
Abstract
The present study investigates the role of N-methyl-D-aspartate (NMDA) receptors in a model of transient focal cerebral ischemia in normotensive rats. The left middle cerebral artery and both common carotid arteries were occluded for 60 min. Preliminary studies indicated that this gave reproducible infarctions of the cortex and striatum. These infarctions were the result of severe ischemia followed by complete reperfusion after clamp removal, as showed by striatal tissue Po2 monitoring. Microdialysis indicated that glutamate concentration increased immediately after occlusion and returned to the baseline value 40 min after clamp removal. MK-801 (1 mg kg−1 i.v.), an antagonist of the NMDA glutamatergic receptor, reduced the cortical infarct volume by 29% (p < 0.001) and the striatal infarct volume by 14% (p < 0.05) when given just prior to ischemia, but had no neuroprotective activity when given 30 min after the onset of ischemia. This short therapeutic window for MK-801 suggests that NMDA receptors play only a transient role in reversible focal ischemia in rats.
Occlusion of the middle cerebral artery (MCA) is rarely permanent in human stroke (Mohr et al., 1986; Yamaguchi et al., 1987). Therefore, new models of transient focal cerebral ischemia in rats have recently emerged as a way of reproducing this clinical situation and of studying the role of reperfusion in the development of infarcts induced by such strokes (for review see Macrae, 1992). Reversible MCA occlusion can be induced by several techniques. The most recent is the topical application of endothelin-1 (Dawson et al., 1994), but the most widely used remain (a) intraluminal occlusion, in which a nylon filament is introduced into the external or common carotid artery until it reaches the origin of the MCA (Longa et al., 1989; Nagasawa and Kogure, 1989; Memezawa et al., 1992; Yang et al., 1994), and (b) extravascular occlusion, most commonly by a microclip (Buchan et al., 1992; Hiramatsu et al., 1993; Takagi et al., 1994). Extravascular occlusion was used in the present study. MCA clipping was associated with occlusion of both common carotid arteries (CCAs) to reduce the variability of infarct volumes (Hiramatsu et al., 1993; Lin et al., 1993; Muszynski et al., 1993).
The durations of MCA and CCA clipping have varied according to the studies from 45 min (He et al., 1993) to 3 h (Goto et al., 1993), inducing infarct volumes of very different sizes. The first part of the present work therefore used various durations of ischemia (45, 60, and 90 min) to determine the more suitable time for pharmacological studies. This was defined as the occlusion time leading to reproducible and large infarct volumes. Once the duration of artery occlusion had been determined, recirculation after this period of ischemia was studied by monitoring the tissue Po2.
Because glutamate is implicated in the development of the infarction after stroke, we also monitered the changes in the extracellular concentration of this amino acid using the microdialysis technique. Finally, the effect of MK-801, a noncompetitive antagonist of the N-methyl-D-aspartate (NMDA) type of glutamatergic receptor, was examined. The neuroprotective effect of this compound, initially reported by Park et al. (1988), has been demonstrated by many authors in models of permanent MCA occlusion (for review see Dezsi et al., 1994). But there have been few studies in transient focal ischemia. Yang et al., (1994) and Dawson et al. (1994) described a reduced cortical infarct volume in normotensive rats given MK-801 prior to the onset of ischemia, whereas Xue et al. (1994) reported that MK-801 given after the onset of ischemia had no effect in spontaneously hypertensive rats (SHRs). The present study therefore examines in normotensive rats the effect of MK-801 (1 mg kg−1) given intravenously just before or 30 min after the onset of ischemia.
MATERIAL AND METHODS
All experiments were performed in strict accordance with NIH and French Department of Agriculture guidelines (license no. 01352).
Surgery
Male Sprague-Dawley rats (300–350 g) were anesthetized with chloral hydrate (400 mg kg−1 i.p.) and allowed to breathe spontaneously. Anesthesia was maintained until the end of physiological variable monitoring by required doses of chloral hydrate. The tail artery was cannulated to monitor MABP, arterial blood gases, and pH. The CCAs were isolated through a ventral midline neck incision, and a loose ligature was placed around them. The left MCA was exposed via a temporal craniotomy. Transient focal ischemia was induced by occluding the left MCA with a Zen-type clip (13 × 0.4 mm; Ohwa Tsusho Co., Tokyo, Japan) and the CCAs by tightening the loops. The microclip was placed on the MCA at a site proximal to the lenticulostriate arteries to induce both cortical and striatal infarctions. At the end of ischemia, the microclip occluding the MCA was removed and recirculation within the CCAs was allowed. Reperfusion in the three arteries was checked under microscope. During the surgical procedure and until recovery from anesthesia, body temperature and temporal muscle temperature were maintained at 37–38°C by means of a heating blanket. Physiological variables were measured just prior to ischemia, halfway through ischemia, and 30 min after reperfusion.
