
Abstract
The DNA repair enzyme, poly(ADP-ribose) polymerase-1 (PARP1), contributes to cell death during ischemia/reperfusion when extensively activated by DNA damage. The cell death resulting from PARP1 activation is linked to NAD+ depletion and energy failure, but the intervening steps are not well understood. Because glycolysis requires cytosolic NAD+, the authors tested whether PARP1 activation impairs glycolytic flux and whether substrates that bypass glycolysis can rescue cells after PARP1 activation. PARP1 was activated in mouse cortical astrocyte and astrocyte-neuron cocultures with the DNA alkylating agent, N-methyl-N ′-nitro-N-nitrosoguanidine (MNNG). Studies using the 2-deoxyglucose method confirmed that glycolytic flux was reduced by more than 90% in MNNG-treated cultures. The addition of 5 mmol/L of α-ketoglutarate, 5 mmol/L pyruvate, or other mitochondrial substrates to the cultures after MNNG treatment reduced cell death from approximately 70% to near basal levels, while PARP inhibitors and excess glucose had negligible effects. The mitochondrial substrates significantly reduced cell death, with delivery delayed up to 2 hours after MNNG washout. The findings suggest that impaired glycolytic flux is an important factor contributing to PARP1-mediated cell death. Delivery of alternative substrates may be a promising strategy for delayed treatment of PARP1-mediated cell death in ischemia and other disorders.
Keywords
Poly(ADP-ribose) polymerases (PARPs) take part in DNA repair and other cell processes (D'Amours et al., 1999; Pieper et al., 1999). PARP1 is the most abundant and best characterized member of the PARP family. When activated by DNA strand breaks or kinks, PARP1 consumes NAD+ to form poly(ADP-ribose) polymers on specific acceptor proteins. These acceptor proteins include histones, DNA polymerases, DNA ligases, and PARP1 itself (Burzio et al., 1979; Zahradka and Ebisuzaki, 1982; D'Amours et al., 1999). This process appears to facilitate DNA repair (Burzio et al., 1979; Zahradka and Ebisuzaki, 1982; D'Amours et al., 1999); however, excessive PARP1 activation can contribute to cell death when DNA damage is extensive. PARP inhibition has been shown to markedly reduce cell death resulting from oxidative stress (Schraufstatter et al., 1986), alkylating agents (Szabo and Dawson, 1998; Ha and Snyder, 1999), and ischemia (Eliasson et al., 1997; Szabo and Dawson, 1998).
The cell death resulting from PARP1 activation involves consumption of cytosolic NAD+ and ATP depletion (Gaal et al., 1987; Pieper et al., 1999), but the intervening steps in this process are not well characterized. One mechanism by which cytosolic NAD+ consumption by PARP1 could cause ATP depletion is through blockade of the NAD+-dependent, glyceraldehyde-3-phosphate dehydrogenase step of glycolysis (Szabo and Dawson, 1998; D'Amours et al., 1999; Sheline et al., 2000). Blockade at this step would limit both production of glycolytic ATP and the flux of glucose carbon into the mitochondrial tricarboxylic acid (TCA) cycle. If blockade of glycolytic flux by NAD+ depletion is a major factor in PARP1-mediated cell death, it should be possible to rescue cells after PARP1 activation by supplying substrates that enter the TCA cycle by routes other than glycolysis. Here we show that pyruvate, α-ketoglutarate, and other substrates prevent cell death when administered at time points after PARP1 activation. The findings suggest that delivery of mitochondrial substrates may be a useful strategy for rescuing cells after extensive PARP1 activation.
METHODS AND MATERIALS
Materials
Reagents were purchased from Sigma Chemical Co (St. Louis, MO, U.S.A.), except where noted.
