
Abstract
As one of the leading causes of death within both the developed and developing world, stroke is a worldwide problem. Risk factors can be identified and controlled at the level of lifestyle changes; however, genetic components of stroke have yet to be identified. The identification of such genetic components is critical in the understanding, diagnosis, and treatment of stroke in the future. This review focuses on the genetic determinants of stroke in both human and experimental systems. Mendelian disorders, candidate genes, and twin studies provide evidence for a strong genetic component of stroke. Genome-wide scanning in both human and animal models has led to the identification of regions of the genome that contain genes for stroke susceptibility and sensitivity. Animal models of stroke allow for environmental control and genetic homogeneity, not possible within a human population, and therefore are essential for the dissection of this complex, multifactorial disorder. Future genetic and genomic strategies and their role in ultimate causative gene identification are discussed.
CLASSIFICATION OF STROKE
There are three main categories of stroke: subarachnoid hemorrhage (SAH), intracerebral hemorrhage (ICH), and ischemic stroke. An account of the pathophysiology of each type of stroke is given in an excellent, detailed review by Bamford et al. (1991). The present review will focus largely on a detailed discussion regarding genetic aspects of ischemic stroke.
RISK FACTORS
Risk factors are thought to play an important role in the pathogenesis of stroke. Many of the known risk factors, such as hypertension, diabetes, or hyperlipidemia, are themselves complex, polygenic traits, and this complicates the investigation into genetic aspects of stroke. Hypertension was found to increase the chances of primary ICH and cerebral infarction in a number of studies (Shaper et al., 1991; Longstreth et al., 1992; Juvela et al., 1993). Diabetes has been found to be associated with ischemic stroke (Burchfiel et al., 1994), as were low apolipoprotein E levels (Couderc et al., 1993). Increased levels of serum lipoprotein A and intermediate-density lipoproteins, and high levels of high-density lipoproteins and cholesterol, were found to be associated with ischemic stroke (Pedro-Botet et al., 1992). In 1989 two studies showed that smoking increases the risk of cerebral infarction (Donnan et al., 1989; Shinton and Beevers, 1989), as does alcohol (Monforte et al., 1990; Longstreth et al., 1992). It is clear, however, that genes and environment interact in at least the final disease pathway, as illustrated by early twin and family studies and more recent molecular investigations (Fig. 1).
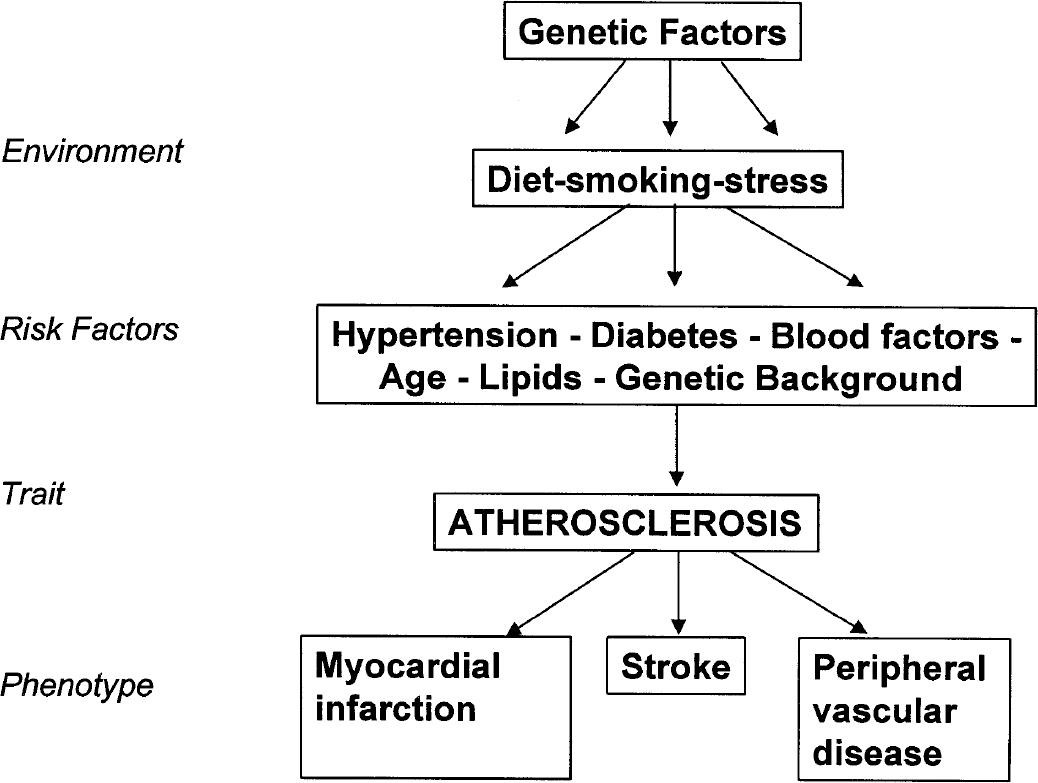
A theoretical scenario to illustrate multiple interactions between genetic and environmental factors in the outcome of a phenotype such as stroke. (Reprinted with permission from Alberts MJ. Genetic aspects of cerebrovascular disease. Stroke 1991;22:276–280.)
GENETIC ASPECTS OF STROKE IN HUMANS
