
Abstract
Preconditioning with sublethal ischemia attenuates the detrimental effects of subsequent prolonged ischemic insults. This research elucidates potential in vivo cross-tolerance between different neuronal death-generating treatments such as kainate administration, which induces seizures and global ischemia. This study also investigates the effects of a mild epileptic insult on neuronal death in rat hippocampus after a subsequent, lethal epileptic stress using kainic acid (KA) as a model of epilepsy. Three preconditioning groups were as follows: group 1 was injected with 5 mg/kg KA before a 6-minute global ischemia; group 2 received a 3-minute global ischemia before 7.5 mg/kg KA; and group 3 was injected with a 5-mg/kg dose of KA before a 7.5-mg/kg KA injection. The interval between treatments was 3 days. Neuronal degeneration, revealed by the silver impregnation method and analysis of cresyl violet staining, was markedly reduced in rats preconditioned with a sublethal ischemia or a 5-mg/kg KA treatment. Labeling with terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′triphosphate-biotin nick-end labeling and DNA laddering confirmed the component of DNA fragmentation in the death of ischemic and epileptic neurons and its reduction in all preconditioned animals. The current study supports the existence of bidirectional cross-tolerance between KA excitotoxicity and global ischemia and suggests the involvement of adenosine A1 receptors and sulfonylurea- and ATP-sensitive K+ channels in this protective phenomenon.
Under some circumstances, both transient global or focal ischemic attacks generate cerebroprotective events that enhance the brain's resistance to detrimental effects of subsequent, longer episodes of ischemia (Glazier et al., 1994; Heurteaux et al., 1995; Kirino et al., 1991; Kitagawa et al., 1990; Liu et al., 1992). This phenomenon has been termed ischemic preconditioning (Murry et al., 1986). Thus, beneficial effects of ischemic tolerance to stroke (focal ischemia) are demonstrated by the observation that brief episodes of global ischemia significantly attenuate infarct size in animals subsequently treated with permanent middle cerebral artery occlusion (Simon et al., 1993). In addition, tolerance to focal ischemia induced by prior exposure to transient focal ischemia has been described in rats (Chen et al., 1996). Other cellular stresses, including cortical spreading depression (Kawahara et al., 1994; Matsushima et al., 1996), oxidative stress (Ohtsuki et al., 1992), hypoxia, or hyperthermia also induce protection against ischemic neuronal damage. Similar to ischemic tolerance, hippocampal kindling or bicuculline-induced seizures also have been shown to increase the resistance of the brain to the neuronal injury associated with a second epileptic challenge. This has been termed epileptic tolerance (Kelly and McIntyre, 1994; Sasahira et al., 1995).
The molecular and cellular processes underlying neuronal degeneration remain poorly understood. However, neuropathologic states such as ischemia and epilepsy have been shown to share histologic, biological, and metabolic correlates. Several studies demonstrate extensive neuronal degeneration in selective cellular layers of the hippocampus in both conditions (Lothman and Collins, 1981; Pulsinelli et al., 1982; Smith et al., 1984; Sperk et al., 1983). Global ischemia and kainic acid (KA) (at doses producing epileptic seizures) also greatly enhance neuronal activity and induce sustained depolarization and extensive intracellular Ca2+ entry, which potentiate the release and action of endogenous excitatory amino acids, activation of caspases, and nitric oxide production, leading to excitotoxic cell death by necrosis, apoptosis, or both (Choi, 1988; Huang et al., 1994; Namura et al., 1998; Siesjö and Wieloch, 1985).
In light of these shared biochemical and histopathologic features, the first objective of the current study was to characterize the potential existence of in vivo cross-tolerance between an excitotoxic KA treatment and global ischemia through assessment of necrotic and apoptotic neuronal death in these animal models. Our study also investigates the effects of a mild epileptic insult on neuronal death after a subsequent, lethal epileptic stress in rat hippocampus using KA as a model of epilepsy. Global forebrain ischemia was produced by four-vessel occlusion (Pulsinelli and Brierley, 1979). The glutamate analogue KA was administered systemically at doses shown to evoke behavioral seizures known resembling human temporal lobe epilepsy (the most common form of adult human epilepsy) (Lothman and Collins, 1981; Nadler, 1981; Pulsinelli and Brierley, 1979). Using these rat models, we investigated the effect of preconditioning by sublethal ischemia or KA-induced seizures on subsequent neuronal degeneration after a more severe ischemic insult or KA treatment. The hippocampus was selected for assessment of neuronal damage, since this brain structure is among the most vulnerable to both ischemia- and KA-induced neuronal death. Time-dependent changes in necrotic and apoptotic neuronal death were determined using cellular DNA fragmentation combined with analysis of neuronal density and silver impregnation technique. Although the physiologic mechanisms underlying ischemic tolerance remain unknown, it is acknowledged that a minimal time interval is required between stimuli (in the different models) for the protective effects to become manifest. This phenomenon suggests the involvement of a biological cascade. In this context, adenosine agonists and ATP-sensitive K+ channel (KATP) openers mimic some of the cerebroprotective effects of preconditioning. Furthermore, these effects are reversed by adenosine antagonists and KATP blockers, suggesting that adenosine release and activation of KATP channels through adenosine A1 receptors may constitute an early step of ischemic cerebral preconditioning (Abele and Miller, 1990; Heurteaux et al., 1995; Perez-Pinzon et al., 1996). Thus, the second objective of this study was to assess the possibility that adenosine and KATP channels participate in the biological cascade of protective events associated with KA-induced epileptic tolerance or cross-tolerance between KA-induced epilepsy and global ischemia.
MATERIALS AND METHODS
Animals
Experiments were performed on 375 male Wistar rats weighing 250 to 300 g (Charles River Breeding Laboratories, St. Aubin, France). The animals, maintained on a 12:12 hour light-dark cycle, were given food and water ad libitum and were acclimated to the animal facility for at least 1 week before drug treatment or surgery.
Preconditioning procedures
The preconditioned animal groups were as follows. Group 1 was administered KA (5 mg/kg intraperitoneally) before 6-minute global ischemia (KA5-I6). Group 2 received a 3-minute global ischemia before KA administration (7.5 mg/kg intraperitoneally) (I3-KA7.5). Group 3 was injected with a KA (5 mg/kg intraperitoneally) before a second challenge with KA (7.5 mg/kg intraperitoneally) (KA5-KA7.5). The time interval between treatments was 3 days. Rats were killed at 3 and 7 days after the last treatment, respectively. Arterial occlusions of 3 and 6 minutes' duration were chosen for sublethal ischemia and lethal ischemic episode, respectively, based on previously published findings. The doses of 5 and 7.5 mg/kg KA were selected based on optimal neuroprotection obtained from a dose-response profile in an earlier pilot study (data not shown). The first epileptic stress was chosen so that it did not induce neuronal death, and 5 mg/kg appeared to be the optimal concentration in this model. A concentration higher than 5 mg/kg of KA for this first epilepsia would produce neuronal damage in CA1 and CA3 pyramidal layers. The second epilepsia must be severe enough to induce neuronal death by itself. A concentration of 7.5 mg/kg of KA is enough to substantially damage hippocampal neurons. However, doses of KA for the second epileptic stress cannot exceed 10 mg/kg. Thus, neuronal protection conferred by preconditioning is related to the intensity of the second lethal ischemic or epileptic episode. Saline-injected or sham-operated animals treated 3 days before KA7.5 injection or 6-minute ischemia were used as controls. Behavioral effects of KA were monitored for 4 consecutive hours after injection, and seizure activity was controlled with administration of diazepam (7 mg/kg intravenously) after that monitoring period. The severity of limbic seizures was determined using a modification of the behavioral scale proposed by Zhang et al. (1997). Briefly, the epileptic convulsion profile of animals was classified according to six distinct stages characterized as follow: stage 1, immobilization, staring, mouth and facial movements; stage 2, wet dog shakes; stage 3, forelimb clonus, head nodding, chewing, and yawning; stage 4, rearing, increased forelimb clonic jerks, salivation and frothing at the mouth; stage 5, hemorrhagic foaming, rearing and falling; and stage 6, intense agitation, jumping, and circling, often leading to death within 68 hours after KA administration. Stage 0 corresponded to animals showing no abnormal behaviors.