Cell cultures
Astrocyte cultures were prepared from cortices of 1-day-old Swiss-Webster mice (Simonsen, Gilroy, CA, U.S.A.) as described previously (Swanson et al., 1997; Ying and Swanson, 2000), and were plated into 24-well Falcon culture plates. The astrocytes were maintained in Eagle's minimal essential medium containing 5 mmol/L glucose and supplemented with 5% fetal bovine serum (Hyclone, Ogden, UT, U.S.A.), 2 mmol/L glutamine, 100 nmol/L sodium selenate, and 200 nmol/L α-tocopherol. The cultures were used for experiments at 20 to 30 days in vitro or as a plating surface for neurons at days 14 to 20 in vitro. For astrocyte-neuron cocultures, neurons were dissociated from fetal mouse forebrain cortices (Ying et al., 1999) and were plated on the astrocyte layers at a density of 1 × 106 cells/cm2. The cultures were maintained in astrocyte-conditioned medium, with partial medium exchanges twice weekly. Astrocyte-neuron cocultures were used when the neurons were 13 to 17 days in vitro.
Experimental procedures
Experiments were initiated by replacing the culture medium with a balanced salt solution (BSS). The BSS contained (in mmol/L) KCl, 3.1; NaCl, 134; CaCl2, 1.2; MgSO4, 1.2; KH2PO4, 0.25; NaHCO3, 15.7; and glucose, 2. The pH was adjusted to 7.2 while the solution was equilibrated with 5% CO2 at 37°C. Osmolarity was verified at 290 to 310 mOsm with a Wescor vapor pressure osmometer (Logan, UT, U.S.A.). Drugs were added from concentrated stocks prepared in BSS immediately before use and were adjusted to pH 7.2 when necessary. Exposures to N-methyl-N ′-nitro-N-nitrosoguanidine (MNNG), hydrogen peroxide (H2O2), or N-methyl-d-aspartate (NMDA) were performed at 37°C in a 5% CO2 atmosphere. Exposures were terminated after 60 minutes (MNNG or H2O2) or 20 minutes (NMDA) by complete exchange with fresh BSS. When used, the PARP inhibitors benzamide, 3-aminobenzamide, or nicotinamide were added prior to the addition of MNNG, NMDA, or H2O2. Posttreatment with pyruvate or other compounds was begun by adding these compounds directly to the BSS (containing 2 mmol/L glucose) either immediately after washout of the MNNG, NMDA, or H2O2, or after intervals of up to 5 hours. Cultures were then maintained in BSS until biochemical or cell death studies were performed.
Poly(ADP-ribose) immunostaining
Immunostaining was performed according to the method of Burkle et al. (1993) with minor modifications. Neuron-astrocyte cocultures were fixed in ice-cold 10% trichloroacetic acid, dehydrated in ethanol, air dried, and placed in blocking buffer. Anti-poly(ADP-ribose) monoclonal antibody (Trevigen, Gaithersburg, MD, U.S.A.) was added at 1:2,000 dilution and incubated at 4°C overnight. After washing, the cultures were incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG (Molecular Probes, Eugene, OR, U.S.A.) at 1:500 dilution for 1 hour at room temperature, then counterstained with propidium iodide. The cultures were photographed with a Leica confocal laser microscope. Neuronal nuclei were located above the astrocyte layer and thereby discernible from the astrocyte nuclei during confocal imaging.
ATP and NAD+ assays
For ATP assays, cells were lysed in boiling buffer containing 100 mmol/L Tris, 4 mmol/L EDTA, pH 7.75. Fifty microliters of cell lysates were mixed with 50-μL aliquots of luciferase/luciferin mixture provided with an ATP bioluminescence assay kit (Roche Diagnostics GmbH, Mannheim, Germany), and photon emission was detected with a luminometer. ATP concentrations were calibrated against ATP standards and expressed as nanomoles per milligram protein. Total NAD+ (NAD+ plus NADH) was measured by the recycling assay of Szabo et al. (1996) with minor modifications as previously described (Ying et al., 2001). Results were calibrated with NAD+ standards and were normalized to protein content as determined by the bicinchonic acid method.