The strongest evidence suggesting a genetic component to stroke in humans comes from the data generated by twin studies. An important study by Brass et al. (1992) compared concordance rates between monozygotic and dizygotic twins. For monozygotic twins the concordance rate was found to be 17.7%; it was only 3.6%, however, for dizygotic twins. This study included very few pairs of twins (7 pairs for monozygotic twins and 1 pair of dizygotic twins), and it was therefore difficult to estimate the degree of heritability to a reliable level. The subtype of stroke occurring was not investigated in this study. This important factor, outlined earlier, is needed to gain insight into the possibility of different genetic components acting in different subtypes of stroke. Recently, Bak et al. (2002) used a large, population-based twin register and nationwide death registries to identify the risk of stroke death for monozygotic twins as being twice that of dizygotic twins in a Danish population. Thirty-five of 351 monozygotic twins (10%) and 34 of dizygotic pairs (5%) were concordant for stroke death. Diagnosis misclassification was also a major concern in this study.
As well as twin studies, family studies have been used to confirm the genetic component of stroke. One large study (Liao et al., 1997) of more than 30,000 subjects showed that risk factors such as smoking, history of diabetes, hypertension, and coronary heart disease did not alter stroke occurrence in people who had stroke events in previous generations of their family. The Framingham offspring study found that a family history of transient ischemic attack or stroke significantly increased the chance of an occurrence of stroke or transient ischemic attack in the offspring when compared with offspring with no history of cerebrovascular events (Kiely et al., 1993). It was found that both maternal (n = 2074; RR = 1.4; 95% CI, 0.60 to 3.25) and paternal (n = 1762; RR = 2.4; 95% CI, 0.96 to 6.03) histories were associated with an increased risk of stroke. A smaller study of 90 patients who had a cerebral infarction found that 47% of them had a family history of stroke, whereas in the control group this was only 24% (Graffagnino et al., 1994).
It is important to note that stroke occurrence between populations of differing ethnic origin is different. A study by Gross et al. (1984) attempted to identify all persons from an area of southern Alabama who had a stroke in 1980 and were hospitalized. Data were gathered on disease onset, clinical course, laboratory results, history of risk factors, and outcome. The age-adjusted incidence rates for initial stroke were 109 per 100,000 for white Americans and 208 per 100,000 for black Americans. Population-based studies by Friday et al. (1989) also indicated black Americans had a statistically significant higher, age-adjusted rate of stroke than white Americans. Stroke death rates in men between the ages of 35 and 74 were studied during the period of 1984 to 1990 in a variety of countries (Thom and Epstein, 1994). This study showed a large demographic variation throughout the world. In Europe, the World Health Organization MONICA project showed that stroke incidence and stroke mortality followed the same pattern of geographical variation (Thorvaldsen et al., 1995). The reason for these differences in stroke rate and stroke mortality observed throughout the world remains unknown. A close look at the different environmental influences, the different severities, and the subtypes of stroke, as well as the differences in health care delivery systems, may influence the interpretation of results.