Forebrain ischemia was performed by four-vessel occlusion as previously described (Pulsinelli and Brierley, 1979). Briefly, rats were deeply anesthetized by inhalation of 2% halothane mixed with 30% oxygen and 70% nitric oxide and their body temperature maintained at 37°C with a heating blanket during and in the hours after surgery. The vertebral arteries were irreversibly occluded by electrocoagulation, and a small-diameter silicon tubing was looped around the carotid arteries to facilitate subsequent occlusion. On the following day, both common carotid arteries were clamped with microvascular clamps for 3 or 6 minutes, depending on the treatment condition, in awake and spontaneously ventilating animals. Rats lost their righting reflex within 1 minute of carotid clamping. Cessation of electroencephalographic activity was confirmed during ischemic insults.
Drug treatments
All drugs were freshly mixed on the day of experimentation. Kainic acid (Sigma, St. Quentin, France) was dissolved in NaCl 0.9% solution and injected intraperitoneally (5 or 7.5 mg/kg). (−)Cromakalim (Beecham Pharmaceuticals) was first dissolved in ethanol at a concentration of 10–1 mol/L and then diluted in NaCl 0.9% solution to reach final concentration of 2 mmol/L (10 nmol/5 μL injected). (−)Cromakalim was administered intracerebroventricularly 30 minutes before KA (7.5 mg/kg) injection. Glibenclamide (Pfizer) was first dissolved in NaOH 0.1 N at a concentration of 10–1 mol/L and then diluted to final concentration of 4 mmol/L in NaCl 0.9% solution (20 nmol/5 μL injected). Glibenclamide was intracerebroventricularly injected 30 minutes before the initial ischemia, KA injection (5 mg/kg intraperitoneally), or R-phenylisopropyl-adenosine (R-PIA, Research Biochemicals International) (25 μg/kg intravenously) injection. The R-PIA, dissolved in ethanol at a concentration of 10–1 mol/L and then diluted to final concentration of 10–4 mol/L in NaCl 0.9% solution, was intravenously (25 μg/kg) administered immediately preceding KA treatment (7.5 mg/kg intraperitoneally). 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX, Research Biochemicals International) was dissolved in ethanol at a concentration of 10–1 mol/L, then diluted to final concentration of 10–3 mol/L in NaCl 0.9% solution and injected (250 μg/kg intraperitoneally) 2 hours before the first ischemia or KA (5 mg/kg) injection. A volume of 0.5 mL/kg and 1 mL/kg were injected for R-PIA and DPCPX, respectively. The pH of the solutions was adjusted to 7. With the exception of (−)cromakalim, which was injected once daily during the experimental duration, all drugs used as pretreatments were administered only once. Control experiments were performed with intraperitoneal, intracerebroventricular, or intravenous injection of NaCl 0.9% under the same procedural conditions as those highlighted for the different drugs. A pilot study revealed no significant differences between control rats injected with saline and those injected with vehicle containing NaOH or ethanol at the concentrations used for dilution of the drugs. The same observations also were obtained from earlier studies using similar vehicles.
Tissue preparation
At the end of the experiment, animals were deeply anesthetized by inhalation of 2% halothane mixed with 30% oxygen and 70% nitric oxide, and the brains were quickly extracted and fresh frozen in isopentane at −45°C. Sections (12 μm) were cut on cryostat (Leica) and postfixed by immersion in 4% paraformaldehyde/10–2 mol/L phosphate buffer (PBS) for 30 minutes. Slides then were dehydrated in ethanol baths (50%, 70%, and 100%), air dried, and stored at −70°C until use. For each brain studied (n = 6 per time point and treatment), 6 sections were placed on 3-aminopropylethoxysilane-coated slides, and 10 slides (randomly chosen) per rat were used in each stage of analysis. A neuropathologist, blind to the experimental conditions, performed the histologic assessment using light microscopic study.
Analysis of neuronal density on cresyl violet-stained sections
The neuronal density of the hippocampal CA1 and CA3 subfields was determined by the method of Kirino et al. (1986). Analysis of neuronal density was performed on coronal sections of the dorsal hippocampus stained with cresyl violet and corresponding to brain sections located between 3.14 and 4.16 mm posterior to bregma (Paxinos and Watson, 1986). The total linear length of the CA1 and CA3 sectors was measured using a digitizer. The number of living neurons in the stratum pyramidale within CA1 and CA3 subfields was counted using a Leica Aristoplan photomicroscope at a magnification of ×400. Neurons that had shrunken cell bodies with surrounding empty spaces were excluded. The neuronal density of CA1 and CA3 sectors (i.e., the number of intact pyramidal cells per 1 mm linear length of the CA1/CA3 stratum pyramidale observed in each 12-μm section) was quantified. Thus, a mean value for each hippocampal CA1/CA3 substructure was obtained from 20 bilateral measurements on 6 sections per animal, for the 6 animals in each of the experimental groups. In KA-treated rat groups, selected rats for neuronal density assessment in the CA1 and CA3 hippocampal layers included three animals reaching stage 4 of convulsion and three animals reaching convulsion stage 5. The neuronal density for a given animal represents the average of both the right and left hippocampal neuronal cell densities. Neuronal density values are expressed as mean ± standard error of the mean. Data analysis was performed by two-factor (experimental condition and brain region) analysis of variance followed by Tukey's w test for multiple comparisons. A P value of < 5% was considered statistically significant.
Silver impregnation technique
The silver staining method (Gallyas et al., 1990b) was used to visualize degenerating argyrophilic cells. For this procedure, frozen sections were dehydrated by successive immersion for 5 minutes in 1-propanol baths (50%, 75%, and 100%). Slides then were esterified at 56°C for 16 hours in 1-propanol containing 4 mL of distilled H2O and 0.8 mL concentrated sulfuric acid per 100 mL. The next day, slides were rehydrated in 50% and 25% 1-propanol and washed twice in distilled H2O. Slides then were acetylated in two successive 10-minute distilled H2O baths containing 1.5% acetic acid and in the silicotungstate developer for 7 minutes. The developer was prepared 3 minutes before use by mixing one part of stock solution A and one part of stock solution B. Solution A contained 100 g Na2CO3 (anhydrous) in 1 L of distilled H2O, whereas solution B contained 2.0 g AgNO3, 20 g tungstosilicic acid, and 3.5 mL of 40% formaldehyde, dissolved in 1 L of distilled water in the given order. Sections then were quickly dipped in distilled water and dried and coverslips were applied.
DNA nick end labeling of tissue sections
Coronal cryostat sections (12 μm) from rats killed 72 hours after the last treatment were used and processed according to the terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′triphosphate-biotin nick-end labeling (TUNEL) method (Gavrieli et al., 1992). Briefly, sections were rehydrated in ethanol (95%, 70%, and 50%) followed by PBS and bathed in 0.3% H2O2 in methanol to inactivate endogenous peroxidase. Sections then were permeabilized in 0.3% Tween/ PBS and washed twice in PBS before application of the TUNEL reaction mixture (Boehringer-Mannheim, Meylan, France). Positive control was obtained by preincubation of a section with DNase I (20 mg/mL) for 15 minutes at 37°C before incubation with biotinylated dUTP. In negative control slides, DNA polymerase was omitted from the incubation with biotinylated dUTP. All slides were incubated in a humid chamber at 37°C for 2 hours. Sections then were washed twice in PBS and incubated overnight at 4°C with the secondary anti-fluorescein-peroxidase conjugate. On the following day, sections were washed three times with PBS and labeling revealed with 3′-diaminobenzidine and nickel chloride (DAB-Ni). After a final rinse in distilled H2O, coverslips were applied to the sections.