Quantification of ischemic damage
The rats were killed with an overdose of sodium pentobarbital 24 h after ischemia. Their brains were removed and frozen in isopentane (–40°C). Cryostat coronal brain sections (50 urn thick) were cut at 1-mm intervals and stained with cresyl violet. Infarction appeared well demarcated: lesioned areas were unstained (white) tissue, which was easily contrasted from areas of viable tissue, which stained violet. Right and left hemisphere areas and striatal and cortical areas of infarction were measured on each section using an image analyzer (Imstar, Paris, France). Differences between left and right hemisphere areas of each section indicating the development of edema, areas of infarction were corrected forbrain edema by multiplying the necrotic area by the ratio of total left hemisphere area to that of the total right hemisphere (Golanov and Reis, 1995). The distances between respective coronal sections were used to calculate a linear integration for the cortical and striatal necrotic volumes. To evaluate the contribution of brain edema to the effect of a treatment, edema, expressed in percentage as the difference between left and right hemisphere volumes (Golanov and Reis, 1995), was calculated in control and treated groups.
Study design and experimental paradigms
Experiment 1: influence of duration of ischemia on infarct volume. Three different durations of occlusion were used: 45, 60, and 90 min.
Experiment 2: recirculation after 1-h ischemia. Recirculation after ischemia was checked by monitoring striatal tissue Po2 (Pto2) by a Polarographic method derived from the technique of Crockard et al. (1976). Briefly, a platinum electrode and a silver reference electrode were chronically implanted in the left striatum 1 week before induction of ischemia. During and after ischemia, the Po2 electrode was polarized at −700 mV and currents were continuously recorded, reflecting the change in Pto2.
Experiment 3: monitoring striatal glutamate concentration by microdialysis during 1-h ischemia. Animals were anesthetized with chloral hydrate (400 mg kg−1 i.p.). A vertical microdialysis probe, prepared as described by Robinson and Whishaw (1988), was inserted into the left striatum at 0 mm anterior and 3.5 mm lateral to the bregma and 7 mm ventral to the skull (Paxinos and Watson, 1986). This probe was perfused with Ringer solution (125 mM NaCl, 2.5 mM KCl, 1.18 mM MgCl2, 1.26 mM CaCl2) at a constant flow rate (1.25 μl min−1). After a 1-h recovery, samples were collected every 20 min. The first three samples were used to determine basal glutamate concentrations; the animals were then anesthetized with chloral hydrate, and ischemia was induced as previously described. Three samples were collected during ischemia and three during the first hour of reperfusion; then samples were collected each hour for 3 h. Glutamate concentrations in the dialysates were determined by HPLC with fluorescence detection after automated precolumn derivatization with o-phthaldialdehyde (Lindroth and Mopper, 1979).
Experiment 4: effect of MK-801 on 1-h ischemia. The right external jugular vein was cannulated for MK-801 (1 mg kg−1) or saline intravenous injections. In a first experiment, MK-801 or saline was injected just before the onset of ischemia. In a following experiment, MK-801 or saline was injected 30 min after the onset of ischemia.
Data expression and statistical analysis
Infarct volumes are given as means ± SD (mm3). Variability was calculated as SD/mean (%). A two-tailed unpaired Student t test was used to compare the values for two groups of animals. Statistical analysis of more than two groups of animals was by one-way analysis of variance, with subsequent individual comparisons by Scheffé test. Tissue Po2 is given as a percentage of the basal value (mean ± SD) and glutamate concentrations as means ± SD in micromolar. In these latter experiments, a two-tailed paired Student t test was used to compare test and basal values.
RESULTS
Experiment 1
Cortical and striatal infarct volumes (individual values and means ± SD) in rats submitted to different durations of ischemia are shown in Fig. 1. The striatal infarct volumes of rats submitted to 45, 60, or 90 min of ischemia were not significantly different (respectively: 40 ± 17, 46 ± 5, and 51 ± 5 mm3). The cortical infarct volume in rats submitted to 45 min of ischemia was 135 ± 93 mm3. The cortical infarct volumes in rats submitted to 60 or 90 min of occlusion were 212 ± 34 and 217 ± 32 m3. They were not significantly different from each other, but both were significantly greater than the cortical infarct volume in rats submitted to 45 min of occlusion (p < 0.05). The variability was also lower in rats submitted to 60 and 90 min of ischemia than in rats submitted to 45 min of ischemia for both the cortical infarct volume (16 and 15% vs. 70%) and the striatal infarct volume (10 and 10% vs. 39%). The MABP, arterial blood gases, and pH in the three groups of animals remained within the physiological range before and during ischemia and during reperfusion (Table 1).
