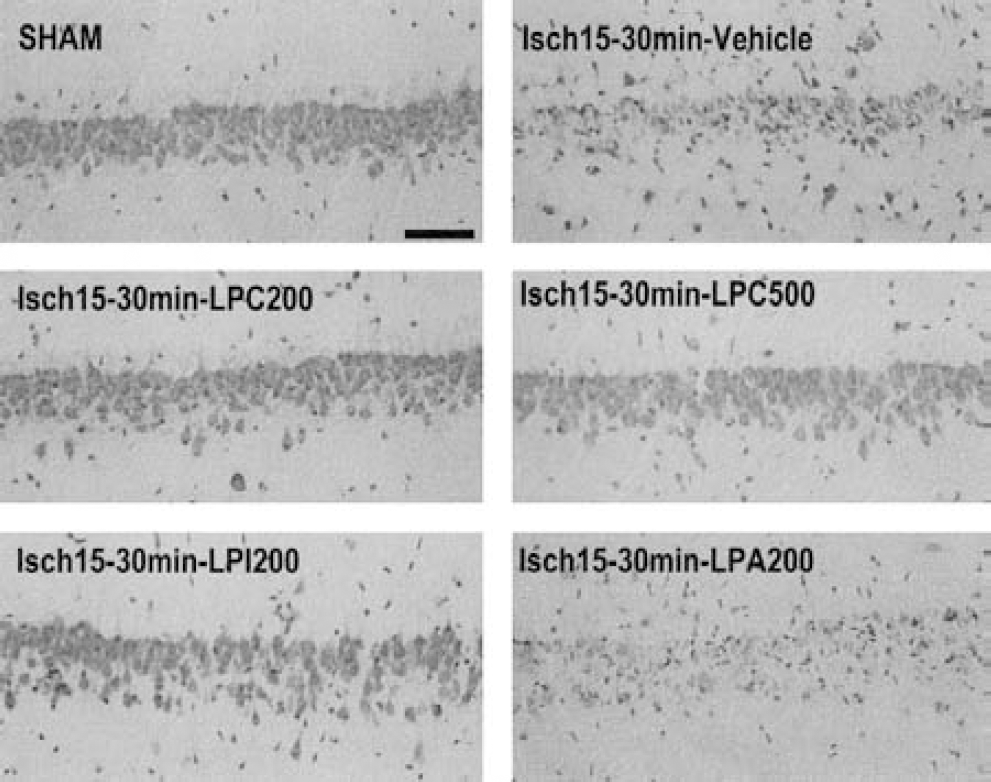
Representative photomicrographs highlighting the effects of different LPLs (lysophosphatidylcholine [LPC], lysophosphatidylinositol [LPI], and lysophosphatidic acid [LPA]) injected 30 minutes following the end of the ischemic period on morphologic changes at the CA1 pyramidal cell layer assessed using cresyl violet staining in rats killed 7 days following ischemia. The LPLs were injected intravenously. LPC200 corresponds to LPC (C14:0) administration at a dose of 200 nmol/kg. LPI200, LPA200, LPC200, and LPC500 correspond to LPI, LPA, or LPC treatment at a dose ranging from 200 to 500 nmol/kg. Sham corresponds to sham-operated rats intravenously injected with vehicle and vehicle to rats submitted to 15-minute global ischemia and intravenously injected with vehicle 30 minutes after the end of ischemia. Scale bar = 100 μm.