Analysis of DNA fragmentation
Genomic DNA was extracted from control, ischemic, epileptic, or preconditioned hippocampi (isolated 3 days after ischemia). Fifty to 100 mg of tissue was lysed in 1 mL of TRIzol reagent (GIBCO BRL, Pontoise). After addition of 0.2 mL of chloroform, the samples were centrifuged at 12000 × g for 15 minutes at 4°C. After precipitation in ethanol and a series of washes, the DNA was solubilized in 8 mmol/L NaOH. Residual RNA was removed by incubation with RNAse A (10 mg/mL) at 37°C for 30 minutes. DNA (3 μg) was 3′end-labeled with [α32P]dCTP and terminal deoxynucleotidyl-transferase enzyme, electrophoresed on 1.5% agarose gel, and autoradio-graphed on Kodak X-OMAT film.
RESULTS
Evidence of mutually protective actions of KA epileptic preconditioning and global sublethal ischemia on hippocampal (necrotic and apoptotic) cell death
Behavioral responses associated with the different models of tolerance. After systemic KA administration, rats exhibited progressive and dose-dependent stereotyped behavioral seizures. This KA-induced epileptic behavioral profile was monitored for 4 hours after injection using the six distinct stages in the scale described by Zhang et al. (1997) (see Materials and Methods). To enable direct comparisons of the different treatment effects, only KA-injected rats displaying comparable seizure activity and duration were included for the histopathologic study. Thus, animals that had reached convulsion stages 4 or 5 (except for passive controls) were selected to better control for reduced neuronal death not specifically attributable to reduced convulsions but rather resulting from the treatments per se. Table 1 (part 1) highlights recorded behavioral profiles for the different controls and tolerance models. No abnormal behavior was observed in passive control groups (saline-injected and sham-operated rats). In contrast, most animals of the active control groups, KA5 (5 mg/kg) and KA7.5 (7.5 mg/kg), reached stages 4 and 5 with most rats displaying grade 4 seizures after a systemic KA5 injection, whereas an equal proportion of KA7.5-injected rats reached grade 4 or 5 seizure levels. Seizure progression was initially characterized by staring spells and wet dog shakes occurring within the first 30 minutes after KA injection, followed by overt signs of motor limbic seizures lasting approximately 90 minutes and differing in intensity and duration in a dose-dependent manner. Preconditioning with KA5 appeared to be associated with slight attenuation of seizure activity induced by KA7.5 (P = 0.04). Thus, a greater proportion of KA5-preconditioned rats reached only stage 4 seizures (instead of stage 5) on administration of KA7.5 3 days later (Table 1, part 1). In contrast, ischemic preconditioning failed to alter the severity of subsequently induced KA7.5 seizures (P = 0.53). In this model, the development and progression of limbic convulsions appeared comparable in terms of magnitude as that induced by a 7.5-mg/kg dose of KA alone.
Representative behavioral profiles of control animals and experimental rats after their last kainic acid treatment
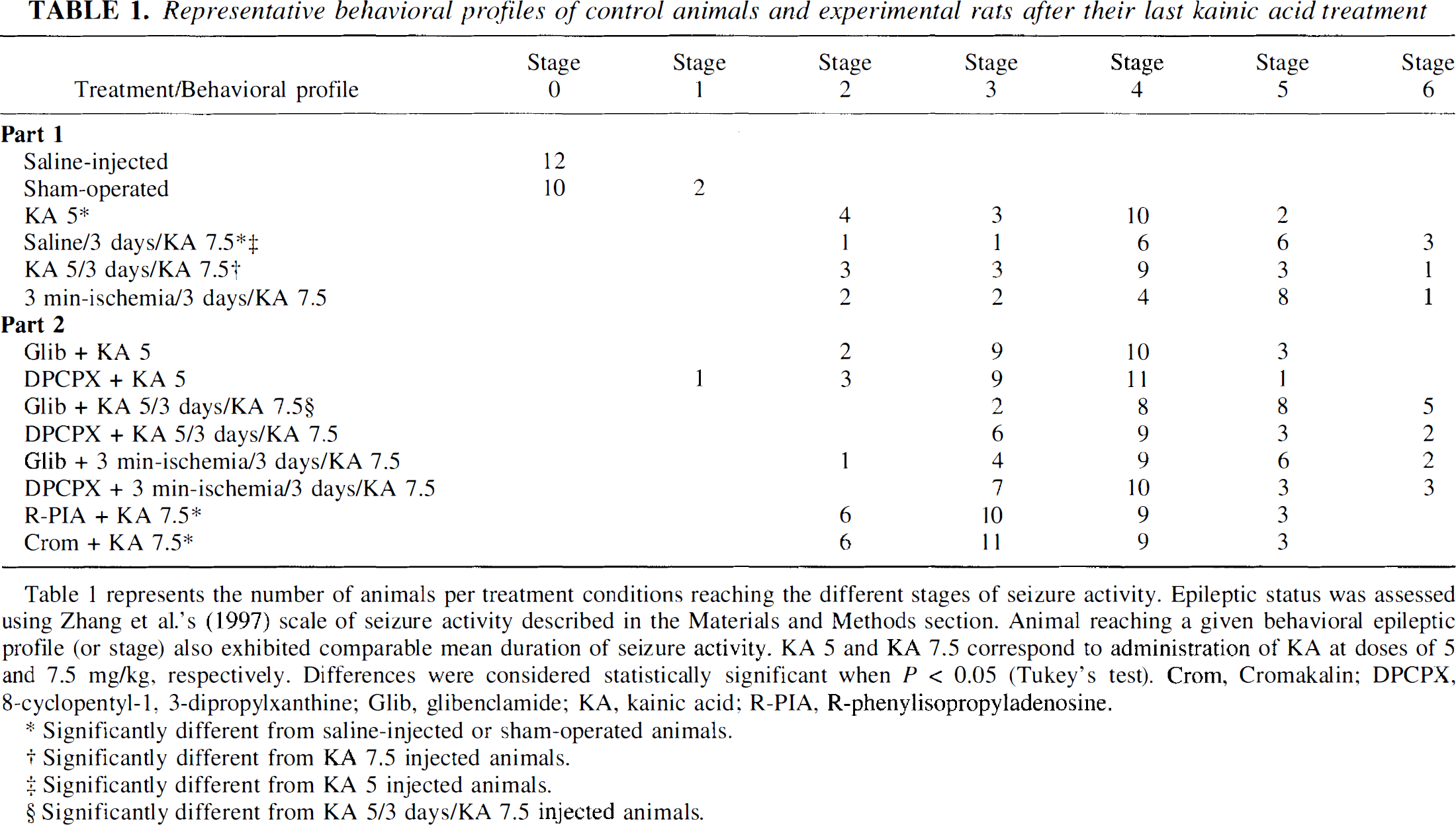
Table 1 represents the number of animals per treatment conditions reaching the different stages of seizure activity. Epileptic status was assessed using Zhang et al.'s (1997) scale of seizure activity described in the Materials and Methods section. Animal reaching a given behavioral epileptic profile (or stage) also exhibited comparable mean duration of seizure activity. KA 5 and KA 7.5 correspond to administration of KA at doses of 5 and 7.5 mg/kg, respectively. Differences were considered statistically significant when P < 0.05 (Tukey's test). Crom, Cromakalin; DPCPX, 8-cyclopentyl-1, 3-dipropylxanthine; Glib, glibenclamide; KA, kainic acid; R-PIA, R-phenylisopropyladenosine.
Significantly different from saline-injected or sham-operated animals.
Significantly different from KA 7.5 injected animals.
Significantly different from KA 5 injected animals.
Significantly different from KA 5/3 days/KA 7.5 injected animals.
