
Abstract
Protein kinase-mediated signaling cascades constitute the major route by which cells respond to their extracellular environment. Of these, three well-characterized mitogen-activated protein kinase (MAPK) signaling pathways are those that use the extracellular signal-regulated kinase (ERK1/2) or the stress-activated protein kinase (p38/SAPK2 or JNK/SAPK) pathways. Mitogenic stimulation of the MAPK-ERK1/2 pathway modulates the activity of many transcription factors, leading to biological responses such as proliferation and differentiation. In contrast, the p38/SAPK2 and JNK/SAPK (c-Jun amino-terminal kinase/stress-activated protein kinase) pathways are only weakly, if at all, activated by mitogens, but are strongly activated by stress stimuli. There is now a growing body of evidence showing that these kinase signaling pathways become activated following a variety of injury stimuli including focal cerebral ischemia. Whether their activation, however, is merely an epiphenomenon of the process of cell death, or is actually involved in the mechanisms underlying ischemia-induced degeneration, remains to be fully understood. This review provides an overview of the current understanding of kinase pathway activation following cerebral ischemia and discusses the evidence supporting a role for these kinases in the mechanisms underlying ischemia-induced cell death.
Cells respond to their extracellular environment through a variety of systems, including growth factor, hormone, and cytokine receptors, linked to common intracellular signaling systems. Protein kinase-mediated signaling cascades constitute the major mediator of such signals. Three well-characterized kinase-signaling pathways are those that use the extracellular signal-regulated kinase (ERK1/2) or the stress-activated protein kinases (p38/SAPK2 or JNK/SAPK) pathways. Each of these pathways is composed of mitogen-activated protein (MAP) kinase kinase kinase (MAPKKK) that, on activation, phosphorylates a MAPKK, then a MAPK. The phosphorylated MAPK then interacts with cellular substrates and/or translocates to the nucleus, resulting in the modulation of transcription factors that in turn leads to a diverse range of biological responses. For full activation, each component of the MAPK cascade has to be phosphorylated at both the threonine and tyrosine residues (Davis, 1993; Seger and Krebs, 1995). Mitogenic stimulation of the ERK1/2 pathway modulates the activity of many transcriptional factors leading to biological responses such as proliferation and differentiation (Boulton et al., 1991; Marshall, 1995; Segal and Greenberg, 1996) (Fig. 1). In contrast, the p38/SAPK2 and JNK/SAPK pathways are only weakly, if at all, activated by mitogens, but are strongly activated by stress stimuli, bacterial lipopolysaccharide (LPS) and cytokines such as interleukin-1 (IL-1) and tumor necrosis factor (TNF) (Freshney et al., 1994; Raingeaud et al., 1995; Rouse et al., 1994), resulting in altered transcription, translation, and activation of factors involved in cell survival and inflammation (Kummer et al., 1997; Lee et al., 1994) (Fig. 1).
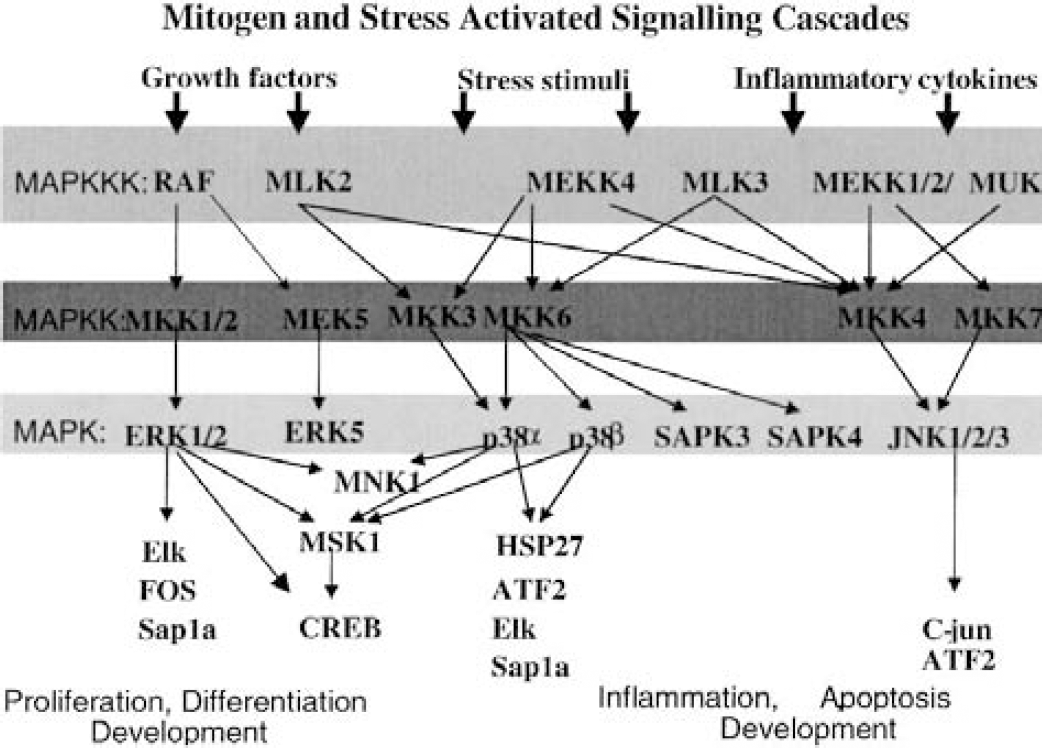
A simplified schematic representation of mitogen- and stress-activated signaling pathways discussed in this article. MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; MEK, MAPK/ERK kinase; SAPK, stress-activated protein kinase; CREB, cyclic-AMP response element binding protein; HSP27, heat shock protein 27.
While these mitogen- and stress-induced pathways were initially identified as independent intracellular signaling pathways activated in response to distinct extracellular stimuli, there is growing evidence of cross talk between them. This suggests that these pathways are components of larger signaling networks (Fig. 1). Both the ERK1/2 and p38/SAPK2 pathways activate common transcription factors such as Elk-1, Sap-1a and cyclic-AMP response element binding protein (CREB) (Cahill et al., 1996; Janknecht and Hunter, 1997; Tan et al., 1996; Traverse et al., 1992; Whitmarsh et al., 1997). Some stimuli can activate CREB by a bifurcated pathway that uses both p38 and ERK1/2 (Xing et al., 1998), whereas other stimuli, such as arsenite, have recently been reported to activate the ERK1/2 pathway through activation of the p38 pathway (Ludwig et al., 1998). Furthermore, ERK1/2 and p38 pathways share common intracellular kinase substrates (Fukunaga and Hunter, 1997; Deak et al., 1998). For example, mitogen- and stress-activated protein kinase-1/2 sits at the junction of p38 and ERK pathways (Deak et al., 1998) and mediates growth factor and stress-induced activation of CREB. It is also becoming increasingly apparent that the diversity of these signaling systems is not yet fully understood. For example, in the case of the Raf/MEK/ERK1/2 pathway MEK-1 (MAPK/ERK kinase-1) was, until recently, believed to be Raf's only substrate, and Raf, MEK-1's only activator. Using knockout (KO) technology, however, it was shown that although the Raf-1 KO mice showed defects in development and protection against apoptosis, the absence of Raf-1 did not compromise the activation of MEK-1 (Hüser et al., 2001). Therefore, while B-Raf may be the major activator of MEK, Raf-1 (C-Raf) may have alternative substrates. Similarly, experiments using Raf-1 defective in MEK1/2 binding were still able to stimulate differentiation of PC12 cells (Pearson et al., 2000). Furthermore, Raf has now been shown to also signal through p97, an ERK5-related kinase, in response to epidermal growth factor but not fibroblast growth factor (Janulis et al., 2001). Together, this suggests that, although the signaling cascades that predominate are well known, alternative signaling pathways are emerging as our knowledge of kinase biology increases. It is possible therefore that under certain intracellular conditions kinases can signal through many alternative routes, and therefore modulation of a single kinase may enhance signaling through one of these alternative pathways.
The study of kinases is further complicated by the fact that the outcome of signaling through any given cascade by a single stimulus can be different depending on signal strength, duration, and location (Marshall, 1995). The complexity of signaling pathways and the diverse array of cellular functions they control dictate the need for very precise control of kinase activation. This regulation is achieved in many ways. For example, each kinase does appear to have substrate specificity (kinases, however, also show spatial distribution in cells, that is, ERKs associate with the cytoskeleton [Reszka et al., 1995] and many have temporal expression through development [see Shaeffer and Weber, 1999 for review]). Another key regulator of signal transduction and its outcomes appears to be scaffolding proteins that tether signaling components into discrete signaling cascades (Pawson and Scott, 1997) and therefore control the fate of the signaling event following activation with a given stimulus. A good example of this is the regulation of JNK activity in the brain by the scaffolding protein JIP-1 (JNK-interacting protein-1), which prevents the phosphorylation of JNK and its translocation to the nucleus (Dickens et al., 1997).
It is beyond the scope of this review to discuss the intricate nature of kinase signaling pathways in detail. It is crucial, however, to appreciate the complexity of kinase signaling pathways when interpreting data obtained from studies reporting kinase activation or modulation of outcome using small-molecule inhibitors in animal models of disease. Indeed, the question is whether it is naive to investigate the activity of any one kinase in isolation. In order to fully understand the consequences of the activation of a given kinase, it is important to understand not only how its activation affects the other components in its direct signaling pathway, but also the crosstalk with other kinase signaling pathways.