Lysophospholipids induce ischemic tolerance against transient global forebrain ischemia
To test the hypothesis that the LPLs described previously as potent neuroprotectors may induce a brain tolerance when injected days before the ischemic event, rats were intravenously injected with 200-nmol/kg LPC or LPI at different times prior to 6-minutes global ischemia, the pharmacologic preconditioning induced neuroprotection depending on the latency between treatment and ischemic insult. As previously described (Blondeau et al., 2000; Blondeau et al., 2001; Heurteaux et al., 1995; Smith et al., 1984), 6-minute ischemia produced severe neuronal damage. Only 21% and 63% of CA1 and CA3 pyramidal cells were preserved, respectively (Fig. 7). Injection of 200-nmol/kg LPC or LPI injected 24, 48, or 72 hours before 6-minute ischemia almost completely inhibited this neuronal loss. The average neuronal densities in pyramidal cell layers of LPC-treated and LPI-treated rats were not significantly different from that of the sham-operated group (Fig. 7). However, when LPC or LPI (200 nmol/kg) was injected as early 6 hours or as long 4 days before 6-minute ischemia, CA1 and CA3 pyramidal cell layers were strongly damaged and showed neuronal densities comparable with the vehicle-injected ischemic group (Fig. 7). The morphologic changes observed in the CA1 pyramidal cell layer 7 days following severe ischemia in rats pretreated with LPC at different times before ischemia are shown in Fig. 8A and confirm the temporal window of effective pretreatment (1–3 days). Similar morphologic changes were observed in CA1 substructure of rats pretreated with LPI (data not shown).
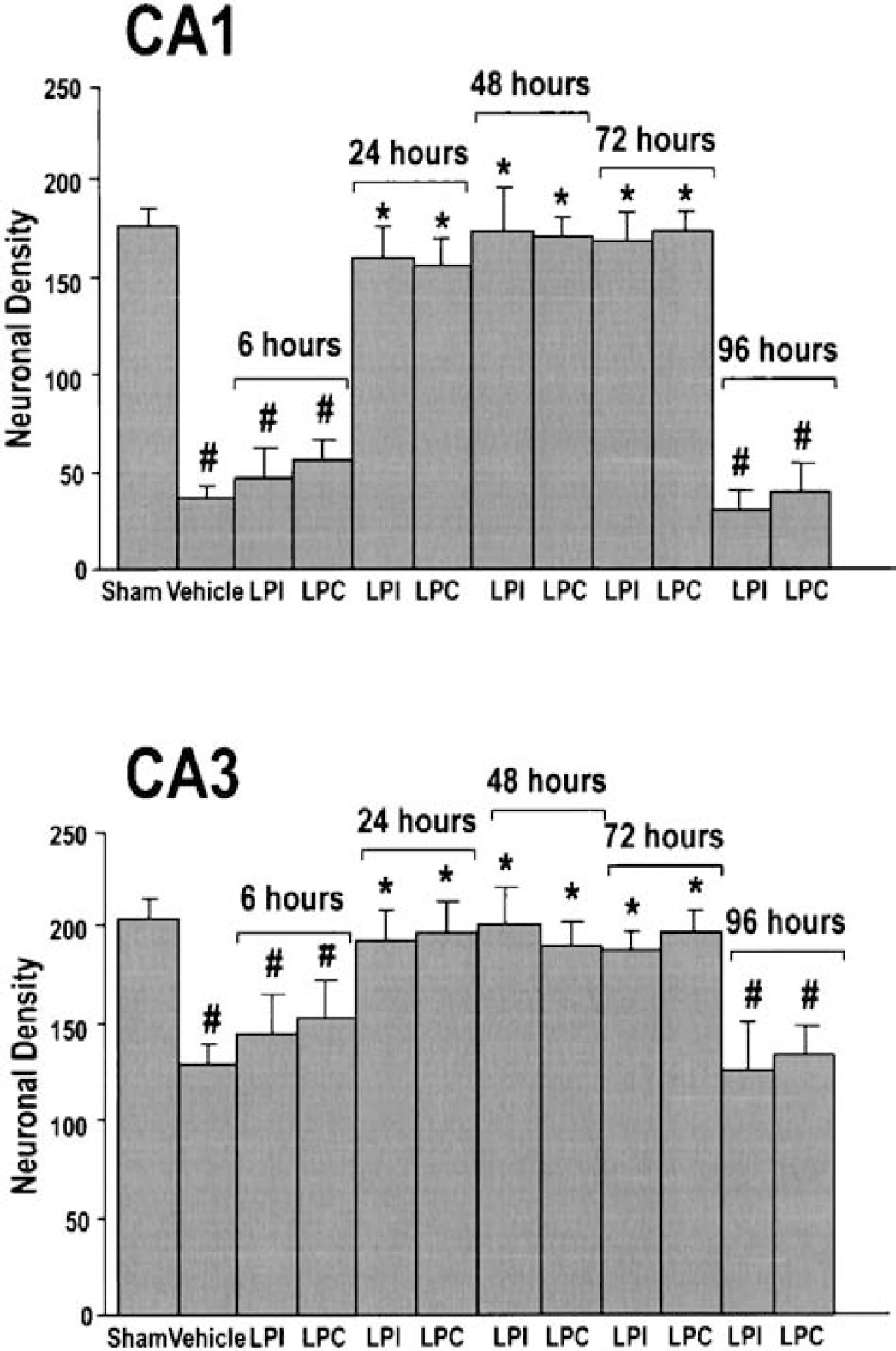