Glucose utilization
The Sokoloff 2-deoxyglucose assay (Sokoloff et al., 1977) was modified for use in cell cultures as described previously (Hertz et al., 1998). After 30-minute incubation in 280 μL BSS containing 5 mmol/L PIPES buffer (pH 7.2), 2.5 μCi of [14C (U)]2-deoxyglucose (340 Ci/mol; ARC, St. Louis, MO, U.S.A.) in 20 μL BSS was added to each culture well. Incubations were continued at 37°C for 40 minutes, then terminated by three washes in ice-cold BSS. The cells were lysed in 0.01 N NaOH/0.5% SDS and 14C was measured by scintillation counting. Prior studies show that more than 90% of the resulting astrocyte 14C signal is attributable to [14C] 2-deoxyglucose-6-phosphate (Hertz et al., 1998), which accumulates in proportion to glucose utilization (Sokoloff et al., 1977).
Cell death determinations
Astrocyte death was quantified in the astrocyte monotype cultures by measuring the lactate dehydrogenase (LDH) activity in cell lysates harvested 24 hours after drug exposures (Swanson et al., 1997). Percent cell death was calculated by normalizing the LDH values to LDH activity measured in lysates from control (wash only) culture wells. Neuron death in the astrocyte-neuron cultures was assessed by propidium iodide fluorescence 24 hours after drug exposures. Neurons were distinguished from the underlying astrocyte layer by their phase-bright, process-bearing morphology, as confirmed by NeuN immunostaining (Ying et al., 1999). Both the fluorescing dead neurons and nonfluorescing live neurons were counted, in a blinded fashion, in four randomly picked optical fields. At least 400 neurons were counted in each well, and results from each well were expressed as percent neuronal death.
Statistical analyses
All data are presented as means ± SD. Data were assessed by analysis of variance followed by the Student-Newman-Keuls post hoc test. P values less than 0.05 were considered statistically significant.