Polygenic inheritance, variable penetrance, gene-environment interactions, genetic and pathologic heterogeneity, and late age of onset are reasons why stroke is particularly difficult to study from a molecular point of view (Alberts, 1991). When considering that subtypes of stroke could be the effect of different genetic-susceptibility genes, correct classification is crucial when undertaking genetic studies. Although several environmental risk factors are shared within subtypes of stroke, both first and second strokes were found to belong more frequently to the same subtype rather than different subtypes (Yamamoto and Bogousslavsky, 1998).
MENDELIAN FORMS OF STROKE
Studies into some mendelian forms of stroke have been successful in identification of the genes responsible. In particular, an autosomal dominant form of stroke (Fig. 2), cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), has been well dissected genetically and the contribution of the gene NOTCH3 has been identified. Linkage study mapped the putative disease gene to a defined region on human chromosome 19 (Tournier-Lasserve et al., 1993; Chabriat et al., 1995). The gene was then cloned by position, the mutation was identified, and its functional role confirmed (Joutel et al., 1996, 1997).
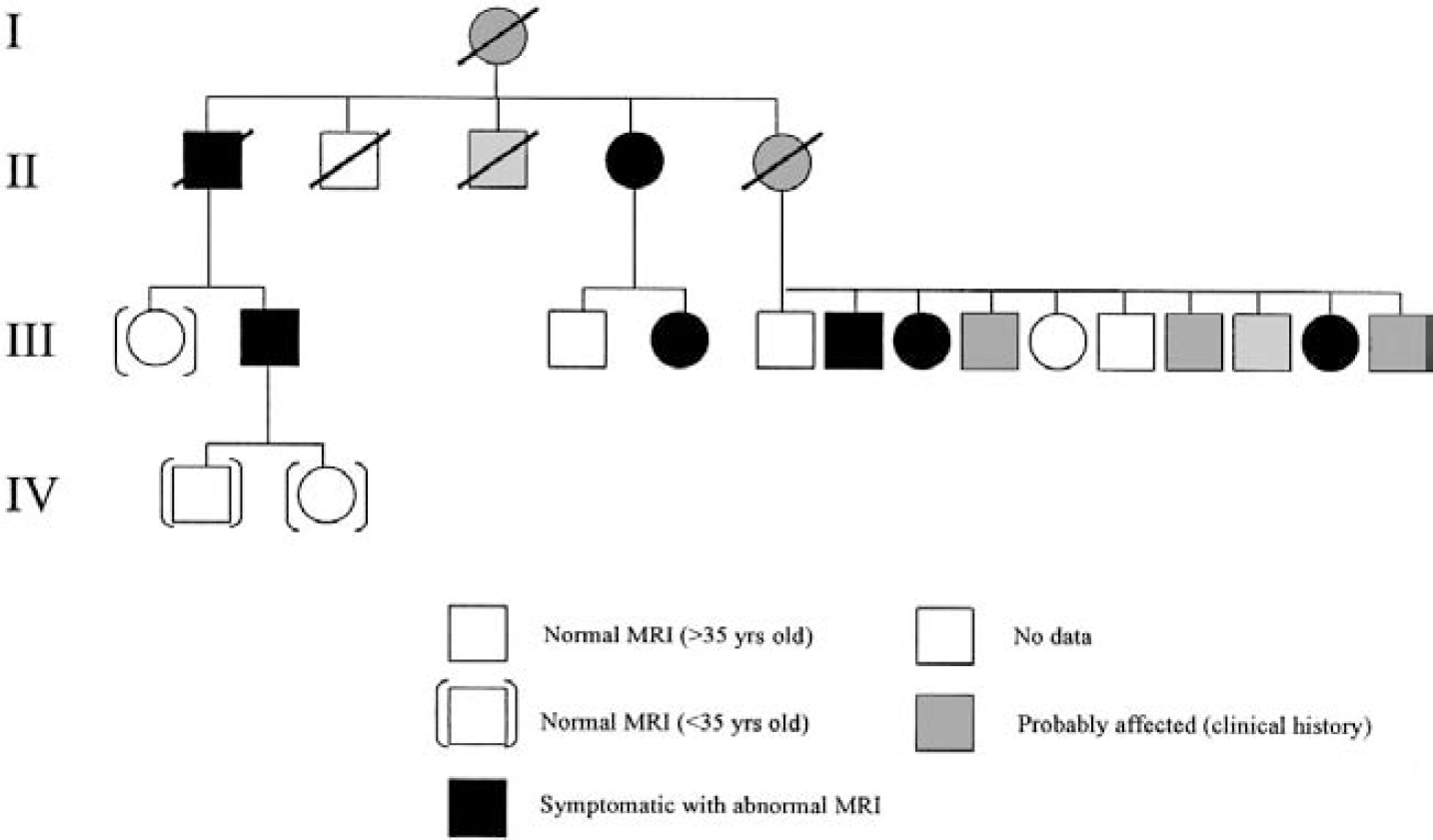
Genealogical tree showing 4 generations of a typical CADASIL pedigree with clinical and magnetic resonance imaging (MRI) status of family members. A circle indicates patient is female, a square indicates patient is male and a strikethrough indicates that patient is deceased. Reprinted with permission from Elsevier Science (Lancet 1995;346:934–939).
A further mendelian disorder associated with stroke is mitochondrial cytopathy, characterized by encephalopathy with stroke-like episodes and lactic acidosis (MELAS). Interestingly, this disorder has been attributed to a mutation of a single nucleotide (commonly an A to G transition) in the patients' mitochondrial DNA (Ciafaloni et al., 1992; Macmillan et al., 1993). Persons who have inherited (Ciafaloni et al., 1992; Macmillan et al., 1993) or co-inherited this mutation (Pulkes et al., 2000) are more likely to be predisposed to stroke occurrence.