Physiological variables in experiments 1 and 2
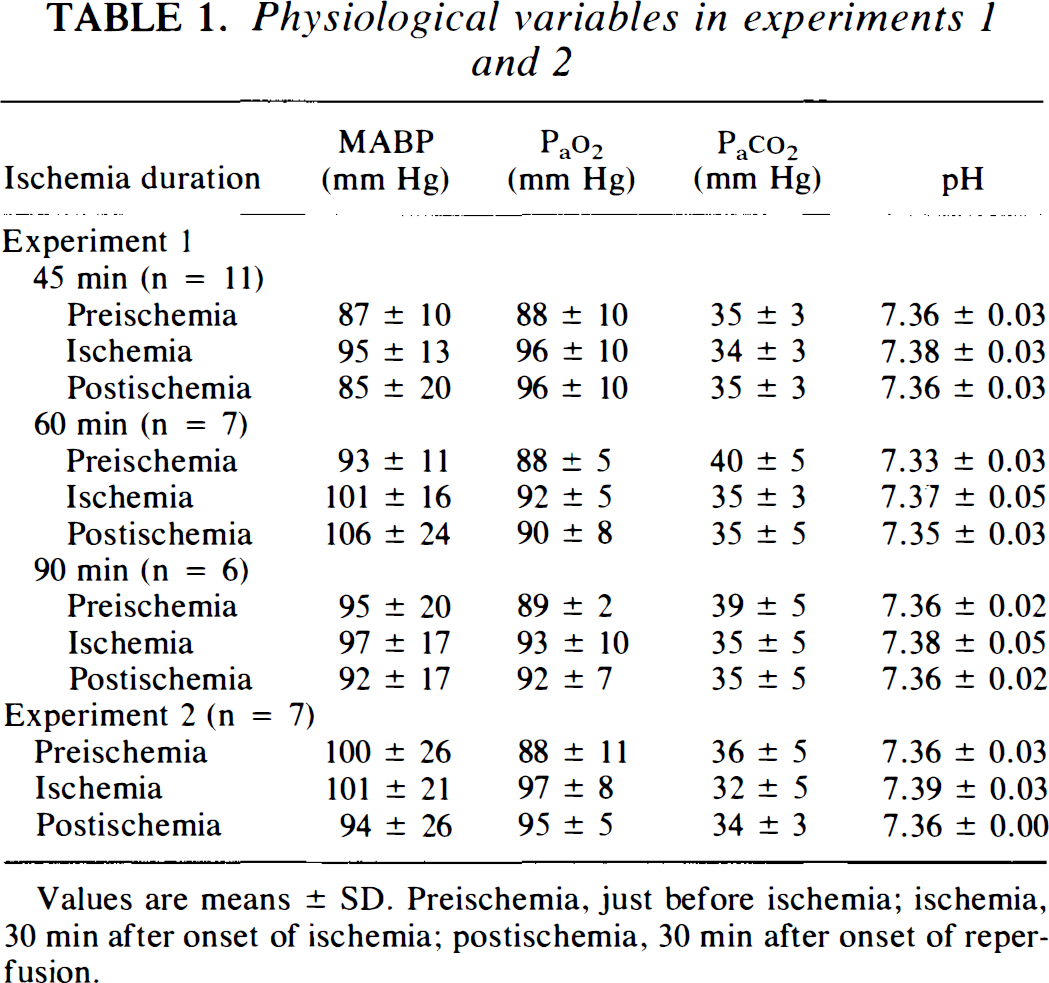
Values are means ± SD. Preischemia, just before ischemia; ischemia, 30 min after onset of ischemia; postischemia, 30 min after onset of reperfusion.