RESULTS
PARP1 activation causes NAD+ depletion, glycolytic blockade, and cell death
To validate neuronal and astrocyte models for studying PARP1-mediated cell death, we first established the effects of PARP inhibitors on cell death induced by three known PARP1 activators: the oxidant H2O2, the excitotoxin NMDA, and the DNA alkylating agent MNNG (Berger, 1985; Szabo and Dawson, 1998). MNNG is commonly used to induce DNA damage and PARP1 activation in experimental settings to avoid nonspecific effects of more physiological PARP1 activating agents (Berger, 1985; Zhang et al., 1994; Eliasson et al., 1997; Ying et al., 2001). The neuronal death induced by each of these agents was substantially reduced by the PARP inhibitor benzamide (Fig. 1A).
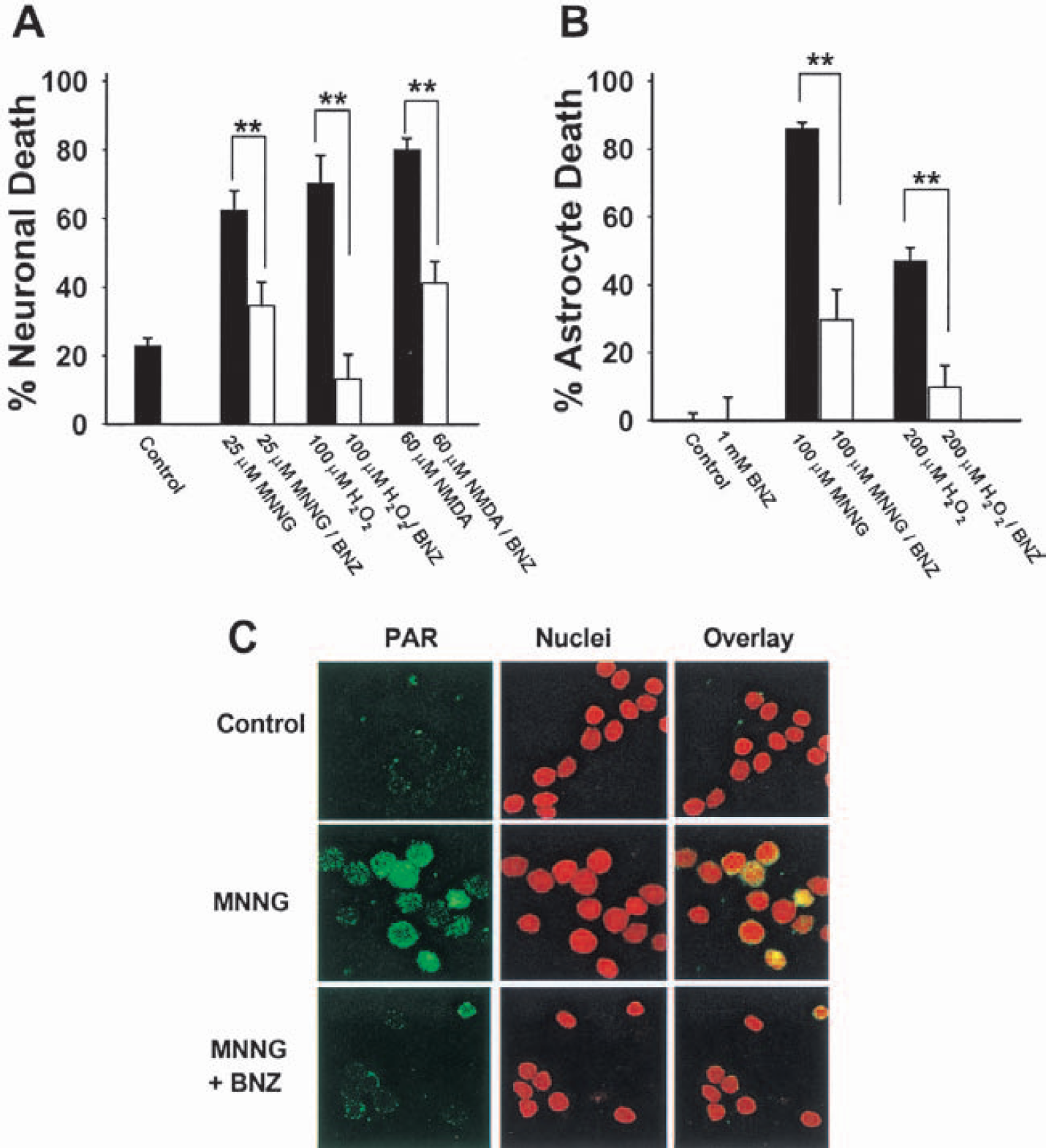
PARP activation contributes to oxidative and excitotoxic cell death.
Benzamide also reduced astrocyte death, which occurred at higher MNNG and H2O2 concentrations than those required to kill neurons (Fig. 1B). Similar results were obtained using other PARP inhibitors, 3-aminobenzamide and nicotinamide (not shown). Figure 1C confirms that MNNG induced poly(ADP-ribose) accumulation in neuronal nuclei, and this too was blocked by benzamide.
Astrocyte monocultures were used for biochemical measures because these, unlike neuronal cultures, provide a near-uniform cell population. PARP activation by MNNG led to a decrease in total cell NAD+ content of the astrocyte cultures (Fig. 2A). Since NAD+ is a necessary cofactor for glycolysis, glycolytic flux after PARP1 activation was measured by the Sokoloff 2-deoxyglucose method (Sokoloff et al., 1977). This approach has been shown to provide an accurate measure of glycolytic rate in diverse settings, including astrocyte cultures (Hertz et al., 1998; Gotoh et al., 2000). As shown in Fig. 2B, glycolytic rate was reduced by more than 90% in astrocytes after exposure to MNNG.