Ischemic stroke can occasionally occur owing to an underlying connective tissue disorder that has caused arterial dissection. In Marfan syndrome an extension of an aortic dissection into the common carotid artery can occur and result in stroke (Spittell et al., 1993). Fabry disease is an X-linked disorder caused by a deficiency in α-galactosidase A. There is a high risk of both stroke and myocardial ischemia associated with this disorder (Crutchfield et al., 1998).
CANDIDATE GENE STUDIES
The basis for human studies into putative causative genes involved in ischemic stroke has taken the form of the candidate gene approach. This involves selecting a functionally relevant gene to study, and then investigating its association with the ischemic stroke phenotype. Candidate genes in stroke research are chosen mainly for their role in stroke risk or vascular reactivity and brain response after insult. Candidate genes in stroke fall into five main groups: renin-angiotensin system, nitric oxide production, lipid metabolism, hemostasis, and homocysteine metabolism.
In the renin-angiotensin system candidate genes are selected owing to their role in vascular tone and endothelial function. The main gene studied is angiotensin-converting enzyme (ACE) gene, specifically an insertion/deletion (I/D) polymorphism present in intron 16 of the gene. This variant, first identified by its association with myocardial infarction (Cambien et al., 1992), has been found to be positively associated with stroke in a number of independent studies (Markus et al., 1995; Nakata et al., 1997; Margaglione et al., 1996; Doi et al., 1997; Kario et al., 1996; Watanabe et al., 1997; Castellano et al., 1995; Hosoi et al., 1996). A meta-analysis of the ACE gene in ischemic stroke also found a significant association between the occurrence of the homozygous deletion (D/D) polymorphism and stroke (Sharma, 1998). Many studies, however, have found no association with this polymorphism and stroke (Sharma et al., 1994; Catto et al., 1996; Pullicino et al., 1996; Ueda et al., 1995; Aalto-Setala et al., 1998; Zee et al., 1999). These conflicting results reflect the problems involved in candidate gene studies in human stroke. A polymorphism in the angiotensinogen gene (M235T) is also thought to play a role in vascular disease and a possible interaction with the ACE gene has been suggested (Nakata et al., 1997). Barley et al. (1995), however, have also shown that the M235T polymorphism is negatively associated with stroke.