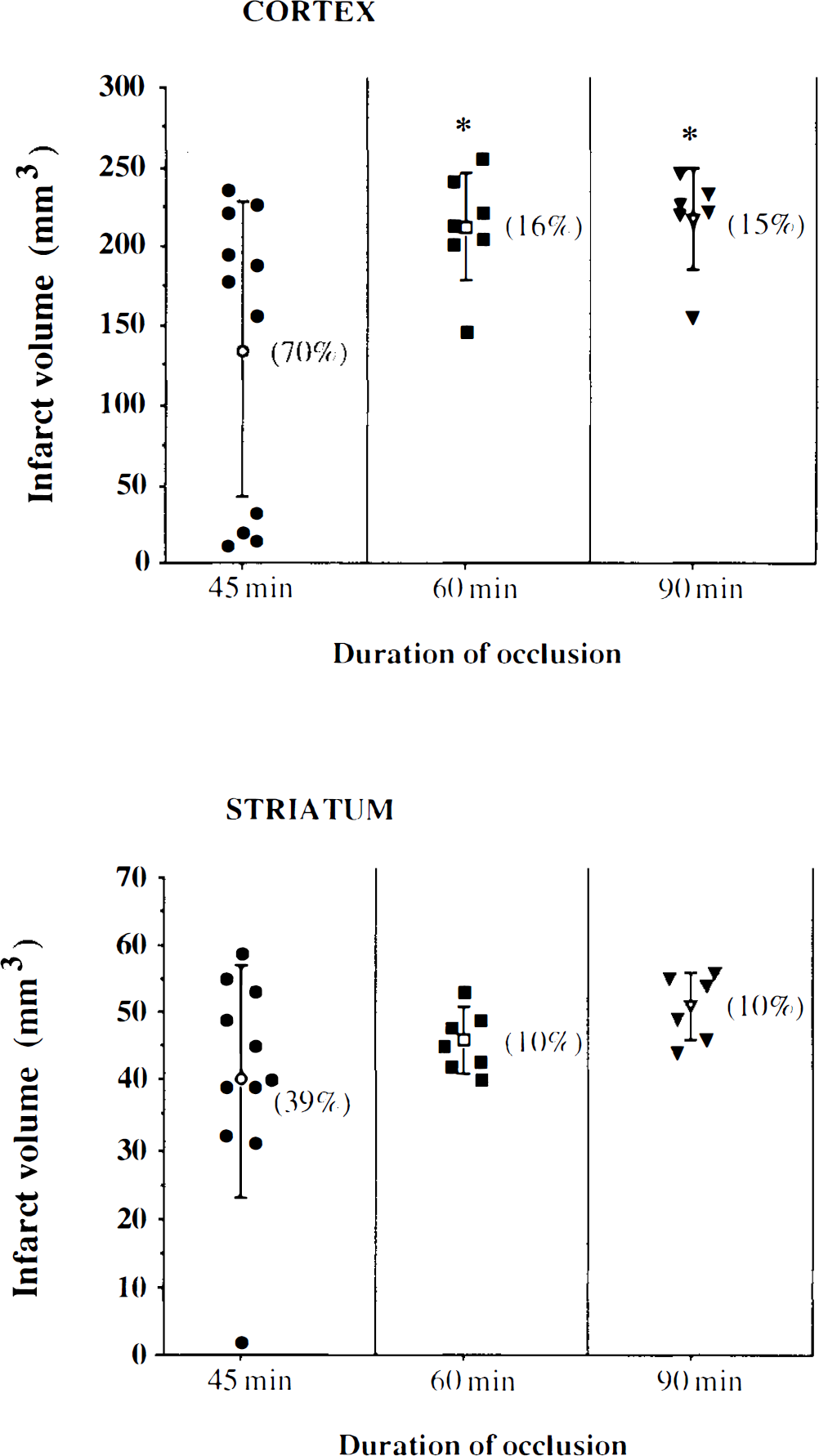
Cortical and striatal infarct volumes in rats subjected to 45-min (circles), 60-min (squares), or 90-min (triangles) ischemia. Open symbols are means ± SD, filled symbols are individual values. For cortical infarct volumes, *p < 0.05 vs. 45-min ischemia by analysis of variance followed by Scheffé test. There was no significant difference between the striatal infarct volumes. Numbers in brackets indicate variability.
Experiment 2
The striatal tissue Po2 during ischemia and reperfusion is shown in Fig. 2. Tissue Po2 was reduced to 20% of its basal value immediately after occlusion of the MCA and CCAs and remained at this level throughout the ischemia (p < 0.001). After the onset of reperfusion, Pto2 increased to values higher than preischemic baseline (149 ± 36 and 139 ± 36% of the basal value, 10 and 20 min after reperfusion; p < 0.05) and had returned to the basal value 1 h after the onset of reperfusion. The cortical and striatal infarct volumes in this group of animals were 183 ± 27 and 48 ± 12 mm3. The MABP, arterial blood gases, and pH remained within the physiological range before and during ischemia and during reperfusion (Table 1).