Lysophospholipids induce ischemic tolerance against transient global forebrain ischemia. This figure represents the quantitative analysis of CA1 and CA3 cell survival obtained with lysophosphatidylcholine (LPC) and lysophosphatidylinositol (LPI) injected at different times (6, 24, 48, 72 and 96 hours) before 6-minute ischemia. Histograms represent the neuronal density assessed in cresyl violet-stained sections per 1-mm linear length of hippocampal CA1 and CA3 pyramidal layers in the different experimental groups submitted to 6-minute global ischemia. Data represent the mean values ± SEM from counts of two brain sections per slide and 10 slides per rat (n = 5 per group). Both LPI and LPC were intravenously injected at a dose of 200 nmol/kg. LPC corresponds to LPC (C14:0). Sham corresponds to sham-operated rats injected with vehicle and vehicle to ischemic rats injected with vehicle 72 hours before the onset of 6-minute ischemia. Differences were considered statistically significant when P < 0.05 (Tukey test). *Significantly different from ischemic animals, #significantly different from sham-operated animals.
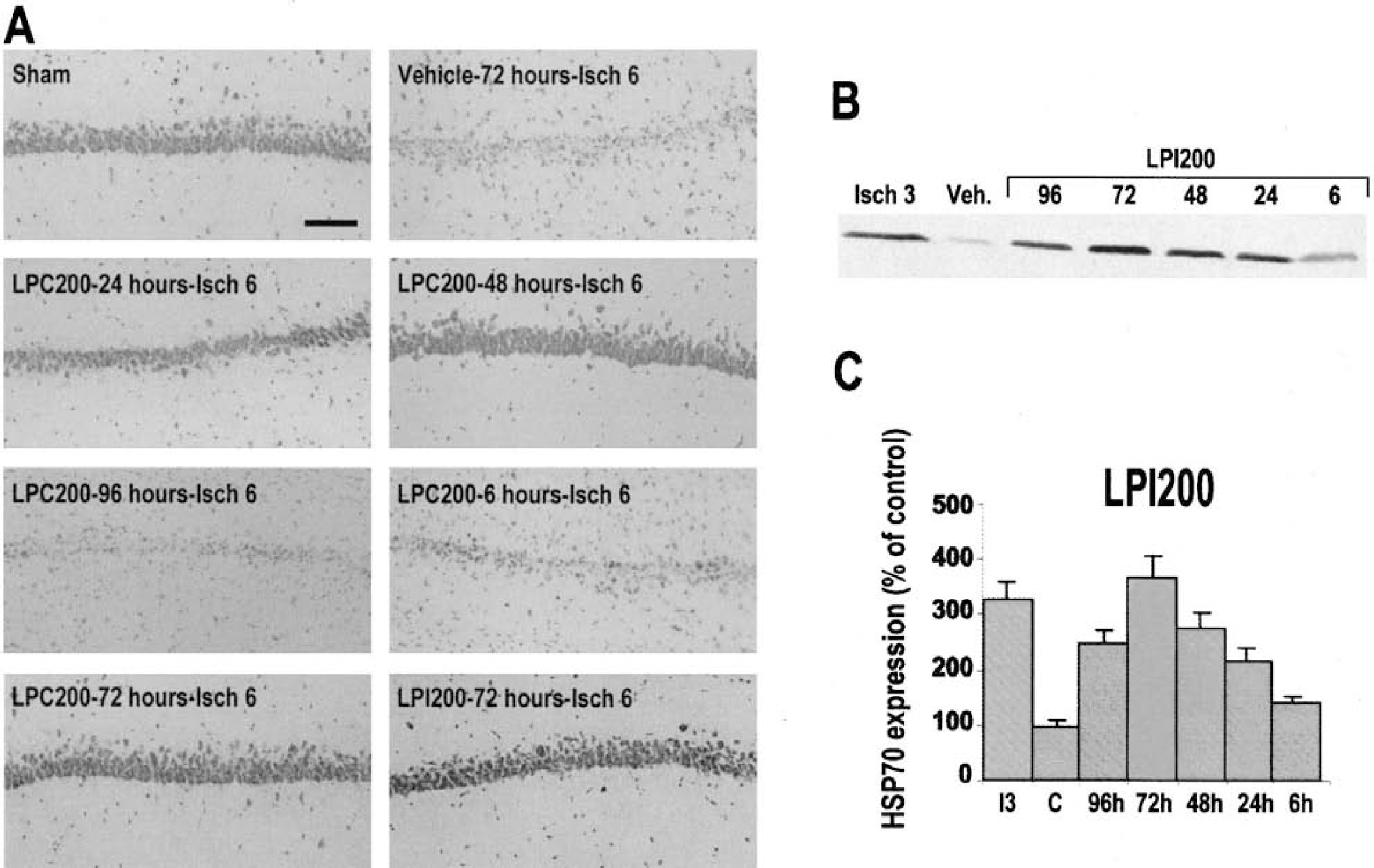
Lysophospholipids (LPLs) induce ischemic tolerance against transient global forebrain ischemia.
Several lines of evidence indicate that the development of cerebral tolerance in vivo coincides with the induction of expression of the 70-kDa HSP70 within the time window of protection (Blondeau et al., 2000; Blondeau et al., 2001; Kirino et al., 1991). To confirm the establishment of a pharmacologic preconditioning by a pretreatment with LPLs, the current study investigated the potential temporal profile of HSP70 synthesis in hippocampus following pretreatment with LPLs at different times before ischemia. Figure 8B depicts the induction of HSP70 protein in the hippocampus at 6, 24, 48, 72, and 96 hours following LPI treatment assessed using Western blot analysis. HSP70 protein remained weakly detectable in protein extracts from hippocampi of rats injected with vehicle at any time following injection or with 200-nmol/kg LPI at 6 hours after injection. The administration of 200-nmol/kg LPI led to a progressive accumulation of HSP70 protein detected in hippocampi 24 hours following LPI treatment with a maximal expression level reached at 72 hours.