The gene-encoding endothelial nitric oxide synthase (eNOS) is a potential candidate gene for stroke, because it is an important mediator of endothelial function. An association between Glu298Asp polymorphism in the endothelial constitutive nitric oxide synthase gene and ischemic events has been observed (Elbaz et al., 2000). There are also a number of studies, however, indicating no association with this polymorphism and stroke (MacLeod et al., 1999; Markus et al., 1998). Further evidence to support the role of endothelial nitric oxide in stroke comes in the form of knockout mice that are deficient in endothelial nitric oxide synthase. These mice were found to be highly sensitive to focal cerebral ischemia (Samdani et al., 1997).
Apolipoprotein E, a glycoprotein that mediates the binding of lipid particles to specific lipoprotein receptors, has been a candidate gene for stroke because it is involved in neuronal membrane maintenance and repair. Contrasting results, however, have been obtained in a number of studies (Couderc et al., 1993; Nakata et al., 1997; Kessler et al., 1997; Margaglione et al., 1998).
A number of candidate genes in the area of hemostasis and thrombosis have been proposed. The homozygous AA genotype of the G-A 455 polymorphism in the B-fibrinogen gene leads to increased levels of fibrinogen and has been found to be more prevalent in cases of large vessel stroke (Kessler et al., 1997). Since increased fibrinogen leads to atherosclerosis and prothrombotic mechanisms, this highlights its potential as a candidate gene. The PIA2 variant in the platelet fibrinogen receptor GP IIIa/IIb has also been found to be an important risk factor in stroke patients younger than 50 years (Carter et al., 1998).
Genetically determined defects in homocystein metabolism can lead to severe hyperhomocysteinemia and atherosclerosis and thus make it a pathway of interest for identification of candidate genes in vascular disease. A genetic variant of methylene tetrahydrofolate reductase (C677T) has been studied extensively in relation to ischemic stroke. The majority of studies, however, have found no association (Markus et al., 1997; Lalouschek et al., 1999; Nakata et al., 1998; De Stefano et al., 1998; Reuner et al., 1998). A meta-analysis also reached this conclusion, finding the C677T polymorphism to be associated with mild homocysteinemia but with no increase in vascular risk (Brattstrom et al., 1998).
GENOME-WIDE SCANNING: EXPERIMENTAL AND HUMAN STUDIES
In order to understand genes involved in stroke and stroke risk it is important to eliminate variability of environmental factors and to select subjects that are prone to stroke. This strategy is not possible in a human population, and therefore studies involving animal models of stroke are essential tools in the process of gene identification in complex, multifactorial disorders such as stroke. One of the most important animal models commonly used to dissect stroke is the spontaneously hypertensive stroke-prone rat (SHRSP). This strain was developed in 1974 in Japan (Okamoto et al., 1974) by selective breeding of a subgroup of the spontaneously hypertensive rat (SHR) strain that developed spontaneous cerebral infarction or hemorrhage. The SHRSP is characterized by early onset of hypertension, and 80% of animals experience stroke when 9 to 13 months of age. There are pathogenetic similarities between strokes in SHRSP rats and humans (Yamori et al., 1976).