Histopathologic changes at the hippocampus. Necrotic neuronal death was assessed by the silver impregnation technique, a method found reliable to visualize morphologically damaged neurons (Gallyas et al., 1990a), and by analysis of neuronal cell density in CA1 and CA3 sub fields on cresyl violet-stained sections. Figure 1 highlights representative argyrophilic stainings observed in the hippocampal pyramidal cell layers 7 days after the last treatment for the different experimental conditions. Table 2 (part 1) summarizes the scores of quantified neuronal damage performed on cresyl violet sections. To enable direct comparisons between controls (KA5 and KA7.5) and preconditioned rats, results reported (histologic illustrations and Table 2) correspond to rats displaying intense seizure activity reaching stages 4 or 5, which normally induce neuronal death in the brain. Silver staining appeared to significantly overlap with damaged cellular areas observed with cresyl violet staining (data not shown). As expected, control (saline-injected and sham-operated) animals exhibited no neuronal damage and argyrophilic staining (data not shown). In contrast, dense staining was observed after global ischemia (sham/3 days/6-minute ischemia) or single KA treatment (saline/3 days/KA7.5). Six-minute global forebrain ischemia induced significant neuronal damage in the CA1 hippocampal sector. Necrosis and cellular degeneration also were apparent in the dentate hilus (CA4) and, to a lesser extent, in the CA3 pyramidal layer (Fig. 1 A). Compared with the sham-operated group, 40% and 80% of CA3 and CA1 pyramidal cell populations showed signs of neuronal damage in the ischemic group, respectively (Table 2). Preconditioning with KA5 before 6-minute ischemia almost completely inhibited neuronal loss in the CA1 pyramidal cell layer and significantly attenuated neuronal damage in both CA3 and CA4 substructures (Fig. 1B). In that preconditioned group, 93% of CA1 and 73% of CA3 cells were preserved (Table 2). Systemic administration of KA7.5 induced severe cellular neuronal injury in the CA1, CA3, and CA4 pyramidal layers (Fig. 1C). Damage sometimes was observed in CA2, although this cellular layer was largely preserved in many animals. In contrast, both KA5 (Fig. 1D) or ischemic (Fig. 1E) preconditioning provided marked hippocampal neuronal protection against KA7.5-induced neuronal loss. The number of argyrophilic bodies was significantly reduced throughout the different hippocampal layers compared with controls. However, although KA preconditioning seemed able to equally protect all hippocampal layers, ischemic preconditioning appeared to be more efficient at inhibiting KA7.5-induced CA1— rather than CA3—neuronal loss. Table 2 shows that saline/3 days/KA7.5-injected rats lost approximately 50% of neurons in both CA1 and CA3 sectors, whereas KA5 or ischemic preconditioning afforded a 78% protection in CA1 substructure and 83% and 61% neuronal protection in CA3 subfield, respectively.
Neuronal density in the hippocampal CA1 and CA3 pyramidal cell layers

Results are expressed as mean ± SEM (n = 6) and represent neuronal densities assessed in cresyl violet stained sections per 1 mm linear length of hippocampal CA1 and CA3 pyramidal layers. A mean value for each CA1/CA3 substructure was obtained from 20 bilateral measurements on 6 sections per animal (n = 6) in each of the experimental groups. No significant differences were found between saline-injected and sham-operated rats and values of these groups were pooled and termed Controls. KA 5 and KA 7.5 correspond to administration of KA at doses of 5 and 7.5 mg/kg, respectively. Differences were considered statistically significant when P < 0.05 (Tukey's test). Crom, cromakalin; DPCPX, 8-cyclopentyl-1, 3-dipropylxanthine; Glib, glibenclamide; KA, kainic acid; R-PIA, R-phenylisopropyladenosine.
