
Abstract
According to the ternary complex model of G-protein linkage to receptors, agonists increase the affinity of the receptors for the G protein. The model predicts that an endogenous agonist's constant of inhibition toward an agonist radioligand is lower than that toward an antagonistic radioligand. The authors hypothesized that competition from endogenous dopamine in striatum of living mice should have a greater effect on the binding of the D2,3 partial agonist N-[3H]propylnorapomorphine than on the binding of the D2,3 antagonist [11C]raclopride. The baseline binding potential (pB(0)), defined as the ratio of bound-to-unbound ligand in the absence of competition from endogenous dopamine, was simultaneously measured in mouse striatum for [11C]raclopride (pB(0) = 8.5) and N-[3H]propylnorapomorphine (p′B(0) = 5.3). The baseline was established by treatment with α-methyl-p-tyrosine and reserpine. Relative to these baseline values in saline-treated mice, the pB of N-[3H]propylnorapomorphine decreased 52% whereas the pB of [11C]raclopride decreased only 30%, indicating greater sensitivity of the former compound to inhibition by synaptic dopamine. Furthermore, amphetamine decreased the pB of N-[3H]propylnorapomorphine to a greater extent (73%) than that of [11C]raclopride (43%) relative to the reserpine condition. For both radioligands, the occupancy of the dopamine receptors by endogenous agonist obeyed Michaelis-Menten kinetics over a wide range of agonist concentrations established by the pharmacologic treatments. The apparent inhibition constant of endogenous dopamine depended on the dopamine occupancy and decreased to a value 1.66 times greater for N-[3H]propylnorapomorphine than for [11C]raclopride at its highest occupancies. The results are consistent with the hypothesis that agonist binding is more sensitive than antagonist binding to competition from endogenous dopamine. Therefore, dopamine agonist ligands may be superior to benzamide antagonist ligands for the estimation of dopamine receptor occupancy by endogenous synaptic dopamine. The analysis of the effect of dopamine occupancy on the inhibition of N-[3H]propylnorapomorphine binding indicated a limited supply of G protein with a maximum ternary complex fraction of 40% of maximum agonist binding capacity.
Dopamine D2,3 antagonist binding sites in living brain can be labeled with [11C]raclopride and other benzamide radioligands for positron emission tomography (PET) or single photon emission computed tomography (SPECT) studies of dopamine receptors. The binding of exogenous ligands to dopamine receptors in vivo normally occurs in the presence of competition from endogenous synaptic dopamine. Consequently, pharmacologic depletion of dopamine increases the binding of benzamide radioligands in the brains of rodents (Young et al., 1991; Inoue et al., 1991), nonhuman primates (Ginovart et al., 1997), and humans (Abi-Dargham et al., 2000) in proportion to the prevailing basal occupancy. Conversely, the stimulation of dopamine release with amphetamine or the blockade of dopamine transport with cocaine results in further displacement of benzamide radioligands from specific D2 binding sites in the striatum of living rats (Young et al., 1991), baboons (Dewey et al., 1993; Carson et al., 1997), and humans (Schlaepfer et al., 1997; Laruelle et al., 1997). Thus, changes in radioligand binding in the brain can be indicative of altered synaptic dopamine concentrations.
The pharmacodynamic response to G-protein–mediated neurotransmission is generally believed to occur in proportion to the quantity of ternary agonist receptor–G-protein complexes (Burstein et al., 1997), as defined by DeLean et al. (1980) and Samama et al. (1993). The affinity of G-protein–coupled receptors for the activating agonist is low when the receptor is dissociated from its G protein and high during an interaction with the guanosine triphosphate (GTP)-free form of the protein (GTP-shift) (Jiang et al., 2001). This property of G-protein–coupled agonist-binding sites affects the interpretation of studies of dopamine release and can be modeled. An extended model proposed by Gjedde and Wong (2001) was based on the original ternary complex model of del Castillo and Katz (1957), according to which the quantity of bound agonist can be expressed as
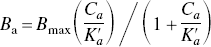
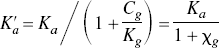
Standard definitions of terms used*

Definitions are those used in publications from Wong et al. (1986) to Gjedde and Wong (2001). Primes refer to properties of agonist radioligand or to results of measurements with agonist radioligand. Double primes refer to properties of GTP-free G proteins. GTP, guanosine triphosphate.