KINASE SIGNALING AFTER CEREBRAL ISCHEMIA
After the onset of cerebral ischemia a multitude of events occur that ultimately result in cell death. Among these, glutamate-mediated excitotoxicity and free radical-mediated injury have been rigorously investigated. The intracellular signaling systems triggered by these events, however, are complex and poorly understood. The relation between MAPK activation and ischemia and reperfusion has been studied in the heart and kidney since the mid 1990s (Aikawa et al., 1997; Bendinelli et al., 1996; Bogoyevitch et al., 1996; Fukunaga and Miyamoto, 1998; Pombo et al., 1994; Shimizu et al., 1998). It is only recently, however, that the effect of cerebral ischemia on MAPKs has been evaluated. Following ischemia many factors are released, including growth factors, cytokines, glutamate, and free radicals, all of which have been shown to stimulate MAPK pathways. In addition to direct activation of MAPKs by these factors, it is possible to envisage that, after injury induced by cerebral ischemia where cellular integrity is compromised, the degree of cellular regulation of kinase activity may be reduced or lost, thus leading to uncontrolled signaling in response to stimulation. For example, it is reasonable to imagine that the breakdown of the cytoskeleton that occurs in these conditions may lead to loss of spatial separation of kinases and also to the disruption of the scaffolding proteins. This may in turn result in aberrant signaling through the kinase pathways and increase crosstalk across the kinase network. The consequence of kinase activation after injury may thus be dependent on the immediate intracellular environment of the individual cell, the cell type, the number of kinase pathways activated at any given time, and the duration of kinase activation.
Although the activation of various kinases, including p38, ERK1/2, and JNK, has now been shown in a variety of different in vitro and in vivo models of brain injury, it remains to be determined unequivocally whether the activation of kinases after the onset of cerebral ischemia is merely a phenomenon of the cell death process or if kinase activation plays a central role in the mechanisms underlying ischemia-induced cell death. Moreover, it remains to be seen whether pharmacological inhibitors selective for a single kinase will provide a novel class of therapeutic agents for the amelioration of ischemia-induced degeneration, or whether the most successful therapeutic agents will be those with a specific “profile” of kinase inhibition.
ACTIVATION OF THE RAF/MEK/ERK1/2 PATHWAY AFTER FOCAL CEREBRAL ISCHEMIA
The Raf/MEK/ERK pathway has received most attention and is therefore the best characterized of the three main MAPK cascades in normal cellular systems. Under normal cellular conditions, the Raf/MEK/ERK pathway is predominantly stimulated by mitogens, which ultimately results in the translocation of ERK1/2 to the nucleus, where it stimulates the transcription factors involved in proliferation and differentiation (Boulton et al., 1991; Marshall, 1995; Segal and Greenberg, 1996). Dominant negative forms of Raf and MEK have been shown to block neurotrophin-induced neuritogenesis, whereas constitutively active forms of these molecules promote neurite outgrowth in the absence of neurotrophins (Segal and Greenberg, 1996; Skaper and Walsh, 1998). This highlights the importance of this kinase pathway in neurotrophic signaling. Recent studies, however, have also shown that the ERK pathway may also play a role in cell death.
Increased ERK1/2 activity has been detected in dissociated neuronal cultures after exposure to NMDA-mediated excitotoxicity (Bading and Geenberg, 1991; Stanciu et al., 2000), seizure-like activity (Murray et al., 1998), exposure to okadaic acid (Runden et al., 1998), or after oxidative stress (Satoh et al., 2000) using phospho-dependent antibodies or ERK1/2-activity assays. More recently data have accumulated showing that phospho-ERK1/2 (pERK1/2) is upregulated in hippocampal organotypic cultures after oxygen glucose deprivation (OGD) (Irving et al., 2001; Namura et al., 2001) or hypoxia (Fig. 2) (Namura et al., 2001). Together, these data suggest that the activation of ERK1/2 may be an event that is central to cell death, stimulated by a variety of different mediators. This apparent dual role of ERK pathway activation suggests that this pathway can be activated in a very compartmentalized manner depending on the cellular circumstances.
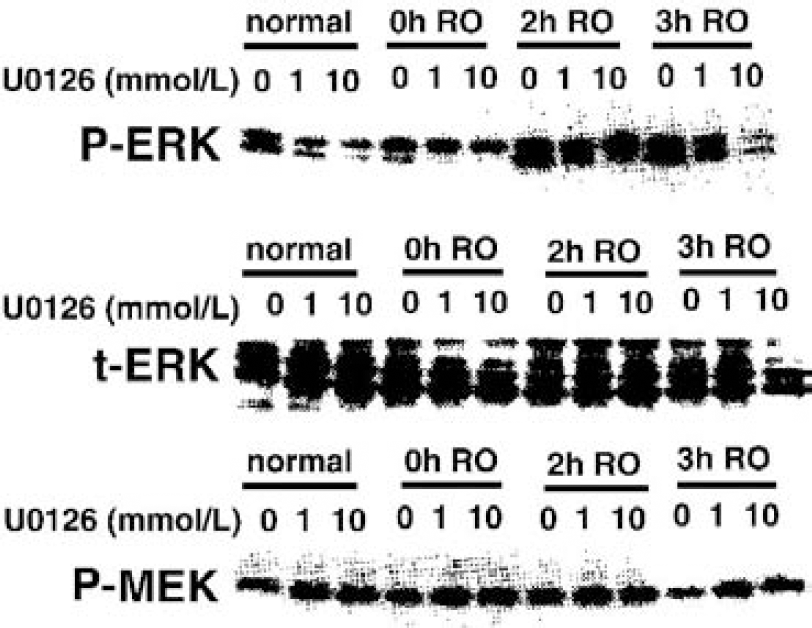
Dose-dependent effects of MAPK/ERK kinase (MEK) inhibitor U0126 on phosphorylation of MEK and ERK in primary cultures of cortical neurons exposed to 9 hours of oxygen deprivation and reoxygenation (RO) for the indicated time. Reprinted with permission from Namura S, Iihara K, Takami S, et al. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci U S A 2001;98:11569–11574. Copyright 2002 National Academy of Sciences U.S.A.
Increased ERK1/2 phosphorylation has also been reported in rodents after transient (Irving et al., 2000, 2001; Alessandrini et al., 1999; Namura et al., 2001), permanent middle cerebral artery occlusion (MCAO) (Irving, 2001; Wu et al., 2000) and global ischemia (Namura et al., 2001; Hu et al., 2000). After permanent and transient (90-minute) focal cerebral ischemia in the rat (performed using the intraluminal thread model) phospho-ERK1/2 (pERK1/2) immunostaining was increased within neurons and glia in “penumbral” regions, that is, those areas where microtubule-associated protein 2 (MAP2) immunostaining was altered but not lost (Irving et al., 2000), 6 and 24 hours after MCAO as detected by immunohistochemistry (Fig. 3), whereas staining was decreased in infarcted tissue (Irving et al., 2000). This result was supported by data obtained by using an ERK1/2 kinase activity assay which showed increased enzyme activity 6 and 24 hours after transient MCAO (Irving et al., 2001) (Fig. 4). Similarly, increased pERK1/2 levels (as detected by Western blotting) were also detected in mouse brain after 3-hour MCAO and 5-minute reperfusion (Namura et al., 2001). After permanent MCAO pERK1/2 levels were maximum approximately 1 hour after occlusion, but could still be detected up to 6 hours in a subset of cells (Fig. 3) (Irving et al., 2000). This is in agreement with studies investigating pERK1/2 levels after pMCAO in mice (Wu et al., 2000). In this study, increased pERK1/2 levels (as detected by Western blotting) became maximal between 30 minutes and 2 hours after ischemia onset, but could still be detected after 6 hours (Wu et al., 2000). In contrast to the studies of Irving et al. (2000), however, this increased pERK1/2 activity was detected predominantly within the necrotic tissue. This discrepancy may reflect differences in the severity of injury produced by the intraluminal thread method of occlusion in rats and mice. Because phosphorylation is an energy-dependent process, it is likely that phosphorylation events will be markedly reduced where blood flow is minimal. Indeed, Namura et al. (2001) reported decreased ERK1/2 activity within the hippocampus during global ischemia, but it rapidly increased after the onset of reperfusion. Owing to the predominantly transient nature of kinase activation, more detailed investigations of the temporal profile of kinase activation and how this relates to blood flow and/or glucose metabolism is required. This will greatly enhance our understanding of how kinase activation relates to ischemia-induced cell stress and death, thus allowing the comparison of kinase activation across a variety of focal ischemia models.