Significantly different from saline-injected or sham-operated animals.
Significantly different from KA 7.5 injected animals.
Significantly different from ischemic animals (6 min).
Significantly different from KA 5/3 days/KA 7.5 injected animals.
Significantly different from KA 5/3 days/6 min-Ischemia group.
Significantly different from 3 min-Ischemia/3 days/KA 7.5 injected animals.
Significantly different from Glib + R-PIA + KA 7.5 group.
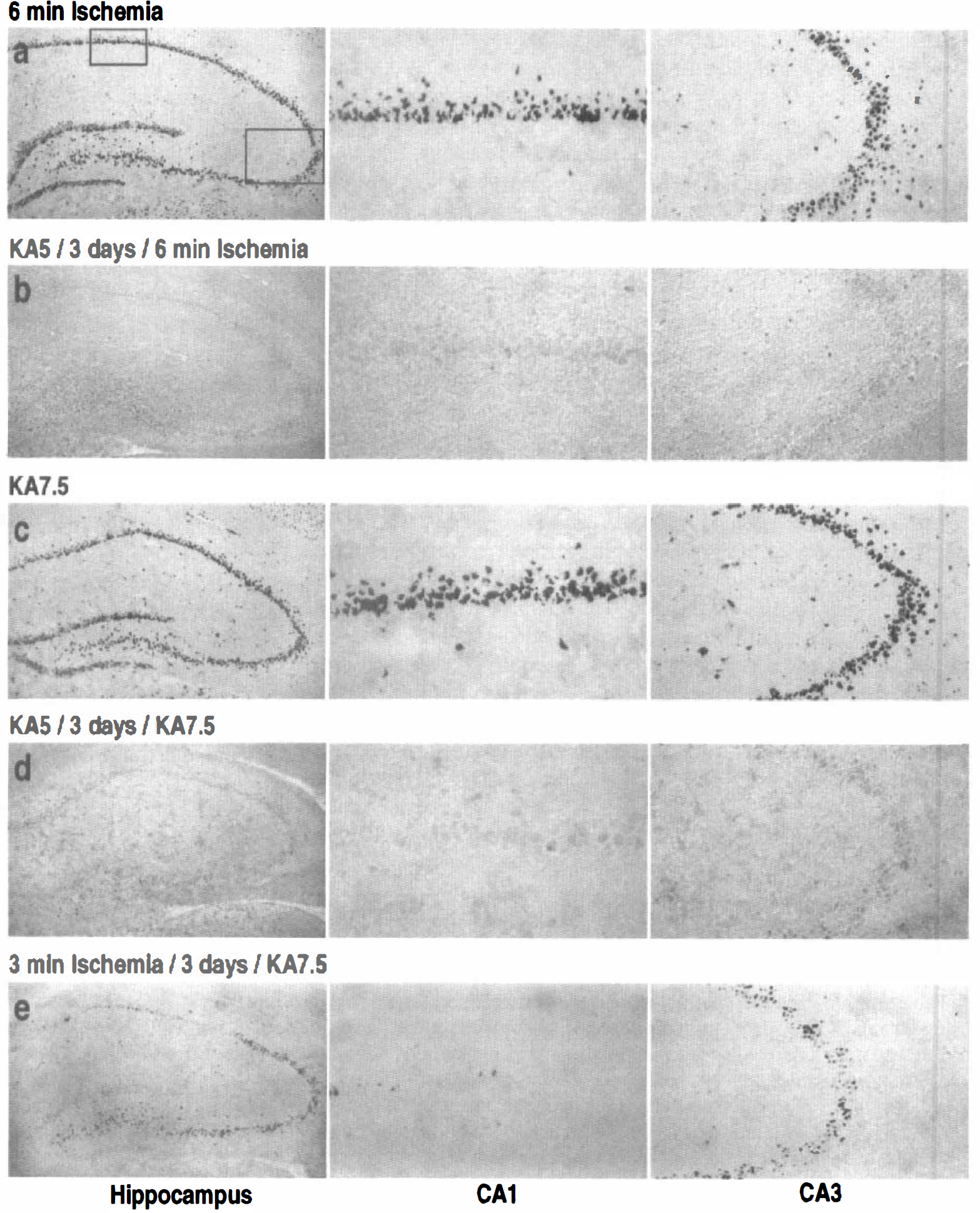
(a-e) Representative photomicrographs highlighting morphologic changes at the hippocampus assessed using silver impregnation in rats killed 7 days after the last treatment. High magnification of the boxed CA1 and CA3 pyramidal subfields are presented for the different treatment conditions. “KA5” and “KA7.5” correspond to administration of kainic acid (KA) at doses of 5 and 7.5 mg/kg, respectively,
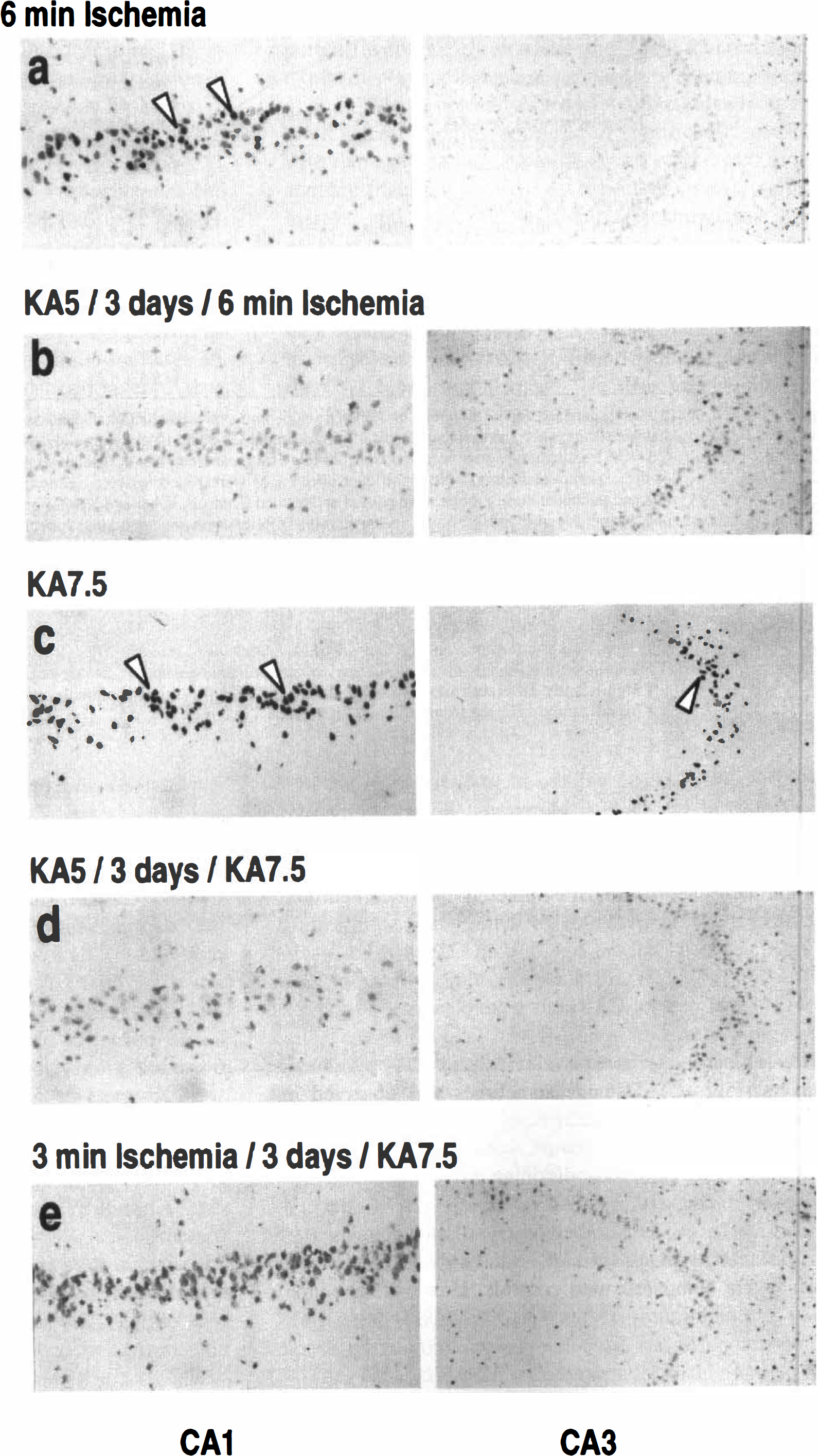
In situ labeling of DNA fragmentation. The DNA strand breaks in cell nuclei were detected by the TUNEL method (Gavrieli et al., 1992) in rats killed 72 hours after the last treatment (Fig. 2). This time point was selected based on results from a pilot study, which included rats killed 1, 3, and 7 days after treatment. No positive DNA nick end-labeled cells were observed in the brains of saline-injected or sham animals (data not shown). After 6-minute global ischemia, TUNEL-positive neurons were observed throughout the CA1 pyramidal layer with an increased concentration of labeled cells in the medial portion. Isolated TUNEL-labeled cells also occasionally were observed in the CA3 and CA4 substructures (Fig. 2A). After KA7.5 treatment, DNA fragmentation was present in many CA1 and CA3 neurons (Fig. 2C), and scattered positive cells also were observed in the dentate hilus. No consistent TUNEL labeling was observed in cells of the CA2 pyramidal layer and dentate gyrus (data not shown). Preconditioning with KA5 completely inhibited DNA fragmentation induced by 6-minute ischemia (Fig. 2B) or KA7.5 (Fig. 2D). Ischemic preconditioning, however, appeared significantly less efficacious in inhibiting KA7.5-induced TUNEL labeling, especially in the CA1 pyramidal layer, where a few TUNEL-positive neurons were observed (Fig. 2E). The presence of “ghost” staining of CA3 pyramidal cells also appeared more pronounced in rats preconditioned with sublethal ischemia and injected 3 days later with KA7.5. Agarose gel electrophoresis of labeled DNA extracted from brain tissue (Fig. 3) showed results consistent with the in situ DNA end labeling. The electrophoretic analysis confirmed the TUNEL method detection of nucleosomal DNA fragmentation and provided further evidence for preconditioning-induced inhibition of apoptotic cell death. No detectable DNA fragmentation was observed in sham-control hippocampi (lane 1), whereas there was clear evidence of a DNA laddering in extracts from ischemic (lane 2) and KA7.5 (lane 4) hippocampi isolated 3 days after injury. Ischemic (lane 5) and KA5 (lanes 3 and 6) preconditionings completely blocked DNA fragmentation induced by KA7.5 or 6-minute ischemia.