The sensitivity of agonist binding to the presence of the ternary complex results in a biphasic displacement of antagonist radioligands by dopamine, with distinct components at nanomolar (DHigh2) and micromolar concentrations (DLow2) (Cresse and Sibley, 1979; Battaglia and Titeler, 1982). Because dopamine receptor agonists interact with the receptors and their G proteins in such a way that the affinity of the receptors toward the agonist increases when the agonist concentration increases, the interaction leads to the prediction that the inhibitory constant of dopamine toward an agonist ligand is lower than the inhibitory constant toward an antagonistic ligand. We tested the prediction by determining the dopamine inhibitory constant toward the agonist ligand N-[3H]prophylapomorphine ([3H]NPA) relative to dopamine's inhibitory constant toward the antagonist ligand [11C]raclopride in the striatum of living mice.
THEORY
Definitions
The treatment assumes that the binding of radioligands is at equilibrium, but substantial changes in competition from an endogenous or exogenous agonist render this assumption invalid. When such changes occur, the calculated binding potentials represent some weighted average of the true binding potential for each of the changing levels of competition. This problem affects all in vivo studies of drug challenges, except those in which the steady state of competitor binding is established (e.g., by protracted infusion of large quantities of an innocuous competitor). No correction for the nonsteady state was possible in the present study.
The theory arises from the common principles of receptor-ligand interaction in vivo described by, among many others, Gjedde and Wong (1990), and the key points are briefly iterated here. The receptor occupancy of a ligand subject to simple competitive binding is a hyperbolic function of the ligand concentration according to the equation (Michaelis and Menten, 1913)
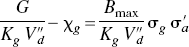
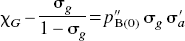
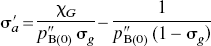
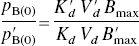
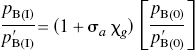
Agonist occupancy
The occupancy of the endogenous or exogenous agonist was calculated from the decline of the binding potential relative to the dopamine-depleted baseline obtained with reserpine plus α-methyl-p-tyrosine methyl ester (AMPT), according to a modification of the previously described equation of Gjedde and Wong (2001)
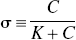
In the present application of the distinct affinities and binding potentials of one specific antagonist and one specific agonist radioligand, for all magnitudes of agonist binding, pB(I) as a function of pB(0) was approximated by the arbitrary relation
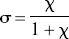
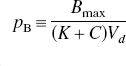
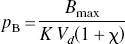
The combination of Eqs. 5 and 6 yielded the solution for the normalized concentration of the competing agonist
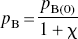
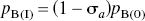
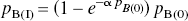
Guanosine triphospate–shift of affinity
The theory of the GTP-shift of affinity yields the prediction that the ratio between the antagonist and agonist affinities depends on the ratio between the concentration of the GTP-free G protein, which forms the ternary complex together with the receptor and its bound agonist. If this ratio declines with increasing agonist concentrations, the explanation may be that the G-protein concentration is too small to render the unbound G-protein concentration essentially independent of the binding. Calculating the quantity of ternary complexes as follows tested this hypothetical explanation:
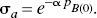
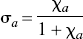
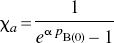
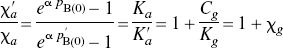
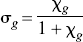
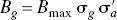
MATERIALS AND METHODS
Male mice (n = 126) of the CD1 strain (Charles River Laboratories, Wilmington, MA, U.S.A.) weighing 25 to 28 g were in experiments approved by the research ethics committee of Johns Hopkins Medical Institutions. In experiment 1, groups of 18 mice were pretreated with intraperitoneal injections of 200 μL saline vehicle, 1 or 10 mg/kg quinagolide (CV 205,502, Novartis), or 10 mg/kg amphetamine sulfate (Sigma Chemicals, St. Louis, MO, U.S.A.) administered 10 minutes before the radiotracer was injected. In experiment 2, groups of 18 mice were pretreated with intraperitoneal saline or reserpine (5 mg/kg) 20 hours before tracer injection, supplemented with 100 mg/kg intraperitoneal α-methyl-p-tyrosine methyl ester administered 1 hour before tracer injection, or with 100 μg/kg subcutaneous nicotine 3 minutes before tracer injection.