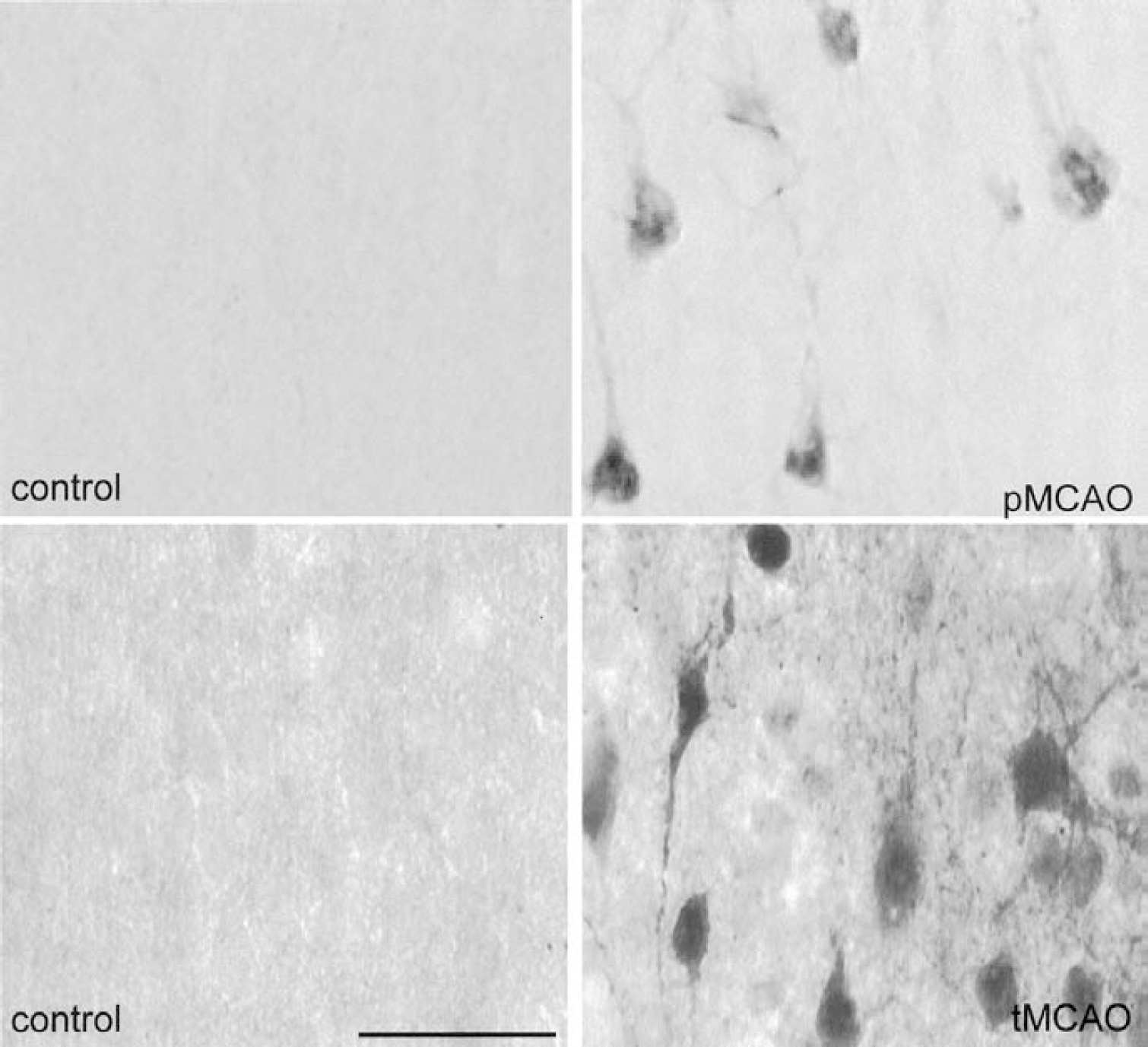
Distribution of phospho-ERK1/2 in the cerebral cortex 6 hours after the induction of permanent middle cerebral artery occlusion (pMCAO) or transient middle cerebral artery occlusion (tMCAO) in the rat. Phospho-ERK1/2 immunostaining was predominantly located within the cytoplasm of the neuropil in histologically normal tissue (control). Six hours after pMCAO and tMCAO, phospho-ERK1/2 staining was decreased within the core of the lesion resulting from MCAO (data not shown). Staining was increased, however, in the cytoplasm of neuronal perikarya within the penumbra (i.e., those areas where the microtubule-associated protein 2 [MAP2] immunostaining was altered but not lost, as described previously [Irving et al., 2000]). Scale bar = 50 μm. Reprinted from Molecular Brain Research, vol. 77; Irving EA, Barone FC, Reith AD, et al.; Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat, pp 65–75, copyright 2000, with permission from Elsevier Science.
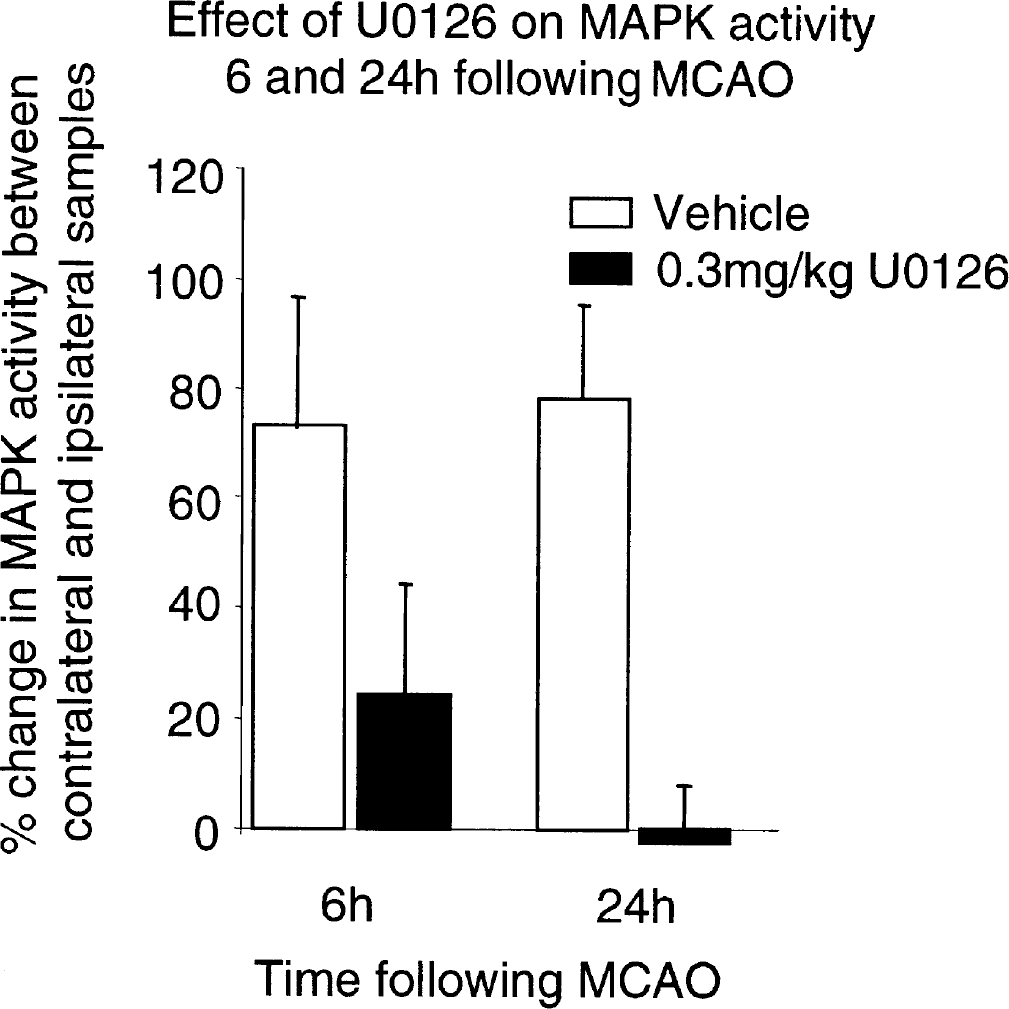
Six and 24 hours after transient middle cerebral artery occlusion (MCAO) in the rat, mitogen-activated protein kinase (MAPK) activity was markedly increased (73 ± 24 and 78 ± 17% respectively) in the ipsilateral cortex compared to the contralateral cortex in vehicle treated animals. Administration of U0126 (0.3mg/kg, intravenously) 1 hour after the onset of ischemia decreased MAPK activity present in the ipsilateral cortex both 6 hours (3-fold) and 24 hours (30-fold) after MCAO compared with vehicle-treated animals. Reproduced from Irving et al. (2001); reprinted with permission of Lippincott Williams & Wilkins.
Activation of ERK1/2 has also been reported after global ischemia in both gerbils (Namura et al., 2001) and rats (Hu et al., 2000). In these models of global ischemia, the neurons of the CA1 are selectively vulnerable to ischemia and show maximal death 48 to 72 hours after the onset of the ischemia. After 15 minutes of ischemia and 30 minutes of reperfusion in the rat, increased pERK1/2 levels were present in the cells of the dentate gyrus (DG), which are spared in this injury paradigm, whereas no activity was detected in the CA1 (Hu et al., 2000). In the gerbil increased pERK1/2 levels were detected in the CA1 10 minutes after 3.5 minutes of global ischemia, but were reduced at 1 hour, at which time activity was predominantly detected in the DG (Namura et al., 2001). The different activity-profile in these two models may again reflect the transient nature of ERK1/2 activation.
The observation that ERK1/2 activity is transiently increased in the ischemic territory before cell death in both focal and global models of ischemia suggests that elevated levels of pERK1/2 may be involved in the mechanism underlying ischemia-induced cell death. The presence of increased pERK1/2 levels in cells that survive, however, and decreased levels in those cells that die after focal and global ischemia may suggest that activation of the ERK1/2 “survival” pathway is required to prevent cells succumbing to ischemia-induced death. Similar observations have been made in other models of brain injury where CREB phosphorylation has been shown to increase in those cells surviving neuronal injury resulting from hypoxic-ischemia in immature rats (Walton et al., 1999) and after transient ischemia in the rat (Fig. 5) (Irving et al., 2000; Tanaka et al., 1999).