Analysis of 1-palmitoyl-lysophosphatidylcholine transport across the blood—brain barrier
To evaluate how much and what form of LPL is entering the brain, 1-[1–14C]-palmitoyl-LPC was intravenously injected according to the protocol of Alberghina et al. (1994). Figure 9A shows the percent distribution of radioactivity of total lipids extracted from brain, liver, spleen, and kidney after short-time tail-vein injection. When the perfusions were carried out for 15 seconds, approximately 0.10% of the perfused 1-[1–14C]-palmitoyl-LPC was entering into the brain. The radioactivity distribution among metabolic products in the organic phase of brain lipid extracts was as follows: unmetabolized LPC (55%), phosphatidylcholine (30%), and saturated fatty acid palmitate (15%). Figure 9B shows that a treatment with 1-palmitoyl-LPC prevented hippocampal neuronal death induced by transient (15-minute) global ischemia. An injection of 200-nmol/kg 1-palmitoyl-LPC given intravenously 30 minutes before 15-minute ischemia (data not shown) or 30 minutes after the onset of 15-minute ischemia (Fig. 9B) induced a potent neuronal protection. Compared with the vehicle-injected rats, where CA1 and CA3 pyramidal cell layers showed severe signs of neuronal damage, the 1-palmitoyl-LPC—treated animals were strongly protected against ischemia. The findings that phosphatidylcholine does not activate TREK and TRAAK channels (Maingret et al., 2000b), saturated fatty acid palmitate does not protect against global ischemia (Lauritzen et al., 2000; Maingret et al., 2000b), and 1-palmitoyl-LPC treatment provides a potent neuroprotection against ischemic insult strongly suggest that the unmetabolized LPL fraction (55% LPC in that case) is responsible of neuroprotective effects.