Mice were briefly immobilized for bolus injection to a tail vein of 200 μL saline containing a mixture of 0.1 MBq [3H]NPA (New England Nuclear; specific activity, 2 GBq/μmol) and 7 MBq [11C]raclopride (specific activity 200 GBq/μmol at time of delivery) synthesized by the method of Ehrin et al. (1987) using a radioisotope generated by the Johns Hopkins GE PET Trace cyclotron. The calculated doses per injection were 35 pmol raclopride and 50 pmol [3H]NPA. Mice were killed by cervical dislocation 5, 10, 20, 30, 45, and 60 minutes after tracer injection, with triplicate determinations at each circulation time.
Brains were rapidly removed and the cerebellar hemispheres and corpora striata were dissected. The left striatum and cerebellar hemispheres were weighed and transferred to plastic vials for immediate measurement of γ emissions using a γ-counter (LKB 1285). Right striata and cerebellar hemispheres were transferred to 20-mL glass liquid scintillation vials to which 1 mL organic base (Solvable;, Packard Instruments, Downers Grove, IL, U.S.A.) was added. After dissolving overnight at 40°C, 10 mL liquid scintillation cocktail (Formula 989, Packard Instruments) was added to each vial and the tritium concentration was measured using a β-radiation counter (Tricarb, Packard Instruments).
Radioactivities were calculated as percentage injected dose per gram tissue. Using the cerebellum radioactivities as the inputs, the binding potentials of [11C]raclopride and [3H]NPA in mouse striatum were calculated for the baseline and drug-treatment conditions in each of the three mice using the reference-region method of Lammertsma et al. (1996). The parameter estimates were subsequently averaged for each group of three mice.
RESULTS