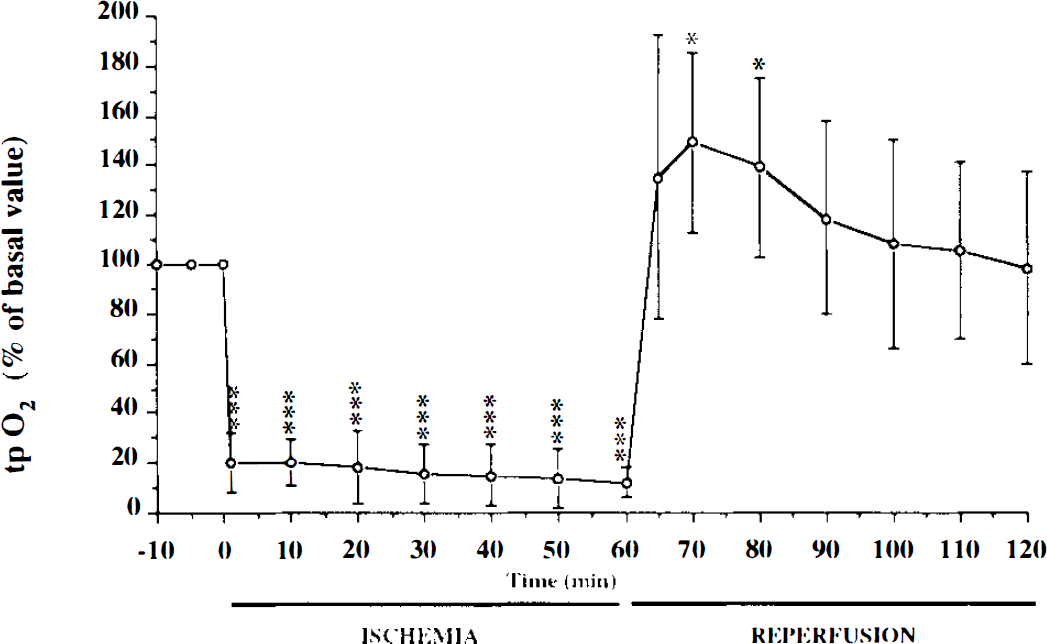
Time course of striatal tissue Po2 (Pto2), expressed as means ± SD, in rats subjected to 60-min ischemia (n = 7). *p < 0.05, ***p < 0.001 vs. basal value, by paired Student t test.
Experiment 3
The changes in the striatal glutamate concentration during ischemia and reperfusion are shown in Fig. 3. The basal striatal concentration of glutamate was 2.68 ± 0.93 μM. Occlusion of the MCA and the CCAs led to a rapid increase in the glutamate concentration, which peaked 40 min after the onset of ischemia (36.09 ± 10.45 μM, p < 0.001 vs. basal concentration, n = 7). After the release of occlusion, the glutamate concentration immediately decreased and did not differ statistically from the baseline value 40 min after initiation of reperfusion (2.48 ± 1.10 μM); it remained at this value throughout out the 4 h following ischemia. The cortical and striatal infarct volumes were 256 ± 48 and 47 ± 8 mm3.
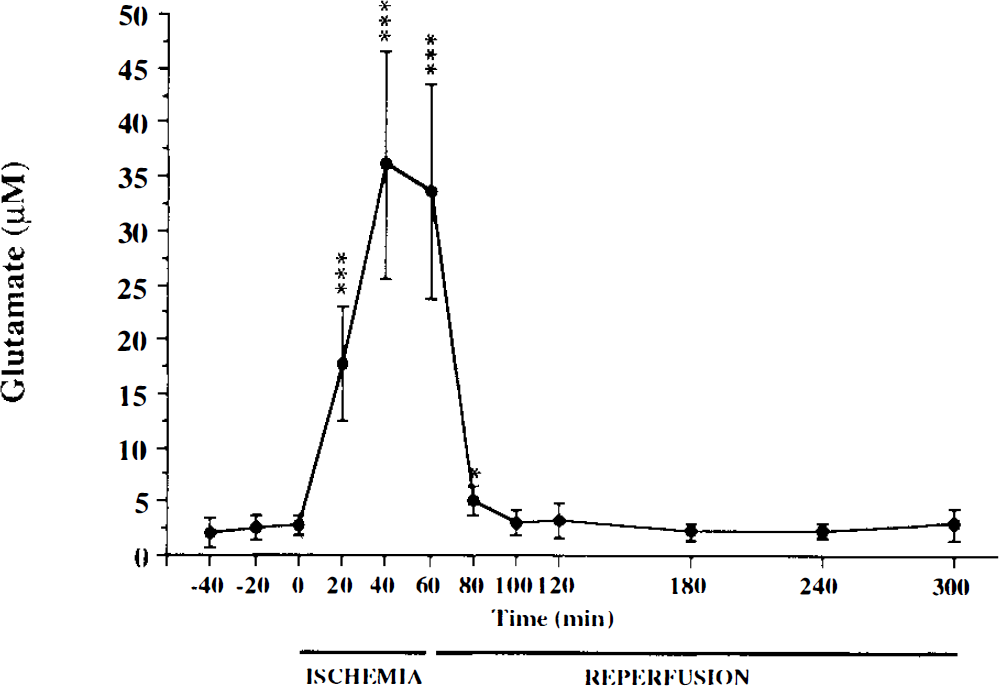
Time course of glutamate concentration in striatal dialysates, expressed as means ± SD, in rats subjected to 60-min ischemia (n = 7). *p < 0.05, ***p < 0.001 vs. basal value, by paired Student t test.