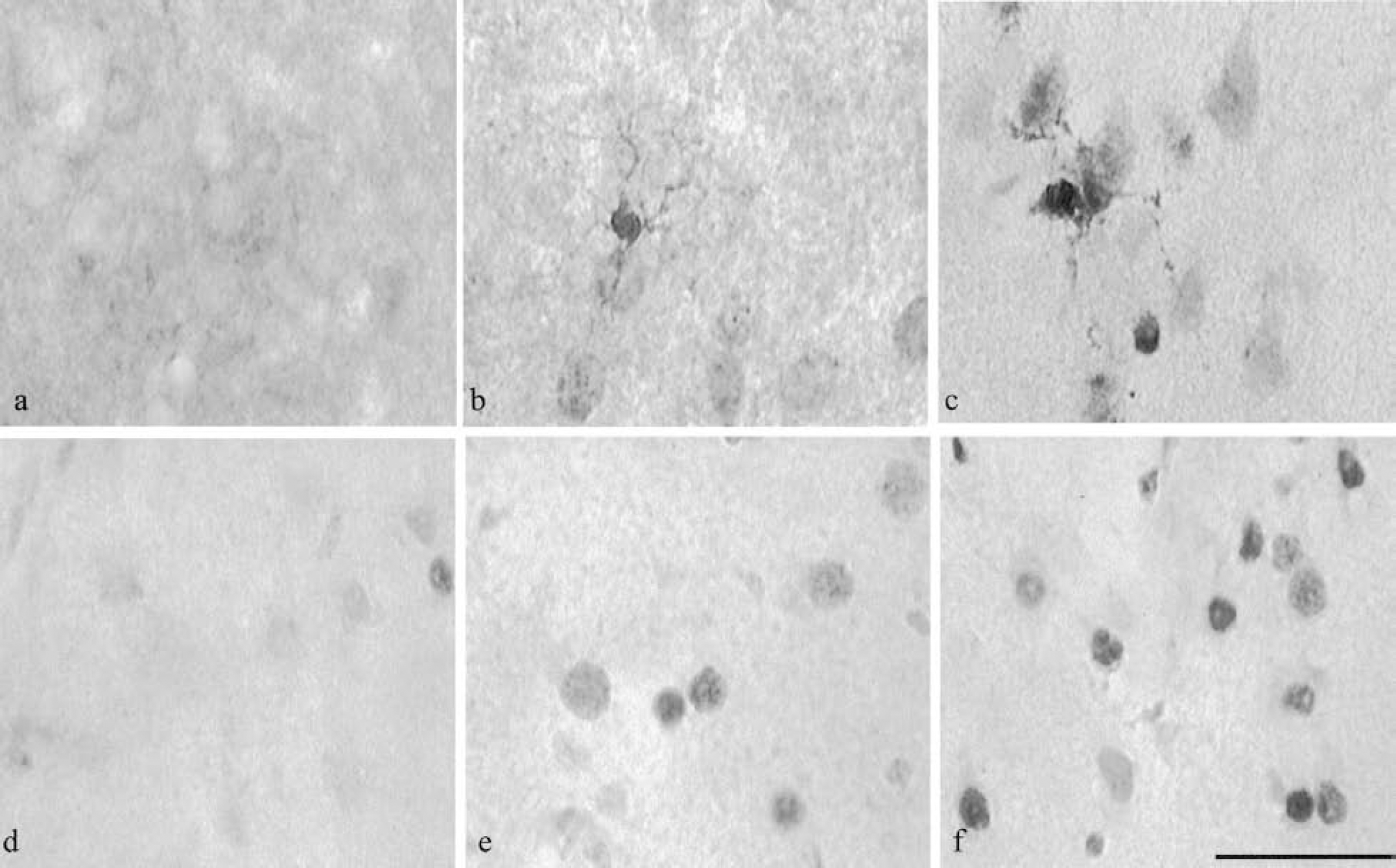
Distribution of phospho-p38
To date there is little information regarding the effects of ischemia on Raf and MEK, the upstream activators of ERK1/2. Interestingly, in primary cultures of cortical neurons exposed to 9 hours of oxygen deprivation followed by 0, 2, or 3 hours of reoxygenation, ERK1/2 activity was markedly increased with time of reoxygenation. In contrast, however, phospho-MEK (pMEK) levels remained unaffected (Fig. 2) (Namura et al., 2001). This suggests that under these experimental conditions, elevated levels of pERK1/2 may not be the result of increased MEK1/2 activity. Furthermore, these results suggest that a loss of normal signaling through the classical Raf/MEK/ERK pathway may occur under conditions of cellular injury. In order, therefore, to fully understand the activation of the Raf/MEK/ERK1/2 pathway and its role in the mechanisms underlying ischemia-induced cell death, the activity of each member of the cascade will have to be profiled after the cessation of blood flow.
THE RAF/MEK/ERK1/2 PATHWAY AND ISCHEMIA-INDUCED CELL DEATH
As discussed previously, the activation of ERK1/2 occurs after a variety of cellular injury paradigms, although the consequence of such activation is currently unclear. Activation after ischemic injury may merely be a consequence of compromised cellular integrity and consequent reduction in the level of signaling regulation. Alternatively, the increased activity may be involved in the progression of ischemic cell death. The advent of specific MEK inhibitors PD-98059 and U0126 (Alessi et al., 1995; Davies et al., 2000; Favata et al., 1998; see Fig. 6) have allowed the investigation of the role of MEK in ischemia-induced cell death. Treatment of cell cultures with PD-98059 or U0126 have been shown to reduce damage in response to various cytotoxic stimuli such as NMDA-mediated glutamate excitotoxicity, okadaic acid, seizure activity, hypoxia, and OGD (Fig. 7) (Bading and Greenberg, 1991; Irving et al., 2001; Murray et al., 1998; Namura et al., 2001; Runden et al., 1998; Satoh et al., 2000; Skaper et al., 2001; Stanciu et al., 2000), suggesting that activation of this pathway is, at least in part, involved in the neuronal death associated with these models of injury.
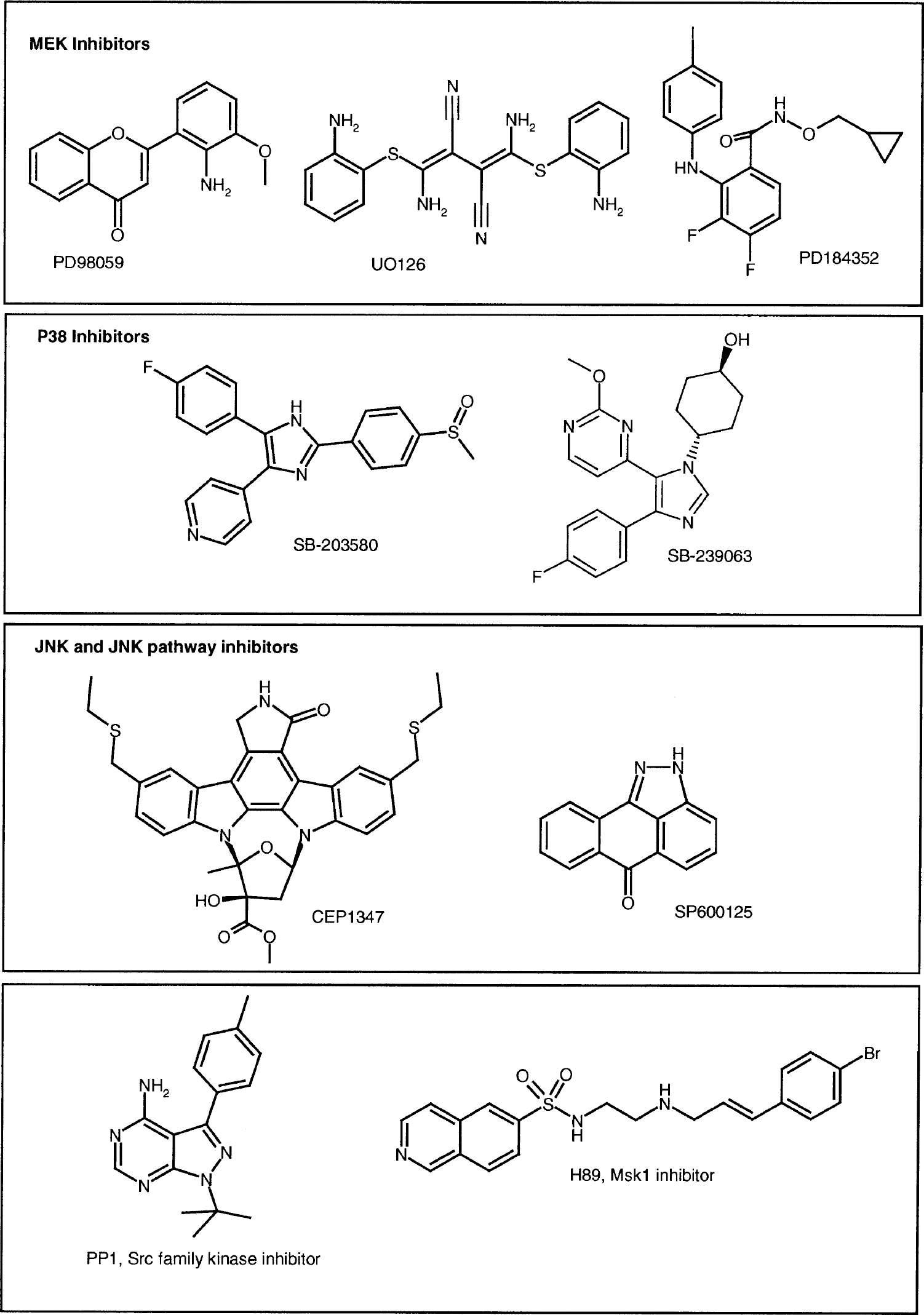
Chemical structures of key inhibitors of the three mitogen-activated protein (MAP) kinase pathways discussed in this article. MEK, MAPK/ERK (mitogen-activated protein kinase/extracellular signal-regulated kinase); JNK, c-Jun amino-terminal kinase.