Rubattu et al. (1996) performed a genome-wide screen in an F2-cross obtained by mating SHRSP and the stroke-resistant SHR, in which latency to stroke on Japanese diet (high salt, low potassium, low protein) was used as a phenotype. This study identified quantitative trait loci (QTLs) on rat chromosomes 1, 4, and 5 (significant LOD scores of 7.4, 4.7, and 3.0 respectively) as being involved in stroke susceptibility. Together, these three regions were thought to account for 28% of the variance in stroke susceptibility.
Our own laboratory has studied a different stroke-related phenotype, the extent of cerebral infarction following permanent distal occlusion of the middle cerebral artery (Fig. 3) (Jeffs et al., 1997). A genome-wide scan was performed in an F2-cross derived from the SHRSP and the normotensive reference strain, WKY rat. A highly significant QTL on rat chromosome 5 was identified with a LOD score of 16.6. It contributed to 67% of the phenotypic variance and was blood-pressure independent.
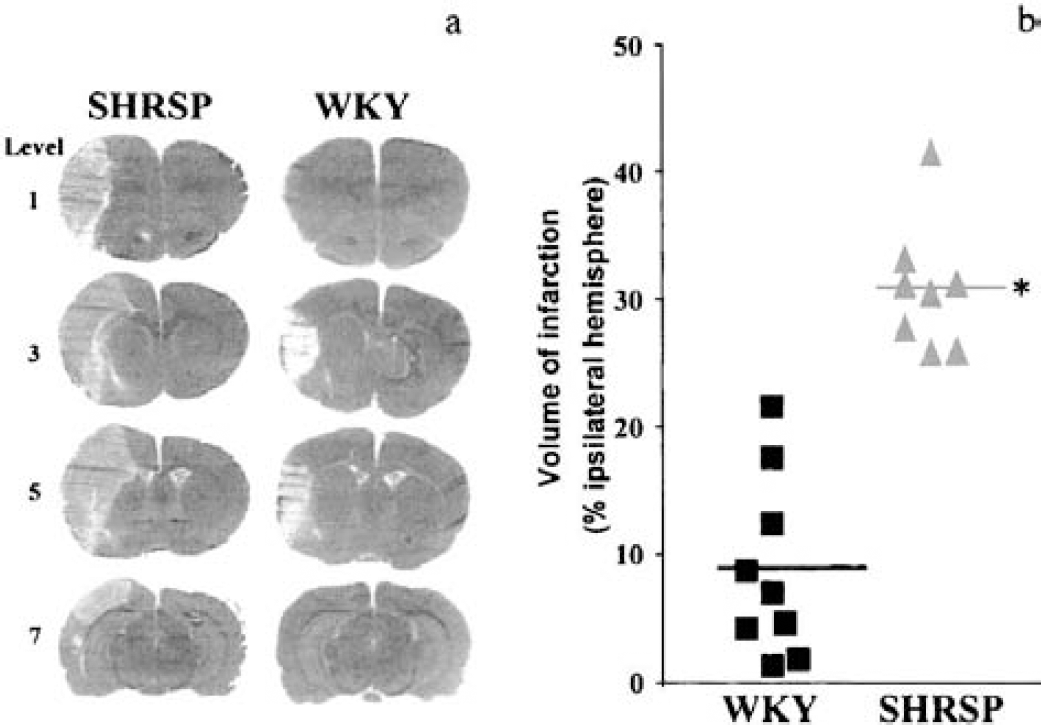
The identification of a region within the rat genome as being involved in the stroke phenotype is, however, only the first step in causative gene identification. The QTL then has to be confirmed as harboring genes that are causative to stroke, conventionally by the production of congenic strains. A congenic strain is one that has had a region of its genome selectively replaced by the same region from another (usually contrasting) strain (Rapp, 2000). If a difference in stroke phenotype is observed when comparing the congenic strain to the respective parental strain, it can be confirmed that the region introgressed contains genes that are involved in stroke. The construction of congenic strains for various cerebrovascular phenotypes is currently being undertaken by a number of groups, and early observations by Rubattu et al. (1999) look promising.