Experiment 4
The effect of MK-801 on infarct volumes in rats undergoing transient focal ischemia is shown in Fig. 4. The cortical and striatal infarct volumes of rats given MK-801 (1 mg kg−1 i.v.) just prior to ischemia were significantly smaller than those of rats given vehicle alone (cortical infarcts: 169 ± 30 vs. 239 ± 30 mm3, 29% reduction, p ≤ 0.001; striatal infarcts: 37 ± 6 vs. 43 ± 6 mm3, 14% reduction, p ≤ 0.05) (Fig. 4A). The MK-801-treated and control groups showed no difference in brain edema (31 ± 4 and 34 ± 2%). The MABP, arterial blood gases, and pH of the two groups of animals remained within the physiological range before and during ischemia and during reperfusion (Table 2).
Physiological variables in experiment 4
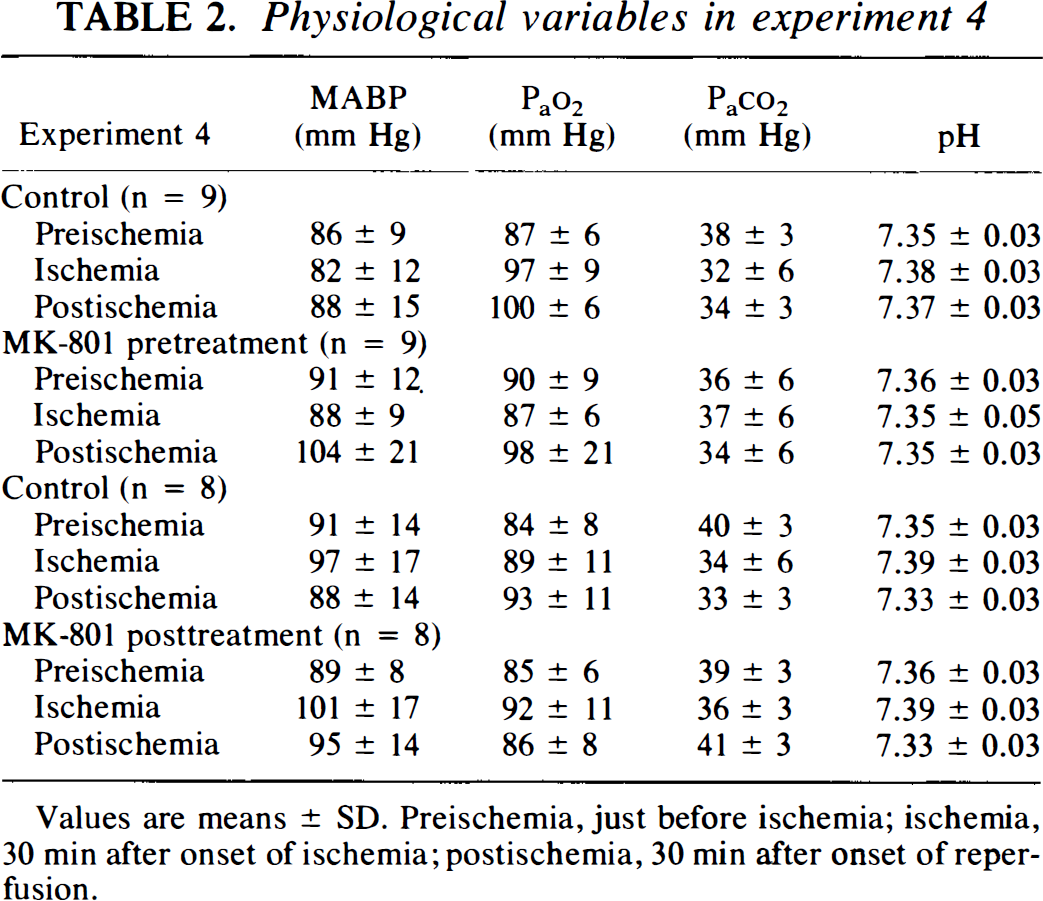
Values are means ± SD. Preischemia, just before ischemia; ischemia, 30 min after onset of ischemia; postischemia, 30 min after onset of reperfusion.