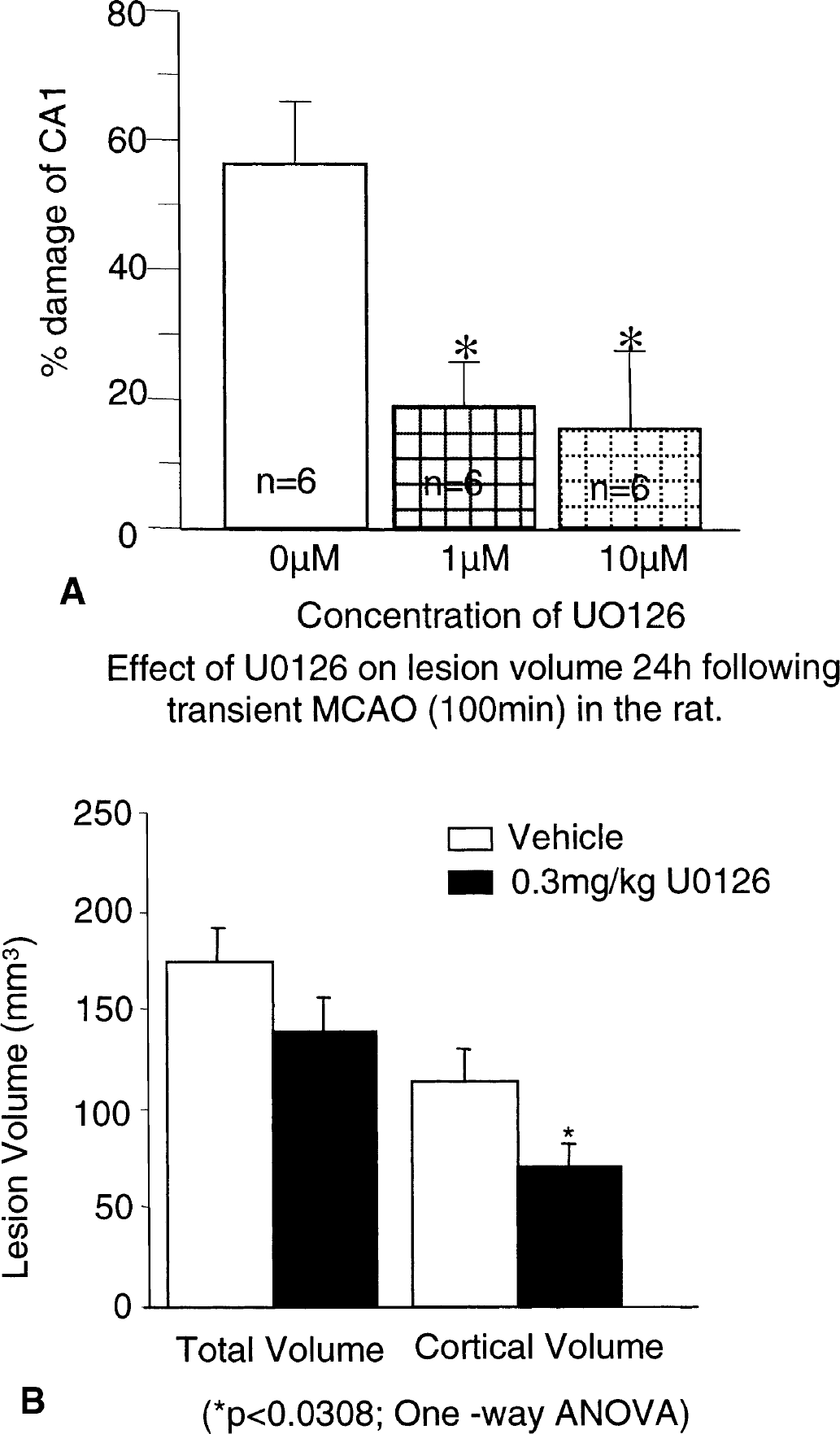
U0126 protects organotypic hippocampal cultures against oxygen glucose deprivation (OGD)–induced cell death.
Furthermore, PD-98059 has been shown to reduce lesion volume by 55% and 36%, 22 and 72 hours, respectively, after transient (2-hour) MCAO in the mouse when administered 30 minutes before injury onset (Alessandrini et al., 1999). More recently, the more potent MEK inhibitor U0126 has been shown to offer significant neuroprotection when administered 1 hour after the onset of transient focal cerebral ischemia in the rat (Fig. 7) (Irving et al., 2001), up to 3 hours after transient ischemia in the mouse (Namura et al., 2001), 1 hour after permanent MCAO in the mouse (Fig. 8) (Namura et al., 2001) and when given before global ischemia in the gerbil (Namura et al., 2001). Together, these data show that MEK inhibitors can prevent cell death induced in a variety of different models, suggesting that MEK activation plays a central role in the cell death seen in these models.
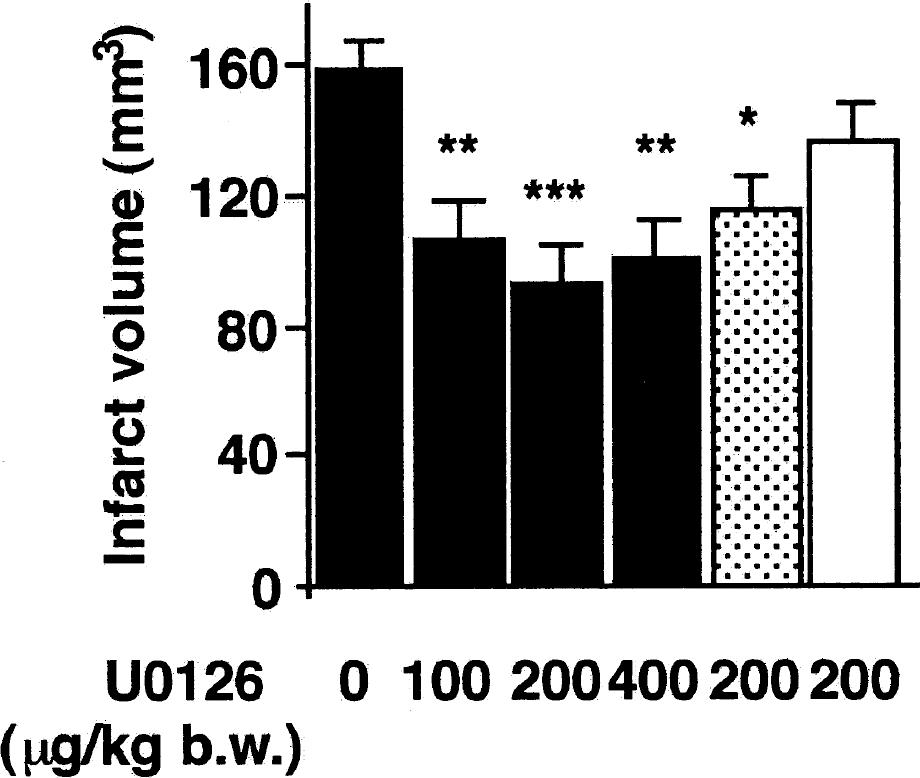
U0126 provides neuroprotection after focal cerebral ischemia in mice. Infarct volume was measured after 24 hours of middle cerebral artery occlusion. U0126 was injected 10 minutes before (filled columns), 1 hour (dotted column) or 3 hours (open column) after the onset of ischemia. *P < 0.05, **P < 0.01, ***P < 0.001, compared with the vehicle-treated group (ANOVA). Reprinted with permission from Namura S, Iihara K, Takami S, et al. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci U S A 2001;98:11569–11574. Copyright 2002 National Academy of Sciences U.S.A.
Caution should be exercised, however, when interpreting data from these studies. First, studies conducted to investigate the neuroprotective properties of U0126 and PD98059 after cerebral ischemia have also assessed the inhibition of ERK1/2 activation. Indeed the neuroprotection offered by U0126 and PD98059 in each case was accompanied by an inhibition of ERK1/2 activation showing successful inhibition of MEK's ability to activate ERK1/2 (Fig. 4) (Alessandrini et al., 1999; Irving et al., 2001; Namura et al., 2001). Most of the studies conducted to date, however, have not investigated MEK activity after stroke (with the exception of Shamloo et al. [1999], who showed increased MEK activation after global ischemia), or have shown that MEK activity is reduced in the presence of the inhibitor. As mentioned previously, in cortical neurones exposed to 9 hours of oxygen deprivation and reoxygenation pERK1/2 levels were increased without significant enhancement of MEK activation (Fig. 2) (Namura et al., 2001). U0126 still protected these cultures, however, after 24 hours of reoxygenation. This suggests that the protection offered by U0126 may not be directly due to MEK1/2 inhibition. Moreover, treatment of these cultures with U0126 (1 or 10 μmol/L) markedly increased pMEK levels (Fig. 2) (Namura et al., 2001). A similar observation has been made using inhibitors of Raf. These studies have shown that Raf appears to counteract its own activation by a novel feedback loop, such that inactivation is always counteracted by reactivation (Hall-Jackson et al., 1999a,b). It is possible that a similar negative feedback loop normally controls MEK activation. Furthermore, the neuroprotection offered by PD98059 after transient MCAO in the mouse was associated with enhanced c-Fos immunoreactivity (Alessandrini et al., 1999). This is surprising because c-Fos activity is, at least in part, controlled by ERK1/2 activation of Elk-1 (Fig. 1). Together, this suggests that the neuroprotection offered by U0126 and PD98059 may not be simply through the direct inhibition of MEK-mediated pERK1/2 activation. It is possible that the inhibition of MEK may stimulate signaling through alternative signaling cascades, and that it may be these signaling events that are indirectly responsible for the neuroprotection seen after cerebral ischemia. This again highlights the importance of investigating not only changes in the target kinase after intervention, but also those kinases known to crosstalk with the individual kinase.
Second, U0126 and PD98059 are non-ATP-competitive inhibitors of MEK (i.e., they bind to MEK at sites independent of the ATP and ERK binding sites) (Favata et al., 1998), and therefore show poor levels of direct inhibition of a variety of kinases including MEK. From the pathways evaluated (Davies et al., 2000; Favata et al., 1998), there does appear to be some selectivity by these compounds for the inhibition of MEK activation in vitro. U0126, however, was originally discovered by screening for small molecules that block AP-1 activation (Favata et al., 1998). AP-1 is a transcription factor downstream of Elk-1, and, therefore, ERK, JNK, and p38 activation (Fig. 1). Interestingly, both U1026 and PD98059 were shown to protect the pluripotent HT29 intestinal cell line against hydrogen peroxide-induced death not through inhibition of ERK1/2 but by the inhibition of JNK (Salh et al., 2000). The doses used in this study were similar to those conventionally used to inhibit MEK activation, suggesting that both U1026 and PD98059 have activity against JNK in a cellular context. Both PD98059 and U0126 have also been shown to have effects on glutamate release from hippocampal synaptosomes (Pereira et al., 2002). In this study PD-98059 was shown to inhibit glutamate release from potassium chloride-stimulated synaptosomes, whereas U0126 potentiated depolarization-induced calcium-independent glutamate release and inhibited calcium-dependent release. In addition, both PD98059 and U0126 have also been shown to prevent the activation of MEK5, and therefore ERK5, albeit at higher concentrations required for MEK, and hence ERK1/2, inhibition (Mody et al., 2001). Although the role of ERK5 in cell death is not yet known, it has been shown to be increased by oxidative stress (Abe et al., 1996). This further supports the concept that some of the neuroprotection offered by U0126 and PD98059 in rodent models of cerebral ischemia may be through mechanisms other than the direct inhibition of MEK signaling.