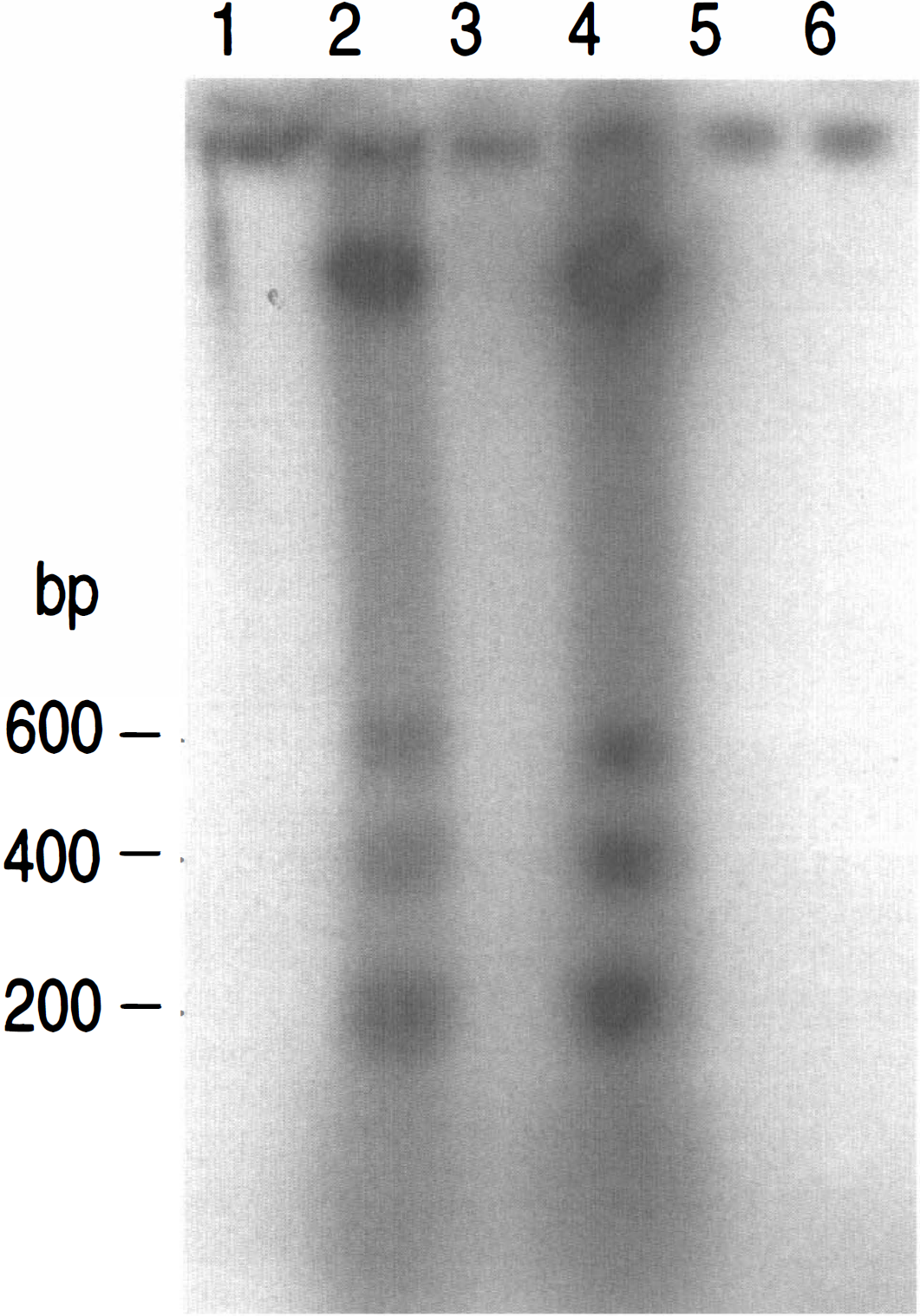
DNA laddering detected by agarose gel electrophoresis in hippocampi 3 days after the last treatment. Control rat (lane 1), ischemic rat (lane 2), rat preconditioned with KA before ischemia (lane 3), epileptic rat (lane 4), rat preconditioned with ischemia before KA (lane 5), and rat preconditioned with KA (5 mg/kg) before KA (7.5 mg/kg) (lane 6). DNA (3 mg) was 3′-end-labeled with [α32P]dCTP and terminal deoxynucleotidyl-transferase enzyme, electrophoresed on 1.5% agarose gel, and autoradiography. A ladder corresponding to 200, 400, and 600 bp is shown in lanes 2 and 4.
Involvement of adenosine, A1 receptors, and KATP channels in the different models of preconditioning
Behavioral responses after the different treatments. Table 1 (part 2) summarizes representative behavioral changes for the different treatments. The control groups treated with either a single injection of KATP antagonist glibenclamide (20 nmol/5 μL) or A1 antagonist DPCPX (250 μg/kg) failed to demonstrate significant behavioral alteration (data not shown). Rats treated with glibenclamide or DPCPX 30 minutes before a single dose of 5 mg/kg KA displayed a behavioral profile that did not significantly differ from that of animals given a single 5-mg/kg dose of KA (P = 0.99 and P = 0.41, respectively). Moreover, glibenclamide also failed to alter the severity of seizures induced by KA7.5 in rats preconditioned with a sublethal ischemia (P = 0.66). In contrast, glibenclamide appeared to significantly enhance seizure activity induced by KA7.5 in rats preconditioned with a KA5 injection (P = 0.003), whereas DPCPX had no effect on the severity of KA-induced seizure activity in rats preconditioned with a 5-mg/kg dose of KA (P = 0.59) or sublethal ischemia (P = 0.42). In contrast, rats injected with R-PIA (25 μg/kg) or (−)cromakalim (10 nmol/5 mL) before KA treatment (7.5 mg/kg) displayed a significant reduction of seizures intensity compared with animals treated with a single dose of 7.5 mg/kg KA (P = 0.0006).
Histopathologic changes at the hippocampus. Figure 4 shows representative effects of adenosine A1 receptor antagonists and agonists on neuronal (necrotic and apoptotic) cell death assessed using silver impregnation and TUNEL stainings in the different experimental conditions. Histologic representations correspond to rats displaying comparable sustained KA-induced seizure activity reaching stages 4 or 5. Table 2 (part 2) summarizes the scores of neuronal damage assessment performed on cresyl violet sections. Pretreatment with glibenclamide or DPCPX failed to potentiate hippocampal neuronal damage induced by a single 5-mg/kg dose of KA (data not shown). However, the deleterious effects of DPCPX (250 μg/kg) injected before KA5 preconditioning (Figs. 4A and 4B) or sublethal 3-minute ischemia (Fig. 4C) were observed in all hippocampal pyramidal cell layers. Numerous argyrophilic neurons appeared in CA1 and CA3 substructures of DPCPX-treated rats, whereas TUNEL-positive neurons were observed mainly in the CA1 sector. Compared with respective preconditioned groups, 55% to 72% of CA1 and 33% to 52% of CA3 pyramidal cells were damaged in animals pretreated with DPCPX injection before the preconditioning stimulus (KA5 treatment or sublethal ischemia, Table 2). In contrast, R-PIA (25 μg/kg) KA7.5 induced neuronal degeneration, a finding consistent with its previously reported neuroprotective properties (MacGregor et al., 1993). Eighty-five percent of CA1 and CA3 cells were preserved (Table 2). Similarly, neuronal damage and apoptotic nuclei were inhibited in both CA1 and CA3 hippocampal sectors of R-PIA-treated animals (Fig. 4D). When administered alone, R-PIA had no intrinsic effect and did not affect hippocampal neuronal damage (data not shown). Figure 5 shows the effects of KATP channels modulators on necrotic and apoptotic cell death. As highlighted, glibenclamide injected before KA5 treatment (Figs. 5A and 5B) or sublethal 3-minute ischemia (Fig. 5C) significantly inhibited the beneficial effects of KA5 treatment before KA7.5 or 6-minute ischemia. Intense argyrophilic staining and numerous TUNEL-positive cells were detected in all sectors of the hippocampus after these treatments (Figs. 5A through 5C). Animals pretreated with glibenclamide before preconditioning stimulus exhibited enhanced neuronal damage in the CA1 (42% to 65%) and in the CA3 (32% to 38%) compared with respective preconditioned groups (see Table 2). Glibenclamide also antagonized the protective effects of R-PIA, as demonstrated by the presence of numerous argyrophilic neurons and intense TUNEL staining in both CA1 and CA3 sectors after KA7.5 administration in animals injected with glibenclamide before R-PIA (Fig. 5D). Compared with rats injected with R-PIA before KA7.5, glibenclamide-pretreated rats exhibited a 66% and 44% loss of hippocampal CA1 and CA3 neurons, respectively. Similar to previously reported findings with ischemic models (Heurteaux et al., 1993; Heurteaux et al., 1995), rats pretreated with (−)cromakalim (10 nmol intracerebroventricularly) before KA7.5 and injected once daily during the recovery period afforded a significant protection (75% and 89% in CA1 and CA3, respectively) against KA-induced hippocampal neurodegeneration (Fig. 5E).