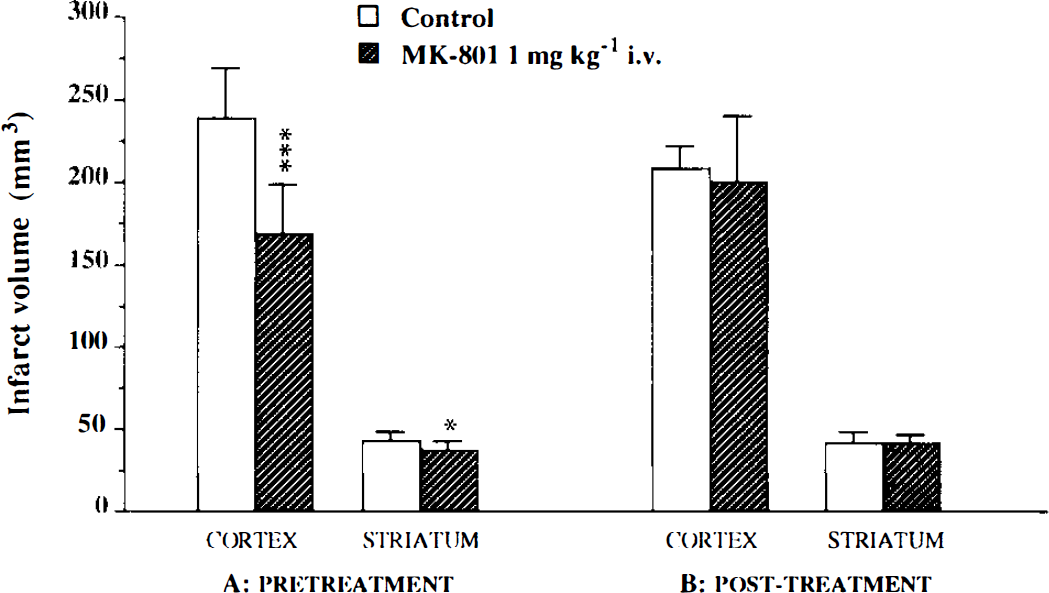
Effect of MK-801 on infarcted volumes (means ± SD) in rats subjected to 60-min ischemia.
Rats given MK-801 (1 mg kg−1 i.v.) 30 min after the onset of ischemia (Fig. 4B) had cortical and striatal infarct volumes (200 ± 40 and 41 ± 6 mm3) that were not significantly different from those of vehicle-treated rats (209 ± 14 and 42 ± 6 mm3). The MABP, blood gases, and pH in both groups of animals were physiological before and during ischemia and during reperfusion (Table 2).
DISCUSSION
One of the most commonly used models of transient focal cerebral ischemia is produced by temporarily occluding one MCA with a microclip. Considering the great anatomic variations in the MCA between rats (Fox et al., 1993) and the possibilities of collaterals opening during ischemia, which may increase the variability of the infarct volume, most of the authors using this model combine occlusion of the MCA with occlusion of both CCAs (Yip et al., 1991; Buchan et al., 1992; He et al., 1993; Hiramatsu et al., 1993; Muszynski et al., 1993). Therefore, in the present study, transient ischemia was induced by MCA and both CCA occlusion.
We first determined the most appropriate duration for ischemia. As argued by Hsu (1993), ischemia should be long enough to induce infarcts large enough to detect any therapeutic efficacy. On the other hand, a very long duration of ischemia could induce irreversible damage much like that induced by permanent ischemia. Occlusion of the MCA and both CCAs for 3 h, followed by 21 h of reperfusion, leads to cortical infarct volumes similar to those of permanent ischemia (Buchan et al., 1992). We therefore used three shorter periods of ischemia (45, 60, and 90 min). An ischemia time of 45 min appeared to be critical since some animals developed cortical infarct volumes similar to those made ischemic for 90 min, while others did not develop any cortical infarction. The cortical infarct volumes produced by this duration of ischemia varied greatly, which was unsuitable for pharmacological studies, since it required many more animals to detect any neuroprotective activity. Ischemia for 60 min induced cortical infarction in all rats, and the volumes were similar to those in the group submitted to 90 min of ischemia. The variation in infarct size was also small. Thus, a 60-min ischemia should allow detection of the neuroprotective effect of drugs with a smaller number of animals. These results are in agreement with those of others (Yip et al., 1991; He et al., 1993; Muszynski et al., 1993). Some authors found less reproducible and less extended infarct volume after 60 min of ischemia, probably because they used a more distal MCA occlusion site, as indicated by the lack of striatal infarction in these studies (Buchan et al., 1992; Hiramatsu et al., 1993). The variation in striatal infarct volume was greater in the 45-min group, but infarct volume was similar to those of 60- and 90-min groups, suggesting that this structure is more vulnerable to ischemia than the cortex.
Another explanation may be that the striatum was not reperfused after clamp removal. Recirculation in MCA is readily seen under the microscope, but it is more difficult in the lenticulostriate artery. The maximum striatal infarct volume by as little as 45 min of ischemia could be the result of a poor reperfusion of this structure. Monitoring of the striatal Pto2, used as a marker of perfusion, indicates that this is not so. Occlusion of the MCA and both CCAs drastically decreased Pto2, but there was a transient hyperoxia immediately after clip removal and then Pto2 rapidly returned to preischemia level. These results are in agreement with those of studies on reperfusion by monitoring the cortical cerebral blood flow (Buchan et al., 1992; Takagi et al., 1994). Thus, reperfusion did occur in the striatum after clip removal, and the maximal striatal infarct volume by as little as 45 min of ischemia reflects the great vulnerability of the striatum to ischemia.