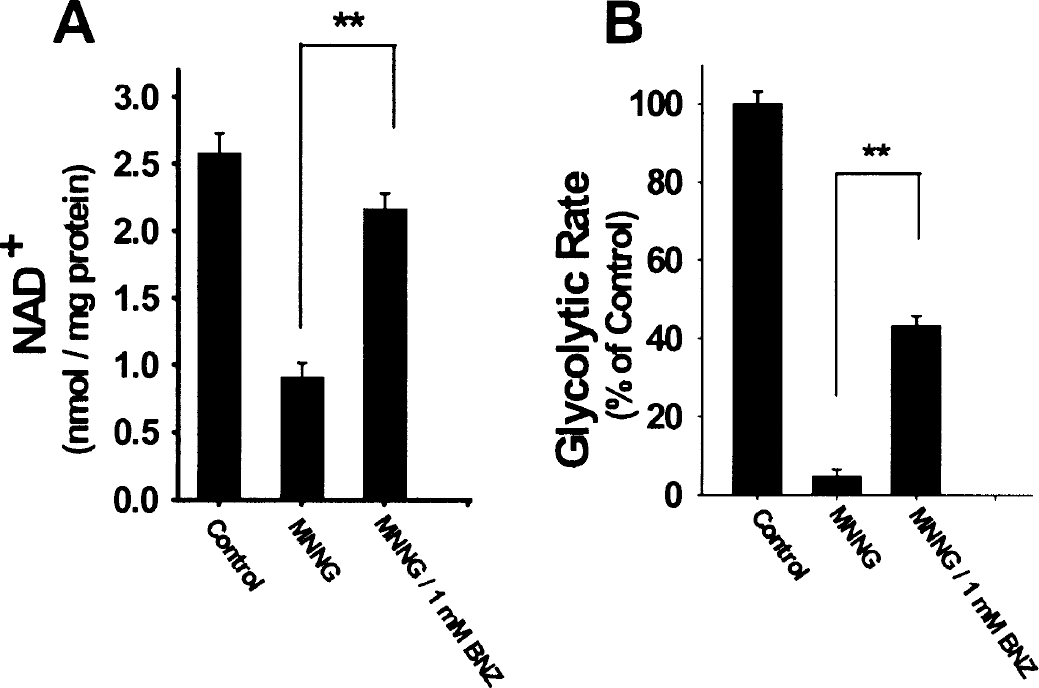
Effects of PARP activation on NAD+ and glycolytic rate.
Delivery of tricarboxylic acid cycle substrates after PARP1 activation prevents PARP1-mediated cell death
Addition of 5 mmol/L α-ketoglutarate or 5 mmol/L pyruvate immediately after MNNG washout almost completely prevented MNNG-induced neuronal death (Fig. 3A). In contrast, 10 mmol/L glucose (in addition to the 2 mmol/L glucose always present in BSS), 10 mmol/L lactate, and 10 mmol/L glutamine (not shown) had little or no effect on neuron death. In astrocyte cultures (Fig. 3B), addition of 5 mmol/L pyruvate or glutamine after washout of MNNG substantially reduced cell death, while lactate and additional glucose were again ineffective. Similar results were obtained when 100 μM H2O2 was used in place of MNNG (not shown). Importantly, glutamine and TCA cycle substrates were effective in reducing cell death when added (and continuously present) after MNNG washout, but not when present only during MNNG incubations (Fig. 3C). Conversely, benzamide reduced cell death when present during the MNNG incubation but had only negligible effect when added after MNNG washout. As shown in Fig. 3D, the effects of glutamine on astrocyte survival measured at 24 hours correlated with an increased astrocyte ATP content measured at 5 hours after MNNG washout. Pyruvate and α-ketoglutarate also increased ATP content when provided after PARP activation (not shown).