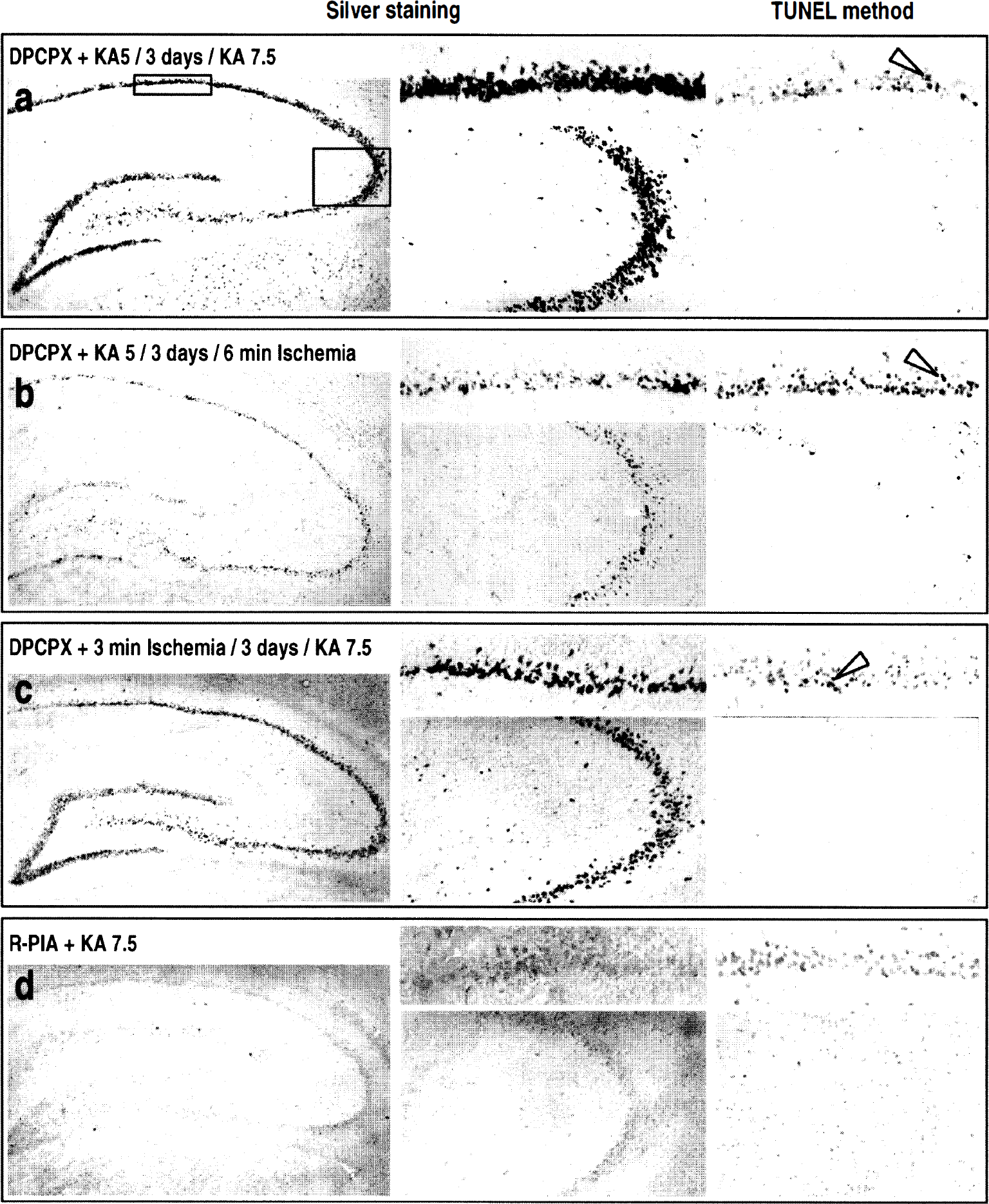
(a-d) Representative photomicrographs showing the effects of selective adenosine A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) (250 μg/kg) and agonist R-phenylisopropyladenosine (R-PIA) (25 μg/kg) on hippocampal morphologic features (silver impregnation staining, left and middle columns) and DNA fragmentation (TUNEL labeling, right column) in rats killed 7 (silver staining) and 3 (TUNEL staining) days, respectively, after the last treatment. The middle column corresponds to high-magnification photographs of CA1 (upper panel) and CA3 (bottom panel) substructures of silver-stained hippocampi for the different treatment conditions. The right column shows high magnification photographs of DNA fragmentation (TUNEL method) in CA1 (upper panel) and CA3 (bottom panel) substructures after the different treatments. “KA5” and “KA7.5” correspond to administration of 5-and 7.5-mg/kg doses of KA, respectively,
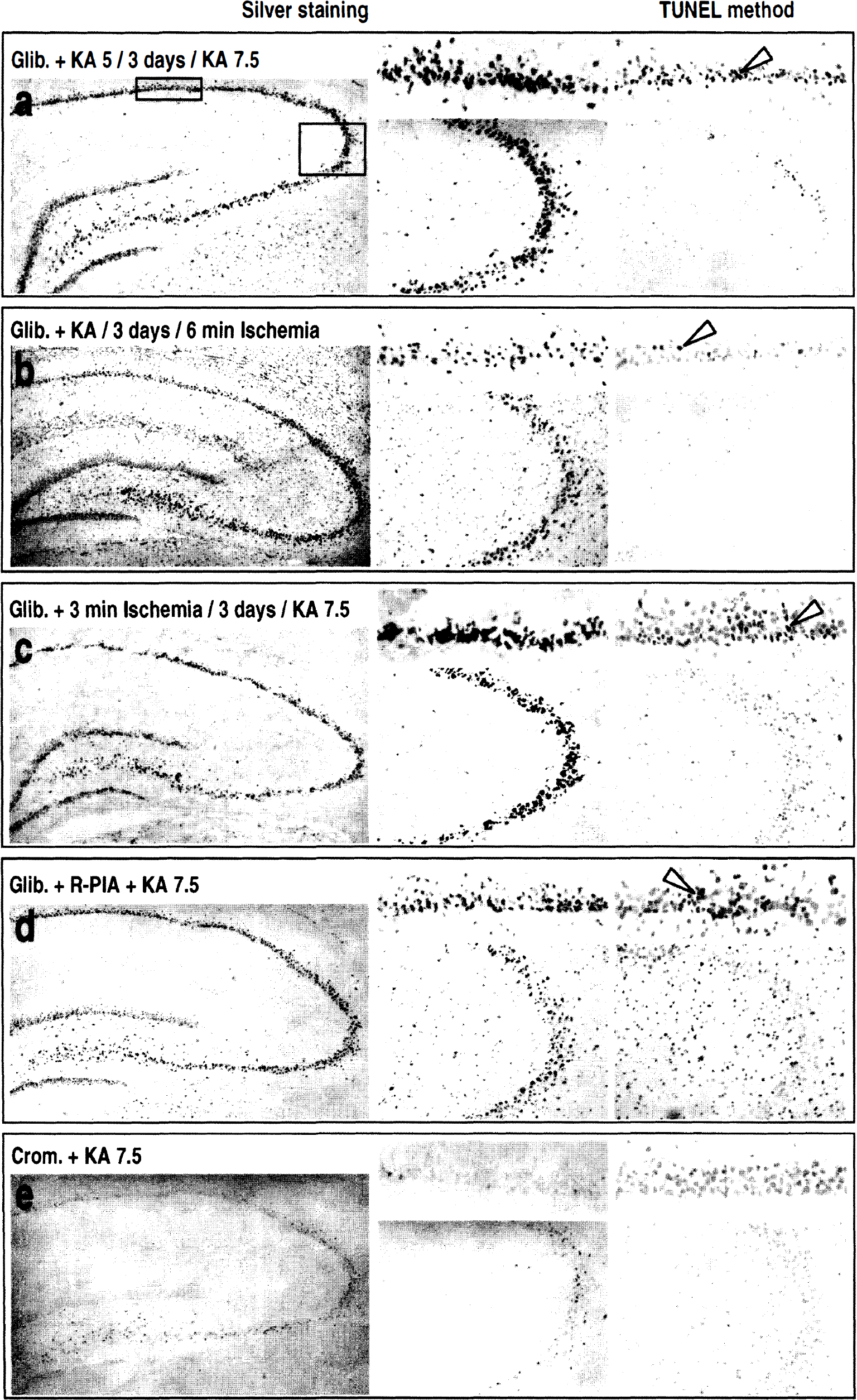
(a-e) Representative photomicrographs highlighting the effects of the KATP channel blocker glibenclamide and the KATP channel opener (−)cromakalim (10 nmol/5 μL) on hippocampal morphologic features (silver impregnation staining, left and middle column) and DNA fragmentation (TUNEL labeling, right column) in rats killed 7 (silver staining) and 3 (TUNEL staining) days after the last treatment. The left and right columns represent high magnification photographs of silver-stained (left column) and DNA fragmentation (TUNEL method, right column) in hippocampal CA1 (upper panel) and CA3 (bottom panel) substructures after the different treatments. “KA5” and “KA7.5” correspond to administration of KA at doses of 5 and 7.5 mg/kg, respectively,
DISCUSSION
The findings reported in the current study represent the first demonstration of the existence of bidirectional cross-tolerance between KA treatment (associated with limbic seizures expression) and global ischemia. In addition to mutually protective actions of KA epileptic preconditioning and sublethal global ischemia on hippocampal neuronal death, our data further characterize epileptic tolerance, using KA as a model of temporal lobe epilepsy. The beneficial effects of KA or ischemic preconditioning on hippocampal damage induced by a subsequent severe global ischemia or a KA7.5 treatment were demonstrated using analysis of neuronal density on cresyl violet-stained sections combined with silver impregnation method and DNA fragmentation assessment. Agarose gel electrophoretic analysis was used to assess the degree of DNA fragmentation in vitro in hippocampal extracts and validated the TUNEL method. The neuroprotection observed in the different preconditioning models appeared of comparable magnitude as that observed after classic ischemic preconditioning. Thus, KA5 preconditioning strongly inhibited neuronal degeneration and apoptosis induced by a 6-minute lethal global ischemic episode. Similarly, ischemic preconditioning efficiently protected CA1 and CA3 pyramidal cell layers against KA7.5-induced neuronal degeneration. Also, KA5 preconditioning significantly attenuated hippocampal degeneration induced by a subsequent lethal KA epileptic challenge.