Occlusion of MCA and both CCAs for 60 min appears to produce severe ischemia that is followed by reperfusion immediately after clamp removal. The small variability in the infarct volumes under these conditions makes this model suitable for studying the neuroprotective effect of drugs in transient focal ischemia.
The accumulation of glutamate and its neurotoxicity in permanent focal ischemia are now well accepted (Hillered et al., 1989; Butcher et al., 1990). However, there have been few studies on the time course of changes in glutamate accumulation in transient focal ischemia (Takagi et al., 1994; Uchiyama-Tsuyuki et al., 1994). The present study examines these changes in the striatum. There was a rapid, sustained increase in the glutamate concentration in the dialysates (maximum 13-fold) during ischemia. This increase is similar to that observed in permanent focal ischemia (Buisson et al., 1993). The glutamate concentration in the sample collected after clamp removal quickly returned to baseline values. These results are consistent with those of Uchiyama-Tsuyuki et al. (1994). We examined the effect of MK-801 (dizocilpine), an antagonist of the NMDA glutamatergic receptor subtype (Wong et al., 1986), to evaluate the role of NMDA receptor overactivation by glutamate.
The neuroprotective activity of MK-801 in permanent MCA occlusion in rats is well demonstrated (for review see Dezsi et al., 1994), even when treatment is given 1 h after ischemia (Hatfield et al., 1992). But there have been few studies on the effects of MK-801 in transient focal cerebral ischemia in rats. MK-801 reduces ischemic damage in normotensive rats when given prior to ischemia (Dawson et al., 1994; Yang et al., 1994), but has no effect in SHRs when given 90 min after the onset of ischemia (Xue et al., 1994). The present study compared the effects of MK-801 given intravenously just prior to or 30 min after the onset of ischemia at a dose of 1 mg kg−1 in normotensive rats. MK-801 significantly reduced the cortical infarct volume by 29% when given before ischemia. There was also a small but significant reduction in the striatal infarct volume (14%). This effect is unlikely to be due to a reduction in brain edema, since there was no difference in edema in the treated and control groups. These results show that activation of the NMDA receptors is implicated in the development of the neuronal damage that occurs in transient focal ischemia. However, MK-801 administered 30 min after the onset of ischemia had no neuroprotective activity. This finding in normotensive rats extends the results obtained by Xue et al. (1994) in SHRs, showing that delayed treatment with MK-801 has no neuroprotective activity. The results of these authors are thus unlikely related to the fact that they used hypertensive rats. It appears, therefore, that the therapeutic window for MK-801 is much smaller in transient focal ischemia than in permanent focal ischemia. The difference between the MK-801 effects in transient and permanent focal ischemia may be explained by the difference in the duration of glutamate accumulation. The microdialysis data from the present study and that of Uchiyama-Tsuyuki et al. (1994) show that glutamate accumulates only, in a 60-min ischemia, during the ischemic period, whereas the glutamate accumulation is sustained for several hours in permanent focal ischemia (Hillered et al., 1989). Thus, in transient ischemia, when treatment is delayed by 30 min, MK-801 is probably present at the brain level at the same time as high extracellular glutamate concentration for too short a period for it to induce any neuroprotection. So in transient ischemia, overactivation of NMDA receptor may play only a transient role, and other phenomena, unrelated to NMDA receptor overactivation, may occur during reperfusion and thus contribute to the ischemic insult.
In conclusion, the model of transient focal ischemia in rats presented here and consisting of a 60-min occlusion of MCA and both CCAs appears to be appropriate for pharmacological studies. This technique induces significant tissular hypoxia followed by reoxygenation, suggesting that the model combines severe ischemia during 60 min and then complete reperfusion. Together they lead to reproducible, large striatal and cortical infarcts. MK-801, an NMDA receptor antagonist, has protective activity only when it is given before ischemia and is ineffective when given as soon as 30 min after the onset of ischemia. Transient ischemia thus gives results that are different from those obtained in permanent focal ischemia, suggesting that the fundamental mechanisms of ischemic insult may be different. As focal ischemia is frequently transient in humans, this raises the question about the benefit of therapeutic strategies intending to prevent NMDA receptor overactivation. As α-amino-2,3-dihydro-5-methyl-3-oxo-4-isoxazolepropanoic acid (AMPA) receptor antagonists reduce infarction in transient focal ischemia in SHRs (Xue et al., 1994), further pharmacological studies are needed to evaluate the influence of glutamate on non-NMDA receptors in our model of transient focal ischemia in normotensive rats.
Footnotes
Acknowledgment:
This work was supported by a grant from Rhône Poulenc-Rorer. We thank Dr. Owens Parkes for editing the text.