Positional cloning is one of the next steps needed to home in on the causative gene. This technique requires a congenic strain that contains a segment from the contrasting strain of approximately 1 to 2 centimorgans (cM) in size and still maintains the phenotype of interest. This task takes several years, requiring many generations of congenic strain construction and genotyping (Rapp, 2000).
One way to “transfer” regions of the rat genome involved in stroke to the respective region in the human genome is to undertake comparative genome analysis. This involves identifying chromosomal regions in the rat known to be involved in stroke and finding the syntenically conserved regions in human. This in silico technique has been successful in the case of Julier et al. (1997) who comparatively mapped a region involved in blood pressure regulation from rat chromosome 10 to human chromosome 17. Others have investigated this same region, and evidence for linkage between essential hypertension and a putative locus on human chromosome 17 has been confirmed (Baima et al., 1999). With the progression of stroke research, it is merely a matter of time before similar strategies will be applied to stroke QTLs.
A more traditional way to identify QTLs and then causative genes in a complex trait in humans is to perform a genome-wide scan. Only one genome-wide scan to identify QTLs involved in stroke has been undertaken. A genome-wide scan in an Icelandic population of 476 stroke patients and 438 relatives identified a stroke susceptibility-locus mapping to human chromosome 5q12 (Gretarsdottir et al., 2002). Broad but rigorous definition of the phenotype was documented and hemorrhagic stroke, ischemic stroke, and transient ischemic attack were all included in order to map a locus for common stroke. The LOD score at the chromosome 5 locus was increased from the initial 2.00 to 3.39 with the genotyping of 45 additional markers over the 45-cM region identified (Fig. 4). Furthermore, linkage analysis undertaken using even higher marker density resulted in a LOD score of 4.40. This study was therefore the first to successfully map a major locus for stroke by combining genealogy, a large population from which patients with broadly defined stroke were selected, and allele-sharing methods.
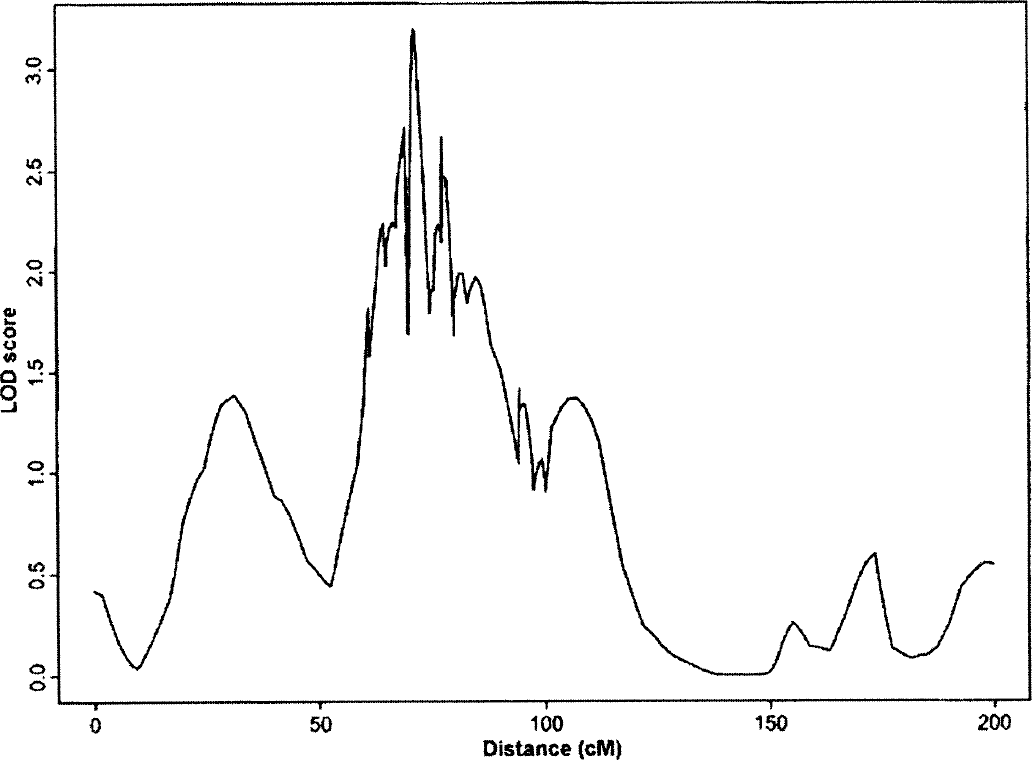
LOD score in graphic form. Genetic distance (in centimorgans [cM]) along the chromosome is on the X-axis; the LOD score is on the Y-axis. A LOD score of 3.39 was obtained after the addition of 45 new markers to the region. The marker order used was from the Marshfield Map (Centre of Medical Genetics, Marshfield Medical Research Foundation). Reprinted with permission from the University of Chicago Press (Gretarsdottir S, Sveinbjornsdottir S, Jonsson HH, et al. Localization of a susceptibility gene for common forms of stroke to 5q12. Am J Hum Genet JID 2002;70:593–603).