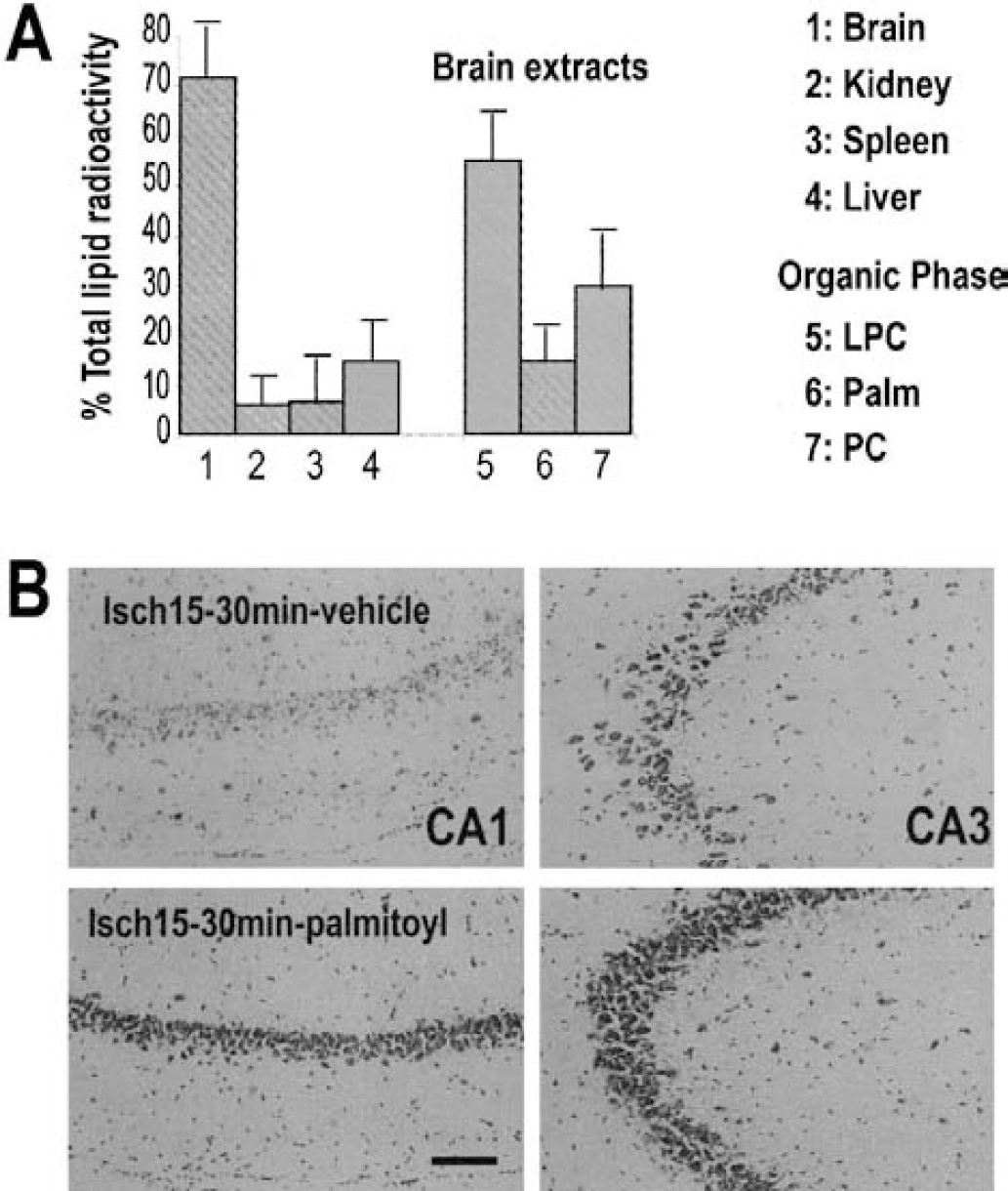
Analysis of 1-[1–14C]-palmitoyl-LPC transport across the blood—brain barrier.