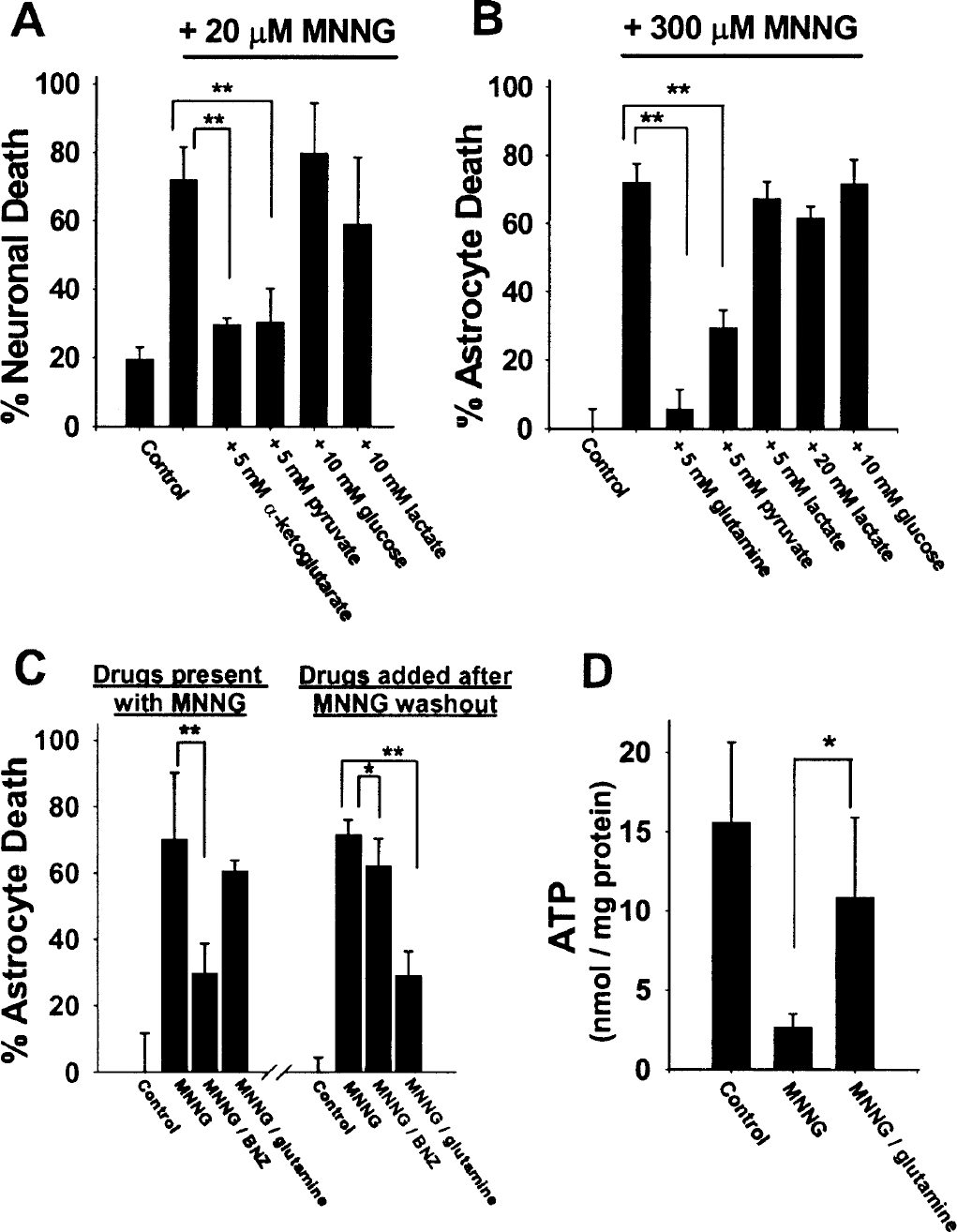
Tricarboxylic acid cycle substrates rescue neurons and astrocytes from MNNG-induced cell death.
Figures 4A and 4B show concentration-dependent effects for selected TCA cycle intermediates and precursors on MNNG-induced cell death. In general, significant cytoprotection was not observed at substrate concentrations below 1 mmol/L, and little or no increased effect was seen at concentrations above 10 mmol/L. Significant cytoprotection was also observed when these compounds were added at more delayed time points, although with diminishing efficacy: α-ketoglutarate reduced MNNG-induced neuronal death when added at 0 or 1 hour after MNNG washout, but significant neuroprotection was not observed at later time points. Similarly, glutamine reduced astrocyte death when supplied at time points up to 2 hours after MNNG washout, but not at later time points (Fig. 4D).