It is clear therefore that further analysis of the consequence of MEK inhibition on kinase activity, both in the Raf/MEK/ERK pathway and also the wider kinase network, will be required to fully understand the precise mechanisms underlying the protection offered by these MEK inhibitors. Owing to the interest in these compounds for cancer research, a large number of tools are becoming available to help elucidate the involvement of the Raf/MEK/ERK pathway in the mechanisms underlying ischemia-induced cell death. PD184352 (Fig. 6) is a more potent and selective MEK inhibitor (Davies et al., 2000) than U0126 (Sebolt-Leopold et al., 1999). Tools for the inhibition of Raf are now also becoming available (Dean et al., 2001; Gaiba et al., 2001; Steadman and Takle, 2001), with interest in their application for prevention of neurodegeneration. It is certainly possible to envisage that inhibition of this pathway through different kinases may have very different outcomes with regard to the pathogenesis of cerebral ischemia. Only when highly selective potent tool compounds become available will the involvement of each individual kinase in ischemic cell death be fully understood.
VASCULAR-MEDIATED NEUROPROTECTION BY MEK INHIBITION
The aforementioned studies center around the assumptions that increased kinase activity in the brain contributes directly to neuronal/glial death and that intervention with small-molecule inhibitors provides direct neuroprotection by blocking kinase activity in the central nervous system (CNS). After ischemic brain injury, however, neuroprotection can be gained either by directly protecting the cellular elements of the CNS or by promoting blood flow into the ischemic regions. Agents such as endothelin receptor antagonists provide significant neuroprotection in rodent models of stroke by enhancing regional cerebral blood flow (rCBF) (Barone et al., 2000; Matsuo et al., 2001). Endothelin-1 (ET-1) is a potent vasoconstrictor and has been shown to induce constriction of rabbit basilar artery vascular smooth muscle cells through activation of the Raf/MEK/ERK1/2 pathway (Zubkov et al., 2000). Furthermore, a role for the Raf/MEK/ERK pathway has been shown in hemolysate-induced constriction of basilar arteries (Zubkov et al., 1999). The Raf/MEK/ERK pathway has also been shown to be activated by other potent vasoconstrictors such as angiotensin II and noradrenaline. Inhibitors of src or PI3-K, however, failed to block hemolysate-induced contraction (Zubkov et al., 1999) but do block endothelin-induced constriction. This suggests that, although ET-1 and hemolysate might operate two different signaling systems, Raf/MEK/ERK1/2 activation is key to the vasoconstriction induced by both. Because ET-1 is released after ischemia, it is possible to speculate that the MEK inhibitors U0126 and PD98059 act at smooth muscle cells to block the effects of ET-1–mediated vasoconstriction, and therefore provide neuroprotection, at least in part, through enhancing rCBF in the ischemic territory. Studies investigating the effects of kinase inhibitors after focal cerebral ischemia have not documented changes in rCBF, but it certainly warrants investigation.
Recently, vascular endothelial growth factor (VEGF) has been shown to increase the permeability of endothelial cell monolayers via activation of ERK1/2, which could be reversed using the ERK1/2 inhibitor, AG126 (Lal et al., 2001). Moreover, the disruption of endothelial cell monolayer integrity and intracellular gap formation by PMA (phorbol 12-myristate 13 acetate) can be reversed by PD98059 (Verin et al., 2000). These results suggest that at least some of the neuroprotection offered by U0126 and PD98059 may also be mediated through reducing vascular permeability and thus edema formation after CNS injury. In further support of this, src (src can signal through the Raf/MEK/ERK pathway) deficiency or blockade of Src kinase (Src kinase is downstream of VEGF receptor activation) with the inhibitor PP1 (Fig. 6) have been shown to provide neuroprotection by reducing edema after focal cerebral ischemia. This reduction in edema was also associated with an increase in rCBF (Paul et al., 2001). PP1 is not specific for Src but is an Src-family kinase inhibitor, potently inhibiting Fyn and Lck (Hanke et al., 1996). Its activity in vivo, however, adds considerable weight to the findings with the Src-knockout animals and suggests that kinase pathways play an important role in controlling vascular permeability. Together, these data suggest that MEK inhibition may provide a very attractive method of intervention following stroke. It is possible to envisage that MEK inhibition may improve outcome through direct cellular protection, increasing rCBF and decreasing edema formation.
p38 PATHWAY ACTIVATION AFTER FOCAL CEREBRAL ISCHEMIA
The role of p38 in ischemia-induced cell death has been reviewed recently (Barone et al., 2001) and therefore will only be briefly discussed in this article. The p38 pathway plays an important role in transducing signals involved in cell survival, apoptosis, and inflammatory cytokine production (Fig. 1) (Kummer et al., 1997; Lee et al., 1994; Lee and Young, 1996; Xia et al., 1995). Sustained activation of p38 has been shown to be associated with neuronal death/apoptosis, and selective p38 inhibitors have been shown to promote survival (Skaper and Walsh, 1998; Kummer et al., 1997; Xia et al., 1995). Nerve growth factor withdrawal causes neuronal apoptosis that is preceded by decreased ERK and increased p38/JNK activities (Xia et al., 1995), suggesting that a balance between ERK and stress-activated kinases is required to mediate cell survival.
The p38 pathway is strongly activated by factors such as TNF-α and IL-1β, which are known to be increased after stroke and have been shown to be involved in the mechanisms underlying ischemia-induced cell death (Freshney et al., 1994; Raingeaud et al., 1995; Rouse et al., 1994). Increased p38 activity was recently reported after cerebral ischemia by Walton et al. (1998), who found that p38 and MAPK-activated protein 2 (MAP-KAP2) activity increased in microglia 4 days after global ischemia. Further investigation of the global ischemia model have supported these findings, in that increased p38 activity was detected in the CA1 region of the hippocampus within 3 to 6 hours of injury onset (Shioda et al., 1998). Similarly, indirect assessment of p38 activity (by investigating the phosphorylation of the transcription factor ATF2, which is phosphorylated by p38 [Bogoyevitch et al., 1996]), showed increased ATF2 activity in the CA1 region of the hippocampus, which was maximal 24 hours after global ischemia (Hu et al., 2000). Together, these studies show that prolonged activation of p38 occurs in the CA1 of the hippocampus after global ischemia and therefore may be involved in the selective vulnerability of these neurons to ischemia. In contrast, however, in a gerbil model of global ischemia p38 activity was shown to transiently increase within 15 minutes in the CA1, with sustained activation being detected only in the CA3 (Sugino et al., 2000).
Experiments conducted in our laboratories have shown that phospho-p38 immunoreactivity was markedly increased in cells with the morphologic appearance of astrocytes within the ischemic “core” and “penumbra” after permanent and transient focal cerebral ischemia in the rat. In penumbralike areas (i.e., in those areas where MAP2 distribution was altered but not lost; see Irving et al. [2000]) the intensity of phospho-p38 staining was also significantly increased within neuronal perikarya as compared with the contralateral hemisphere (Fig. 5) (Irving et al., 2000). To support this, we were also able to show increased p38 activation after permanent MCAO indirectly using biochemical analysis of ATF2. In this study ATF2 activity was detected as early as 1 hour, was maximal between 3 and 6 hours, but was still detectable for 24 hours (see Barone et al., 2001, for review). Studies investigating p38 activation using Western blotting showed a similar temporal profile of p38 activation in that phospho-p38 levels were increased as early as 10 minutes after permanent MCAO in the mouse, which peaked by 30 minutes but were sustained above background levels at 24 hours (Wu et al., 2000). Similarly to findings reported in Irving et al. (2000), the majority of p38 was detected within the ischemic “core” (i.e., those areas where MAP2 staining was lost; see Irving et al. [2000]) early after ischemia onset. Interestingly, this study suggests that the transient activation of p38 is initiated before pERK1/2 activation, which was increased at 30 minutes and peaked at 2 hours after permanent MCAO (Wu et al., 2000). In contrast to ERK1/2 activation, the majority of studies investigating p38 activity following cerebral ischemia describe prolonged activation in brain regions destined to die after the cessation of blood flow. It is therefore possible to speculate that, similar to the in vitro situation (Xia et al., 1995), the fate of the cell depends on the balance between the activation of survival pathways, such as ERK1/2, and cell death pathways, such as p38 and JNK. More detailed studies, however, will be required to determine whether ERK and p38 signaling pathways are activated concurrently or sequentially in any given cell and how this relates to energy status within the ischemic territory.
ROLE OF p38 ACTIVATION IN MEDIATING CELL DEATH AFTER INJURY
In vitro studies have shown that treatment of cardiac myocytes with the p38 inhibitor SB-203580 (Fig. 6) protects these cells from ischemia (Ma et al., 1999). Furthermore, SB-203580 was shown to reduce ischemic cell death in the CA1 region of the hippocampus after global ischemia in the gerbil (Sugino et al., 2000) through the inhibition of p38. This compound, however, has also been shown (for reviews, see Adams et al., 2001; Clerk and Sugden, 1998; Harper and LoGrasso, 2001) to be an inhibitor of cRaf, as well as JNK3 and JNK2, at approximately 10-fold higher concentrations than the p38 IC50s. It has also been reported to be an inhibitor of the activation of protein kinase B (Lali et al., 2000), albeit at somewhat higher concentrations than those required to inhibit p38. More convincing data supporting the theory that p38 inhibition provides neuroprotection after CNS injury was obtained in our laboratory using a newer generation, potent p38 inhibitor, SB-239063 (Fig. 6). In these studies, SB-239063, when administered after the onset of permanent focal cerebral ischemia in the rat, reduced infarct volume and improved functional outcome when assessed 24 hours after occlusion Barone et al., 2001) in both “severe” and “moderate” models of stroke. It is attractive to speculate that this is through inhibition of p38 present in astrocytes, and, therefore, the inhibition of TNF-α and IL-1 release and their subsequent effects on neurones and glia. Indeed, SB-239063 has been shown to decrease TNF-α and IL-1β mRNA levels in the plasma after permanent MCAO (Barone et al., 2001). Furthermore, because cytokine and apoptotic pathways have been shown to converge, it is possible that p38 activation may be key to both (Hara et al., 1997; Sidotidefraisse et al., 1998). In light of this, it is possible that, in addition to the acute neuroprotection offered by p38 antagonists after stroke (Barone et al., 2001), p38 antagonists may also offer protection against the delayed cell death that occurs over time after stroke. Further, selective p38 inhibitors are now being reported (Boehm and Adams, 2000), which will hopefully encourage further investigation of the role of p38 activation in cerebral ischemia. It remains to be seen, however, whether molecules of this type will be suitable for development for use in stroke.