DISCUSSION
Lysophospholipids are important intermediates in the synthesis and degradation of membrane phospholipids. They regulate a variety of biologic processes, including cell proliferation and tumor cell invasiveness, and are involved in inflammation (Moolenaar, 1999; Spiegel and Milstien, 1995). Lysophosphatidylcholine, produced by the action of phospholipase A2 on phosphatidylcholine, promotes inflammatory effects including macrophage activation, increased expression of endothelial cell adhesion molecules and growth factors (Kume et al., 1992; Kume and Gimbrone, 1994; Yamamoto et al., 1991), elicitation of vasoconstriction, and inhibition of endothelium relaxation (Murohara et al., 1994; Okatani et al., 2001), which is why LPLs effects are generally expected to be deleterious. Because LPLs levels increase during cerebral ischemia (Kinouchi et al., 1990; Moto et al., 1991), a deleterious role of LPLs would be expected in this pathology. This study suggests that instead of provoking neurotoxicity and neurodegeneration, LPLs, particularly LPC and LPI, protect vulnerable brain regions against global ischemic injury. Lysophosphatidylcholine and LPI are neuroprotective when they are injected in one single intravenous dose 30 minutes before the onset of ischemia or 30 minutes after the end of ischemia. Lysophospholipids also induce a sustained brain tolerance, which manifests itself several days after LPL injection. A pretreatment with LPC or LPI (200 nmol/kg) intravenously injected 1 to 3 days before severe 6-minute ischemia triggers 98% of cell survival in the CA1 pyramidal layer.
A neuronal protection has been also observed in an in vitro model of excitotoxicity using glutamate treatment. The level of protection is dependent on the concentration of glutamate and time of application of the glutamate treatment. At a concentration of 40-μmol/L glutamate, which causes rapid cell swelling, LPC and LPI (10 μmol/L) were both efficient if applied before, during, and after exposure to glutamate, whereas they had no effect when applied only during and after the toxic insult. However, when LPC and LPI (10 μmol/L) were applied only during and after exposure to glutamate, a strong neuroprotection was obtained at a concentration of 20-μmol/L glutamate, corresponding to the extracellular glutamate concentration observed in ischemic brains. The differences observed in neuronal protection with LPL posttreatment between in vivo and in vitro models give the limits of excitotoxicity models on cell culture. It is well established that, in vivo, glutamate itself is only minimally effective as an excitotoxin when it is directly applied to the brain because both neurons and astrocytes have active glutamate transport systems that are capable of taking up glutamate from the extracellular fluid. These transport systems are not present in the in vitro model used. With regard to transient ischemia, glutamate released into the extracellular space is rapidly cleared during reperfusion, and the extracellular glutamate increases are significant but moderate (Obrenovitch and Urenjak, 1997). With 15-minute ischemia, the limit of protection induced by LPL after treatment is not reached as it is with an in vitro application of 40-μmol/L glutamate. Longer periods of ischemia are necessary to lose the protection by LPLs.