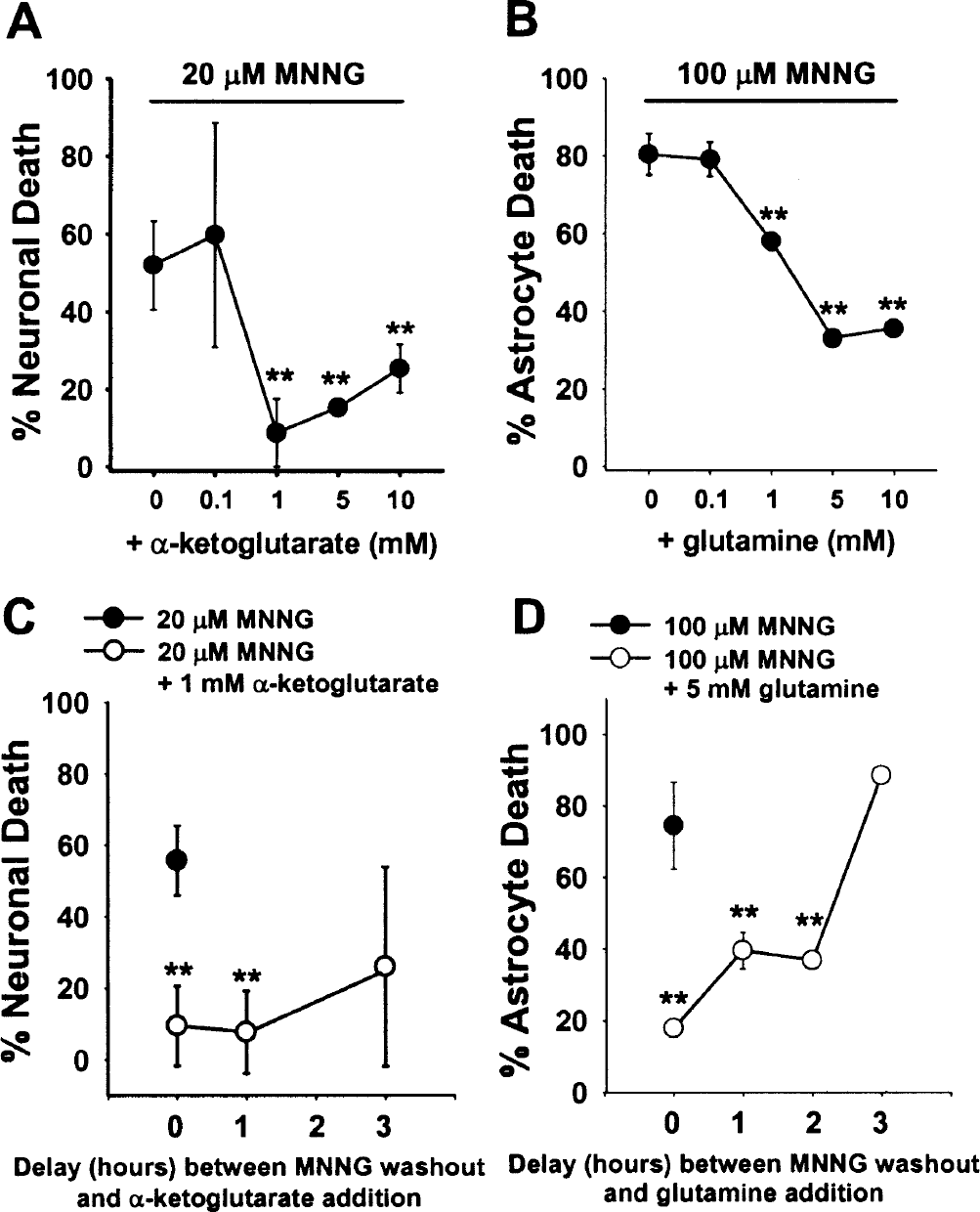
Effects of tricarboxylic acid cycle substrate concentrations and posttreatment delay intervals on cell survival.