Can the protective effects of ischemic or KA preconditioning be attributable to a decrease in the intensity or duration of seizural activity after KA7.5? Some observations suggest that this is unlikely, since ischemic preconditioning afforded comparable neuronal protection to epileptic preconditioning but failed to alter KA-induced epileptic convulsions (Table 1, part 1). Moreover, although KA5 preconditioning did confer a small reduction in the severity of convulsions induced 3 days later by KA7.5, analysis of animals showing similar behavioral symptoms and convulsion intensity is compatible with the notion that protection may be independent of KA-induced behavioral tolerance. Consistent with these findings, Kelly and McIntyre (1994) also demonstrated that hippocampal kindling enhanced the brain's resistance to subsequent neuronal damage induced by KA treatment, despite the presence of more generalized convulsions and development of faster severe limbic status in kindled compared with control animals (Kelly and McIntyre, 1994). Taken together, these findings support the contention that the protection conferred by our preconditioning treatments, against excitotoxicity induced by KA7.5, is largely mediated by physiologic mechanisms outside of those mediating behavioral convulsions per se. Animals preconditioned with KA5 also appeared to be well protected against hippocampal necrosis and apoptosis, which otherwise would be observed consequent to a 6-minute global ischemia. This finding further demonstrates that the observed mutual protection is not seizure-dependent, since KA5 pretreatment equally protects against neuronal death in animals experiencing seizures (KA7.5 treatment) and ischemic animals without seizures (6-minute ischemia).
The cascade of events that underlie the mutually protective actions of epileptic preconditioning and sublethal ischemia and the protective effects of KA5 on subsequent neuronal damage by KA7.5 treatment remain poorly understood. The pharmacologic approach of the current report suggests the involvement of adenosine, adenosine A1 receptors, and KATP channels to the protection observed after ischemic and epileptic preconditioning. Indeed, the beneficial effects of preconditioning treatments (i.e., KA5 or a 3-minute ischemia) appeared to be suppressed by DPCPX, a selective A1 receptor antagonist (Lohse et al., 1987). Moreover, neuroprotective effects of preconditioning procedures also were significantly suppressed by glibenclamide, a potent blocker of KATP channels. Thus, the enhanced resistance against KA7.5-induced hippocampal injury (associated with intense—stages 4 and 5—seizure activity) demonstrated in rats preconditioned with either a 5 mg/kg dose of KA or a short 3-minute ischemia, appeared to be completely inhibited in animals pretreated with DPCPX or glibenclamide before epileptic or ischemic preconditioning.
How can this neuronal protection, afforded by two drugs with distinct targets—the A1 receptor and the KATP channel—be interpreted? Adenosine, a metabolite of adenosine nucleotides, is released during ischemia, after experimental seizures as well as during seizures in epileptic patients (During and Spencer, 1992; Rudolphi et al., 1992b; Winn et al., 1980). It is therefore conceivable that adenosine be released in the different preconditioning events used in the current study (i.e., after seizures induction by KA5 treatment or after a brief 3-minute ischemia). In this context, adenosine also has been shown to indirectly activate KATP channels through the G-protein pathway after binding to A1 receptor. This has not been directly shown in neurons but is well documented in cardiac cells (Kirsch et al., 1990; Kurachi, 1995). On the other hand, evidence exists demonstrating the ability of A1 receptor agonists (e.g., R-PIA) and activators of KATP channels (e.g., (−)cromakalim) to protect against the deleterious effects of brain ischemia (Evans et al., 1987; Heurteaux et al., 1995; Rudolphi et al., 1992b; Von Libitz et al., 1994). This work also demonstrates that both of these substances potently conferred protection against the detrimental effects of KA7.5 in animals expressing intense seizure activity. One particularly intriguing property is the dependence of preconditioning on time. In cardiac preconditioning, a delayed or “second window of protection” against infarction also is observed 24 to 72 hours after ischemic preconditioning, and it also involves the early activation of adenosine A1 receptor and KATP channels (Baxter et al., 1994; Baxter and Yellon, 1997; Bernardo et al., 1999). Is the ability of both adenosine and KATP channels to regulate vasomotor tone centrally involved in the neuronal protection observed in this work? The answer to this question probably is no, since the vasodilating properties of adenosine have been shown to be associated with A2 rather than A1 receptor activation (Edvinsson et al., 1993; Nyce, 1999; Rudolphi et al., 1992a). On the other hand, glibenclamide recently has been reported to exert minimal effect on CBF in normoglycemic rats (Horinaka et al., 1997) and to only partially inhibit dilation of cerebral arterioles in response to hypoxia (Taguchi et al., 1994).
It seems that the important step in the different preconditioning events described in this study is accompushed through activation of neuronal KATP channels by adenosine. However, adenosine, once liberated by preconditioning treatments, also could activate other transduction pathways, independent of that related to the KATP channel, and also is beneficial in neuroprotection. Opening other types of K+ channels also could have a neuroprotective effect. Particularly good candidates would be the G-protein-gated inward rectifiers, which are abundantly present in most regions of the brain (Lesage et al., 1995) and which also are activated by adenosine through the A1 receptors (Lauritzen et al., 1997; North, 1989). However, these particular channels are not blocked by glibenclamide, and their activation cannot be evaluated because they have no potent and specific blockers. Eliminating the beneficial effects of preconditioning by glibenclamide therefore suggests the participation of KATP channels but does not disqualify the activation of other pathways. Furthermore, although indirect activation of KATP channels by adenosine seems to represent an important step in preconditioning-induced cascade of events, it cannot be the sole event leading to a neuroprotection that is maximal only 3 days after the initial protective treatment.
Thus, further research is required to identify other biological processes involved. Nevertheless, the findings reported here provide a better understanding of the nature of brain adaptation against neuronal death induced by ischemia or by excitotoxic compounds like KA, which induce epileptic seizures. Research along these lines also might help subsequent development of new therapeutic strategies capable of modifying the outcome of ischemic and epileptic episodes.
Footnotes
Acknowledgments:
The authors thank G. Jarretou, N. Vaillant, F. Aguila, and D. Doume for skillful technical assistance and I. Lauritzen and C. Widmann for advice and helpful comments.