The molecular target involved in the neuroprotective effects of LPLs remains to be unambiguously identified. However the best candidates we have at hand are the recently cloned TREK-1, TREK-2, and TRAAK channels, which correspond to a new class of background K+ channels with a 2P-domain structure (Lesage and Lazdunski, 2000; Patel and Honore, 2001; Patel et al., 2001), and which are strongly activated by certain LPLs (Lesage et al., 2000; Maingret et al., 2000b). The first reason to consider these channels as potential targets is that there is growing evidence that the activation of K+ channels represents an opportunity for a new neuroprotective strategy against cerebral diseases. Thus, openers of ATP-sensitive K+ channels (KATP) have been described as potent neuroprotectors against ischemia and epilepsy (Abele and Miller, 1990; Heurteaux et al., 1993; Lauritzen et al., 1997; Plamondon et al., 1999), and the same channel type is clearly involved in ischemic and epileptic tolerance (Plamondon et al., 1999; Reshef et al., 1998). The second reason is that PUFAs, which are known to activate background channels such as TREK1, TREK2, and TRAAK channels (Fink et al., 1998; Lesage et al., 2000; Patel et al., 1998), prevent neurodegeneration induced by cerebral ischemia or epilepsy (Lauritzen et al., 2000) and also induce brain tolerance (Blondeau et al., 2001). Other drugs such as riluzole also activate these channels and are neuroprotective (Duprat et al., 2000; Ettaiche et al., 1999; Lang-Lazdunski et al., 1999; Pratt et al., 1992). The third factor suggesting an important role of background 2P-domain K+ channels in neuroprotection is the parallel obtained between levels of cell survival and levels of activation of TREK and TRAAK channels by different LPLs. The LPLs with large polar heads and long acyl chains, such as LPC and LPI, which are known to open background channels, prevent neuronal death induced by ischemia and glutamate treatment.
Conversely, no neuroprotection was obtained with LPA, which is ineffective as an activator of these channels. In fact, LPA has been reported to induce stimulation of neurite retraction, stimulate astrocyte proliferation (Jalink et al., 1993; Keller et al., 1997; Tigyi and Miledi, 1992), and inhibit glutamate uptake by astrocytes (Keller et al., 1996). Furthermore, high concentrations or prolonged exposures to LPA induce necrosis and apoptosis in hippocampal neurons (Holtsberg et al., 1998). The opening of TREK and TRAAK channels by LPLs is expected to lead to a synaptic hyperpolarization (probably via synaptic TRAAK channels (Lauritzen et al., 2000; Reyes et al., 2000) as well as an hyperpolarization of dendrites (probably via TREK1 channels, which are dendritic;Lauritzen et al., 2000), which would reduce glutamate release and decrease NMDA receptor-associated Ca2+-permeable channel activity via a reinforcement of Mg2+ blockade. This study showing the analysis of 1-palmitoyl-LPC transport across the blood—brain barrier provides an additional argument in favor of the activation of TREK and TRAAK channels by LPLs in the neuroprotection observed. When 200-nmol/kg LPC was intravenously injected, 0.1% entered into the brain in the form of 55% unmetabolized LPC, leading to a cerebral LPC concentration in the range known to activate TREK and TRAAK channels (Maingret et al. (2000b). The neuroprotection by LPC and LPI might be due, in addition to their effect on TREK and TRAAK channels, to a direct blockade of NMDA channels by LPLs, which also takes place (Casado and Ascher, 1998). Lysophospholipids are also known as potent chelators and contain chaperonelike activities (Kern et al., 2001). These effects could contribute to the neuroprotective effects of LPLs, particularly at high concentrations.
Further work is clearly needed to understand the molecular mechanisms involved in the neuroprotection induced by LPLs, and particularly to demonstrate in a definitive way the role of TREK and TRAAK background channels. The dominant effect of the activation of TREK and TRAAK channels in LPL-mediated cell survival would be proven if specific blockers of these channels were available, but it is currently unknown whether these channel blockers exist. The knockout strategy is another route. Nevertheless, whatever the exact mechanism of action, the observation that a single intravenous injection of LPLs exerts strong neuroprotective effects, even when these compounds are injected after the ischemic insult, is extremely promising and warrants further clinical investigation. In addition to these observations, we have an interesting new model of ischemic tolerance. This pharmacologic “preconditioning” induced by LPLs may also be a neuroprotective strategy against irreversible brain damage for individuals who are at risk for stroke.
Footnotes
Acknowledgments
The authors thank R. Mengual for his valuable help in lipid experiments, and G. Jarretou, I. Lemasson, F. Aguila, and V. Lopez for their skillful technical assistance.