DISCUSSION
These results show that extensive PARP1 activation leads to a block in glycolysis, and that PARP1-mediated death of both neurons and astrocytes can be prevented by exogenous TCA cycle substrates administered at time points after PARP1 activation. Since the properties of PARP1 are conserved across many cell types (D'Amours et al., 1999; Pieper et al., 1999), the strategy of posttreatment with TCA cycle substrates may be generally applicable.
PARP1, a nuclear enzyme, consumes cytosolic NAD+ but does not have direct access to the mitochondrial NAD+ pool. Since mitochondria contain a substantial fraction of total cell NAD+, the decrease in whole cell NAD+ levels induced by MNNG likely underestimates the extent of cytosolic NAD+ depletion. It is not obvious why benzamide less completely prevented the effects of MNNG on glycolytic rate, but this may be related to the nonlinear relationships between cytosolic NAD+ concentration, NAD+/NADH ratio, and glycolytic rate (Ainscow and Brand, 1999).
These effects of MNNG on NAD+ and glycolysis are consistent with the proposal that extensive PARP1 activation and NAD+ consumption impairs glycolysis by depleting cytosolic NAD+ (Szabo and Dawson, 1998; D'Amours et al., 1999). The failure of lactate to provide a cytoprotective effect further supports this mechanism. Lactate can normally be used as a metabolic substrate by astrocytes, neurons, and other cell types via conversion to pyruvate and subsequent entry into the TCA cycle. However, lactate conversion to pyruvate requires NAD+ as a cosubstrate, and consequently lactate cannot be used as an energy substrate when cytoplasmic NAD+ has been consumed for poly(ADP-ribose) formation. Of note, lactate produced by astrocytes has been proposed as the major endogenous energy substrate used by neurons in brain (Pellerin and Magistretti, 1994). The present findings suggest this metabolic pathway would be nonfunctional after extensive PARP1 activation.
Although not themselves TCA cycle substrates, glutamine and glutamate also were effective in preventing astrocyte death after MNNG exposure. This is not surprising, because both glutamate and glutamine can be rapidly taken up and metabolized to α-ketoglutarate in astrocytes (Yu et al., 1982; Yudkoff et al., 1988; Broer and Brookes, 2001). However, glutamine failed to prevent significant neuronal death. The reason for this is not clear, but may be related to the fact that, unlike astrocytes, only a subpopulation of neurons expresses significant glutaminase activity (Kaneko et al., 1992).
The TCA cycle substrates do not themselves prevent PARP activation by MNNG, as evidenced by the failure of these agents to reduce cell death when present only during, but not after, the MNNG incubations. The PARP inhibitor benzamide had the inverse effect: benzamide prevented cell death when present during MNNG incubations, but had only minor effects when added after MNNG washout (Fig. 3C). The failure of benzamide to reduce cell death when added after MNNG washout indicates that cell death was not substantially attributable to continued PARP1 activation after MNNG washout, as might occur if MNNG induced sustained activation of PARP1 or if MNNG were incompletely removed from the cells.
This study is the first to show that TCA cycle substrates can rescue cells after extensive PARP1 activation. One implication of these findings is that TCA cycle substrates may be effective in preventing cell death at time points after onset of stroke. Pyruvate and glutamine administered after ischemic injury in brain and other tissues have recently been reported to improve cell survival (Khogali et al., 1998; Lee et al., 2001). Although other mechanisms are certainly possible, the present findings suggest that these agents act by circumventing the effects of PARP1 activation.
Footnotes
Acknowledgments
We thank Elizabeth Gum and Jennifer Bergher for expert technical assistance and Dr. Mary B. Sevigny for helpful advice.