FUTURE GENETIC AND GENOMIC STRATEGIES
A number of strategies are available to identify genes that are being differentially expressed during the stroke process. In using these techniques the possibility of a novel transcript or novel functions of known genes involved can be discovered. High throughput expression profiling can be undertaken by a variety of methods, including the use of complimentary DNA (cDNA) microarrays (Schena et al., 1995) or oligonucleotide microarrays (Lockhart et al., 1996; Lipshutz et al., 1999). Another technique used to identify differentially expressed genes, which is less commonly used, is that of differential display (Wang et al., 2000). Proteomics is also another advancing area in which the dissection of multifactorial, complex diseases may benefit. This area concerns protein presence in specific organs or tissues at specific time points (Sironi et al., 2001).
Soriano et al. (2000) identified differentially expressed genes in an animal model of focal ischemia using a custom-made oligonucleotide array. They used a rat chip that contained 750 different probe sets, which was originally intended for bone and cartilage research. Greater than 70% of the gene probes are expressed in the central nervous system, however, which made its use appropriate in this study. A number of genes were identified during this experiment, many of which require further characterization and function determination to deduce their possible roles in stroke. Aitman et al. (1999) showed that microarray technology combined with congenic strategy may lead to the identification of a causative gene. The fatty acid receptor/transporter Cd36 was underexpressed in the SHR strain in microarray experiments. This deficiency was subsequently found to be caused by a deletion event that resulted in the creation of a dysfunctional chimeric protein. More recently, by combining both mouse congenic strains and microarray gene expression analysis, Eaves et al. (2002) have looked at gene expression differences in a mouse model of diabetes. This powerful technique could be applied to stroke, in order to identify underlying genes once congenic strains containing smaller regions have been produced.
A number of genes have been identified by microarray analysis of hippocampal gene expression in rats subjected to global cerebral ischemia at various time points after insult (Jin et al., 2001). Microarray analysis may therefore be useful for elucidating novel molecular mediators of cell death and survival in the ischemic brain. Subtractive hybridization has also been used to look at gene expression changes following permanent middle cerebral artery occlusion in the rat (Bates et al., 2001). As well as identifying previously established ischemic-induced gene products, subtractive hybridization has found unidentified genes to be altered in expression following focal ischemia.
All of these techniques can increase the chance and speed of gene discovery, particularly when applied to experimental models and congenic strains when made available (Eaves et al., 2002). This would allow for the study of gene-environment and gene-gene interactions, as well as genotype to phenotype links, that are critical in order to gain insight into complex traits. The recent advances in the human genome project (Lander et al., 2001; Venter et al., 2001) and the identification of multiple single-nucleotide polymorphisms within the human genome (Hirakawa et al., 2002), combined with bioinformatics, will facilitate the study of multifactorial, complex disorders such as stroke. Ultimately, the identification of causative genes will facilitate early diagnosis, prevention, and treatment of stroke.