ACTIVATION OF THE JNK/SAPK PATHWAY AFTER CEREBRAL ISCHEMIA
Activation of the JNK/SAPK pathway occurs in response to similar stress stimuli that activate p38 and, like p38, is associated with the induction of apoptosis, protection from cell death, proliferation, and differentiation, depending on the cell type studied (Fig. 1). Three JNK isoforms have been detected in the mammalian system; JNK1 and JNK2 are expressed in all tissues, and JNK3 is predominantly found in the CNS (Martin et al., 1996). Studies using KO mice have shown that the three JNK isoforms have different functions in the CNS. Mice lacking both JNK1 and JNK2 are embryonic lethal, which has been attributed to the increased abundance of apoptotic cell death in the forebrain and decreased cell death, thus preventing remodeling in the hind brain (Kuan et al., 1999). Mice lacking JNK3 show decreased sensitivity to kainic acid-induced seizures and cell death in the hippocampus (Yang et al., 1997). JNK activity in the brain is in part regulated by the scaffolding protein JIP-1 (JNK-interacting protein-1), which inhibits the phosphorylation of JNK and its translocation to the nucleus (Dickens et al., 1997).
A number of studies have investigated the activation of JNK and c-Jun after global ischemia; the profile of activation, however, appears to be quite different between experiments. Increased JNK phosphorylation has been reported up to 72 hours after 5-minute global ischemia in the gerbil (Ferrer et al., 1997; Sugino et al., 2000; Tsuji et al., 2000) and 24 hours after 15-minute global ischemia in the rat (Hu et al., 2000). In the gerbil model, this increased JNK activity was associated with increased c-Jun expression (not activity) in the hippocampus, but not in those areas susceptible to global ischemia (i.e., the CA1 region) (Ferrer et al., 1997). In this model c-Jun activity was not detected up to 7 days after reperfusion in the CA1 region of the hippocampus, but was detected in the CA3 region at 6 and 12 hours (Ferrer et al., 1997). Similarly, Sugino et al. (2000) detected increased phospho-JNK levels in the CA1 after 15 minutes of reperfusion in the gerbil, which then decreased and disappeared over time. A sustained phospho-JNK activation, however, occurred in the CA3 fibers and dentate gyrus as early as 6 hours after reperfusion, which, although decreased, was still detectable 72 hours after ischemia. In contrast, increased phospho-c-Jun levels were detected in the CA1 region of the hippocampus 24 hours after 15 minutes of global ischemia in the rat (Gillardon et al., 1999; Hu et al., 2000). In these studies, no increased phospho-c-Jun levels were detected in the dentate gyrus, therefore showing that the temporospatial distribution of phospho-c-Jun but not c-Jun correlates with selective vulnerability of neurones to global ischemia. It is interesting to note that in this study increased phospho-c-Jun levels were not associated with increased phospho-JNK levels (Gillardon et al., 1999). It is possible that an early transient activation of JNK may have occurred, which then results in a sustained prolonged activation of c-Jun. Alternatively, this is another example whereby normal signaling cascades are disrupted after cerebral ischemia. Interestingly, JIP-1 expression was shown not to change at any time after global ischemia (Ferrer et al., 1997). Basal levels of JIP-1 mRNA, however, appeared to be higher in the areas resistant to ischemia, that is, CA2/CA3 compared with CA1 (Gillardon et al., 1999). Together, these studies show that activation of the JNK/SAPK pathway occurs after global ischemia. Further investigation, however, will be required to determine whether or not this activation may be involved in the selective neuronal vulnerability that is characteristic of these models.
Activation of the JNK/SAPK pathway has also been shown to be increased after transient (90-minute) focal cerebral ischemia in the rat (Hayashi et al., 2000; Herdegen et al., 1998). In this model, c-Jun phosphorylation was detected in the ipsilateral cortex and striatum around the necrotic infarcted area. This increased activity became detectable 3 hours after the onset of ischemia, became maximal at 72 hours, and subsequently declined over 5 days (Herdegen et al., 1998). Similarly to c-Jun, JNK activity increased as early as 3 hours after transient MCAO, and further increased up to 72 hours (Hayashi et al., 2000). In contrast to c-Jun activity, however, which was detected around the infarcted tissue, in this study JNK1 activity was detected within the ischemic lesion. This increased activation of JNK-1 was associated with an increase in total JNK expression after 1, 3, 8, or 72 hours of transient ischemia, and also a small increase in JIP-1 expression toward the boundary areas of the lesion after 72 hours (Hayashi et al., 2000). After permanent ischemia in the mouse increased phospho-JNK levels (as detected by Western blotting) were detected as early as 10 minutes after occlusion; the levels peaked at 30 minutes, but remained significantly upregulated up to 6 hours. At 24 hours after MCAO levels were still elevated above background levels. Immunohistochemical studies confirmed that this JNK activity was present within the ischemic lesion (Wu et al., 2000). In this model the activation of JNK was delayed compared with ERK1/2 and p38, and, in contrast to ERK1/2 and p38—which were markedly reduced again 2 hours and 30 minutes, respectively, after occlusion-JNK activity remained markedly upregulated 6 hours after occlusion. The early and sustained activation of JNK within tissue destined to die in these models suggests JNK activation may play a role in the mechanisms underlying ischemia-induced cell death. c-Jun activity, therefore, is upregulated and sustained in vulnerable cells in global ischemia models, whereas, in focal models, JNK is increased in cells destined to die after ischemia onset. The activation of the JNK pathway and its relation to ischemia-induced cell death requires further investigation.
CONSEQUENCE OF JNK/SAPK PATHWAY ACTIVATION IN CEREBRAL ISCHEMIA
Of the three MAP/SAPK pathways, the consequence of JNK pathway activation after ischemia has only more recently been investigated, owing to the recent availability of inhibitory tools. The regulator of JNK activity, JIP-1, has been shown to prevent neuronal cell death caused by growth factor withdrawal, supporting a role for JNK in neuronal apoptosis (Dickens et al., 1997, Kim et al., 1999). Further supporting a role for JNK activation in cell death, hydrogen peroxide has been shown to increase JNK activity (Hashimoto et al., 2002; Salh et al., 2000), and ultimately results in cell death. Under these experimental conditions, cells were protected by PD98059, U0126, and α-synuclein, not through ERK1/2 inhibition, but in a manner dependent on JNK inhibition. Inhibition of JNK by α-synuclein may have been mediated through increased expression of JIP-1 (Hashimoto et al., 2002); the effects of PD98059 and U0126 on JIP-1 expression, however, have not yet been investigated. In addition, cells transfected with JIP-1 were resistant to hydrogen peroxide-induced death (Hashimoto et al., 2002).
There is evidence that JNK activation is also involved in free radical-mediated cell death. The spin trap agent α-phenyl-N-tert-butylnitrone protected the CA1 region of the hippocampus in gerbils after 5-minute global ischemia. This neuroprotection was associated with the inhibition of JNK and p38 activity in the CA1, 6 hours after reperfusion (Tsuji et al., 2000). While part of the activity of p38-inhibitory molecules such as SB-203580 may be explained by inhibition of JNK3 and/or JNK2 (see above), molecules that inhibit the JNK pathway without inhibiting the p38 pathway have now been reported. CEP1347 (Fig. 6) inhibits the JNK pathway at the mixed-lineage kinase level, without inhibiting the p38 or ERK pathways (for review, see Harper and LoGrasso, 2001). This compound has been shown to inhibit cell loss in vivo in a number of settings, particularly those regarded as models of apoptotic cell loss and Parkinson disease (see Harper and LoGrasso, 2001, for review). The evaluation of this compound, however, which is in Phase II clinical studies for Parkinson disease, has not yet been reported in a stroke model. Recently more selective JNK inhibitors have being generated, which include SP600125 (Fig. 6) and SPC9766 (structure not disclosed) (for review, see Harper and LoGrasso, 2001). Although these compounds have mainly been evaluated in rheumatoid arthritis and asthma models of inflammation, SPC9766 has recently been evaluated in a rat model of transient focal cerebral ischemia (2-hour occlusion) (Raymon et al., 2001). In this study, SPC9766 (1 mg/mL), when administered 15 minutes after the onset of ischemia (intracerebroventricularly for 3 hours), resulted in a 29% and 34% reduction in infarct volume 24 hours and 72 hours, respectively, after reperfusion. This decreased lesion volume was also associated with an improvement in cognitive function and a decrease in the number of apoptotic cells present in the infarct region (Raymon et al., 2001). Further molecule types are now being described as inhibitors of JNKs (see Harper and LoGrasso, 2001, for review), which will provide tools to allow further elucidation of the involvement of this pathway in ischemia-induced cell death.
ROLE OF MAPK PATHWAYS IN PRECONDITIONING
Preconditioning is a phenomenon in which brief episodes of sublethal injury result in a resistance to further prolonged lethal injury. Preconditioning can be induced in a variety of different organs including brain, heart, liver, small intestine, skeletal muscle, kidney, and lung (see Bolli [2001], Bonventre [2002], Dawson and Dawson [2000], and Raeburn et al. [2001] for reviews). In the brain preconditioning can be induced by a variety of injuries including brief ischemic episodes, spreading depression, and exposure to excitotoxins and cytokines. Although still far from being fully understood, the mechanisms underlying this phenomenon are beginning to be elucidated (Ishida et al., 1997; Gonzalez-Zulueta et al., 2000; Grabb and Choi, 1999; Gu et al., 2001; Guo et al., 1999; Schulz et al., 2001). In the brain, ischemic tolerance is predominantly mediated by the activation of NMDA receptors, leading to increased intracellular calcium and new protein synthesis (Gonzalez-Zulueta et al., 2000; Grabb and Choi, 1999; Kato et al., 1992). The intracellular mechanisms triggered by this, however, are largely unknown. Several studies have now documented a role for both the mitogen-activated and stress-activated pathways in CNS preconditioning (Gonzalez-Zulueta et al., 2000; Gu et al., 2000, 2001; Kawaguchi et al., 1999; Mabuchi et al., 2001; Shamloo et al., 1999).
Preconditioning may be stimulated in many in vitro and in vivo models of neuronal injury by a variety of different means, all of which may involve altered kinase activation. For the purposes of this review, however, the involvement of kinases in the preconditioning phenomenon will be limited to those involved in protecting cells from ischemia-induced cell death. A brief 5-minute exposure of hippocampal organotypic slice cultures to OGD (i.e., sublethal injury) induces preconditioning and protects cultures from subsequent lethal OGD (Gonzalez-Zulueta et al., 2000). In this study it was shown that the preconditioning stimulus was associated with a marked increased Ras and ERK1/2 activity and that inhibition of Ras, Raf, MEK, or ERK1/2 blocked the neuroprotection offered by the preconditioning stimulus.
The phenomenon of ischemic preconditioning has been well documented in both focal and global models of cerebral ischemia. In models of global ischemia the neuroprotective effect of the preconditioning ischemia (3 minutes of ischemia) is maximal after two to four days of reperfusion (Shamloo and Wieloch, 1999). Phospho-MEK and phospho-ERK have been shown to be increased in the CA1 region of the hippocampus after 3-minute global ischemia, which is sustained up to 5 days after ischemia onset (Shamloo et al., 1999; Gu et al., 2000, 2001). Interestingly, ERK1/2 activation is maximal at 3 days after preconditioning ischemia, which correlates with the interval for inducing tolerance in this model (Gu et al., 2001). Both pERK and pMEK were activated 30 minutes after reperfusion after the second ischemic insult (9-minute ischemia, 2 days after preconditioning stimulus) in nonconditioned and preconditioned brains. One day after the second ischemic insult, however, the increased ERK1/2 and MEK activation returned to normal in those animals that underwent preconditioning ischemia, but was sustained in nonconditioned animals (Shamloo et al., 1999). These data suggest sublethal ischemia induces sustained activation of ERK1/2 signaling, which may be involved in modulating transcriptional events involved in preparing the brain for subsequent injury. Normalization of ERK1/2 signaling after the second lethal injury in those animals that underwent preconditioning may suggest that preconditioning stimulates protection of the CA1 by activating mechanisms that induce regulatory proteins, which downregulate detrimental signaling cascades. This supports the hypothesis that ERK1/2 can have either beneficial or detrimental effects on cell survival, depending on the cellular context. That is, in the noninjured brain, ERK1/2 signaling may promote cell survival, but in the injured brain it may participate in the mechanisms underlying cell death.
In contrast, however, Gu et al. (2001) have shown that after 6 minutes of global ischemia ERK1/2 activation was increased 3 days after the second ischemic challenge in nonconditioned and preconditioned brains. ERK1/2 activation was decreased, however, below sham levels in non-preconditioned animals after 8 and 10 minutes of global ischemia, but was enhanced in preconditioned animals. This study suggests that enhanced ERK1/2 and MEK activation may be involved in the neuroprotection offered by ischemic preconditioning and, therefore, preventing the downregulation of ERK1/2 may be beneficial after ischemic injury. In support of this finding PD98059 (2 ug intracerebroventricularly), when administered 3 days after the preconditioning ischemia and 30 minutes before the lethal ischemic injury, significantly inhibited the protective effect of ischemic preconditioning when animals underwent 6-minute lethal ischemic injury (Gu et al., 2001). This suggests that, under this study design, the activation of ERK1/2 promotes neuronal survival in the both the noninjured and injured brain.
In contrast to ERK1/2 and MEK, JNK was not increased in the CA1 after preconditioning ischemia, but was markedly increased in the CA1 region 3 days after 6, 8, or 10 minutes' global ischemia in the rats without preconditioning (Gu et al., 2001). In those animals receiving the preconditioning stimulus, however, the degree of JNK activation was markedly reduced (Gu et al., 2001). Although PD98059 was shown to have no effect on JNK activity when administered 3 days after 3-minute ischemia, PD98059, as described previously, prevented ischemic tolerance and also the inhibition of JNK activation normally seen 3 days after 6-minute global ischemia with ischemic preconditioning. This suggests that JNK activation is not involved in the protective mechanisms underlying preconditioning but is involved in ischemic cell death.
The aforementioned studies describe ischemia-induced ischemic preconditioning. Ischemic preconditioning, however, has been shown to be induced by factors such as cortical spreading depression (CSD), glutamate, and thrombin (Kobayashi et al., 1995; Mabuchi et al., 2001; Masada et al., 2000; Xi et al., 1999). Although preconditioning can be induced by a variety of different means, kinase pathways, especially the ERK1/2 pathway, do appear to play a central role in this phenomenon. It is well documented that CSD induced 24 hours before focal cerebral ischemia increases the tolerance of the brain to ischemic injury (Kobayashi et al., 1995). Administration of PD98059 before CSD reduced the development of tolerance of the brain to cerebral ischemia (Kawaguchi et al., 1999). In addition, PD98059, when administered along with thrombin, prevented thrombin-induced preconditioning (Xi et al., 2001). Caution is required, however, when interpreting data from these studies. Preconditioning depends on the induction of a sublethal injury, but the injury needs to be severe enough to stimulate the protective mechanisms in the brain. It is possible to imagine that, owing to the neuroprotective properties of U0126 and PD98059 discussed in this review, administration of these compounds before the preconditioning stimulus may merely reduce the level of preconditioning injury below the threshold required to induce preconditioning-mediated protection.
Further investigation of this preconditioning phenomenon will be required to unequivocally confirm the role of kinases in the protective mechanisms underlying ischemic preconditioning, and will greatly enhance our understanding of the role of kinase signaling in cell survival in both the injured and noninjured brain.
DEVELOPMENT OF KINASE INHIBITORS AS THERAPEUTIC AGENTS FOR STROKE
There is thus a growing body of evidence that inhibitors of the ERK, p38, and JNK pathways have efficacy in animal models of cerebral ischemia. The compounds described to date (e.g., U0126 and SB-203580), although relatively selective, do show a degree of activity at other kinases. There is close homology between kinases, and some 400 to 500 are predicted to be encoded by the human genome. There is, therefore, great potential for such compounds to possess an inhibition profile rather than specificity for a single target. Kinase inhibitor selectivity data are defined in isolated enzyme assays, and it is often difficult to translate this into a cellular and subsequently in vivo situation. Owing to the greatly differing Km values for ATP for the different enzymes (and most kinase inhibitors are ATP competitive), apparent selectivity at the enzyme level can be eradicated in a cellular context. Conversely, apparent lack of selectivity at the enzyme level can be overcome by temporal and spatial differences in expression and activation.
When targeting a given signaling cascade careful consideration must be given to the selection of which kinase to target. The very nature of kinase cascades dictates that the inhibition of a kinase toward the top of a cascade may have a much more widespread effect as compared with inhibition of a single downstream kinase. For example, if Raf is inhibited one can imagine that MEK, ERK, Elk-1, and all other kinases normally activated through this pathway will be directly affected. Targeting ERK, however, which is further downstream, will result in a more selective inhibition, and therefore the biological consequence could be very different. More detailed analysis of kinase activation, and the correlation with blood flow, glucose metabolism, and ultimately cell death after the onset of ischemia, will be crucial to understanding the involvement of various different kinases in the progression of ischemic cell death. Such studies should help highlight the kinases playing a central role in disease pathology. More rigorous investigation of the action of the currently available kinase inhibitors will also greatly enhance understanding and provide direction for the development of novel agents. Few studies to date have investigated the effects of small -molecule inhibitors on kinase activity, other than their target kinase in vivo. In experiments where a broader analysis of kinase activation has been conducted the data provide evidence to suggest that inhibition of a given kinase can actually result in the activation or inhibition of another (e.g., treatment of cultures exposed to 9 hours of oxygen deprivation and reoxygenation with U0126 resulted in inhibition of ERK1/2 activity but enhanced MEK activity; Fig. 2) (Namura et al., 2001). In addition, treatment of animals with PD-98059 was also associated with increased c-fos actvity (Alessandrini et al., 1999). This is probably not surprising, owing to the large network of kinase signaling cascades, and highlights the need for evaluation of the precise nature by which these inhibitors are acting in the diseased state. It is possible to envisage the most efficacious neuroprotectant molecules will display a favorable profile of kinase activation/inhibition, rather than just target a single kinase in a selective fashion.
Notwithstanding these challenges, MEK, p38, and JNK inhibitors have all been shown to provide neuroprotection in a variety of stroke models.
Furthermore, a new generation of inhibitors of p38—drugs for the treatment of inflammatory disease, for example—is well advanced, and their potential application to the treatment of stroke is now being investigated. As the level of understanding of the involvement of these MAPK/SAPK pathways in disease grows, and small-molecule inhibitors are identified, the prospect of being able to exploit the MAPK and SAPK pathways for intervention in stroke is enhanced.
In summary, kinase pathways involve a highly complex network of signaling cascades, many of which are activated following the onset of focal cerebral ischemia. Activation of the MAPK/ERK1/2, p38/SAPK2, and JNK/SAPK pathways has been implicated in the mechanisms underlying cerebral ischemia through demonstration of decreased cell death in rodent models of stroke after the administration of kinase inhibitors. It is clear, however, that a greater understanding of kinase signaling in pathologic conditions will be essential to uncovering the full potential for inhibitors of these pathways as a new generation of therapeutic agents for the treatment of stroke.
Footnotes
Acknowledgements
The authors would like to thank Dr. Andrew Parsons, Dr. Andrew Takle, and Dr. Oliver Rausch for their critical review of the manuscript.