Single time–radioactivity curves for [3H]NPA and [11C]raclopride in the striatum of randomly selected single mice in the groups of three identically treated mice are illustrated in Fig. 1. Random results from a mouse pretreated with reserpine and AMPT are shown in Fig. 1A, and random results from mice pretreated with saline (Fig. 1B), amphetamine (Fig. 1C), and the high dose of quinagolide (Fig. 1D) are shown in Figs. 1B to 1D. The lines connect points predicted by the deconvolution of the striatal uptake from the cerebellar uptake according to the reference region model of Lammertsma et al. (1996) for the estimation of binding potential.
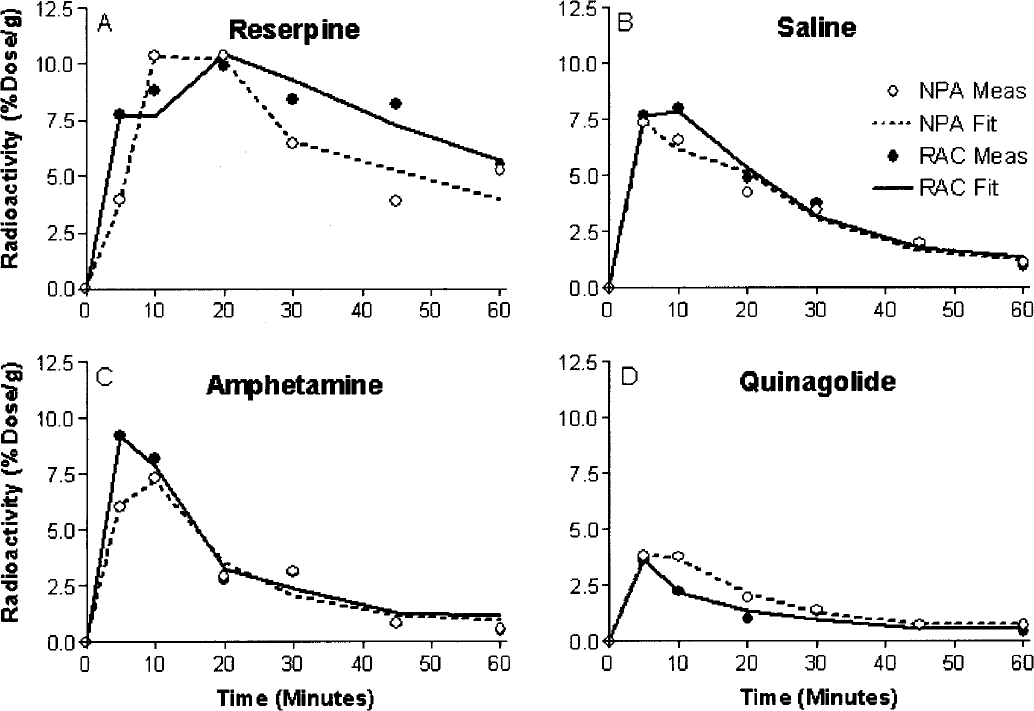
Radioactivity concentrations for [3H]NPA and [11C]raclopride in a single mouse striatum during a 1-hour period after intravenous injection of a single bolus containing 0.1 MBq [3H]NPA and 7 MBq [11C]raclopride to groups of 18 mice pretreated with
The group means of the estimated binding potentials (pB) under the several conditions are summarized in bar-graph form in Fig. 2A. The results of the fits of equation 4 to the data are shown in Table 2. In the baseline condition established by administration of reserpine and AMPT, the mean (± SD) of three estimates of the agonist-depleted binding potentials (pB(0)) were 5.30 ± 0.77 for [3H]NPA and 8.42 ± 1.62 for [11C]raclopride. In two separate estimations of the group mean of the saline-treated condition relative to the baseline (pB(0)), the binding potential of [3H]NPA declined by an average of 52% (pB(I)), whereas the binding potential of [11C]raclopride declined by 30% (pB(I)). Amphetamine reduced the binding potential of [3H]NPA by 73% and the binding potential of [11C]raclopride by 47%. The low dose of quinagolide decreased the binding potential of [3H]NPA by 77% and the binding potential of [11C]raclopride by 63%, and the high dose decreased both binding potentials by approximately 94%. Relative to the saline conditions, the nicotine challenge actually increased the binding potential of [3H]NPA by 10% and did not alter the binding potential of [11C]raclopride.
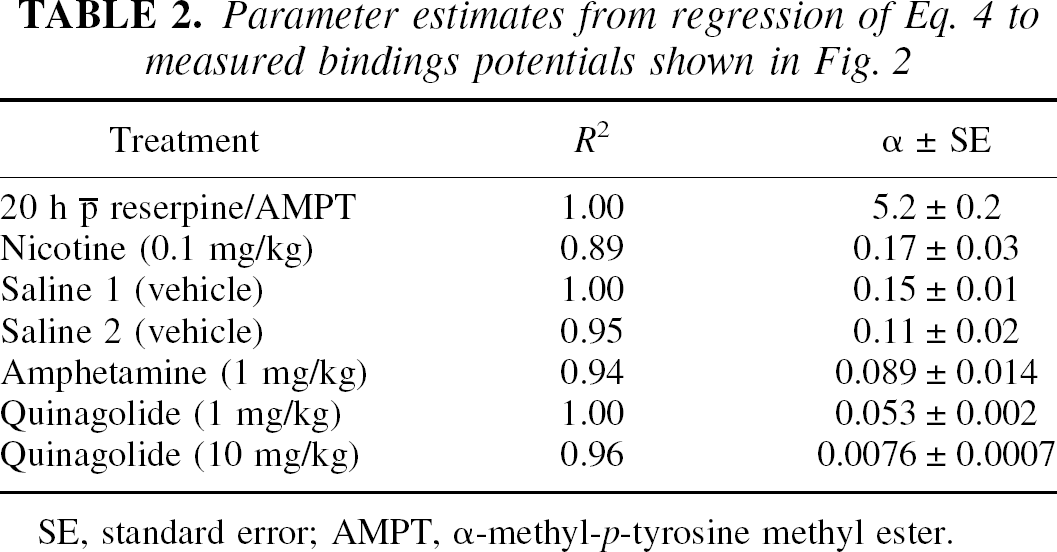
SE, standard error; AMPT, α-methyl-p-tyrosine methyl ester.
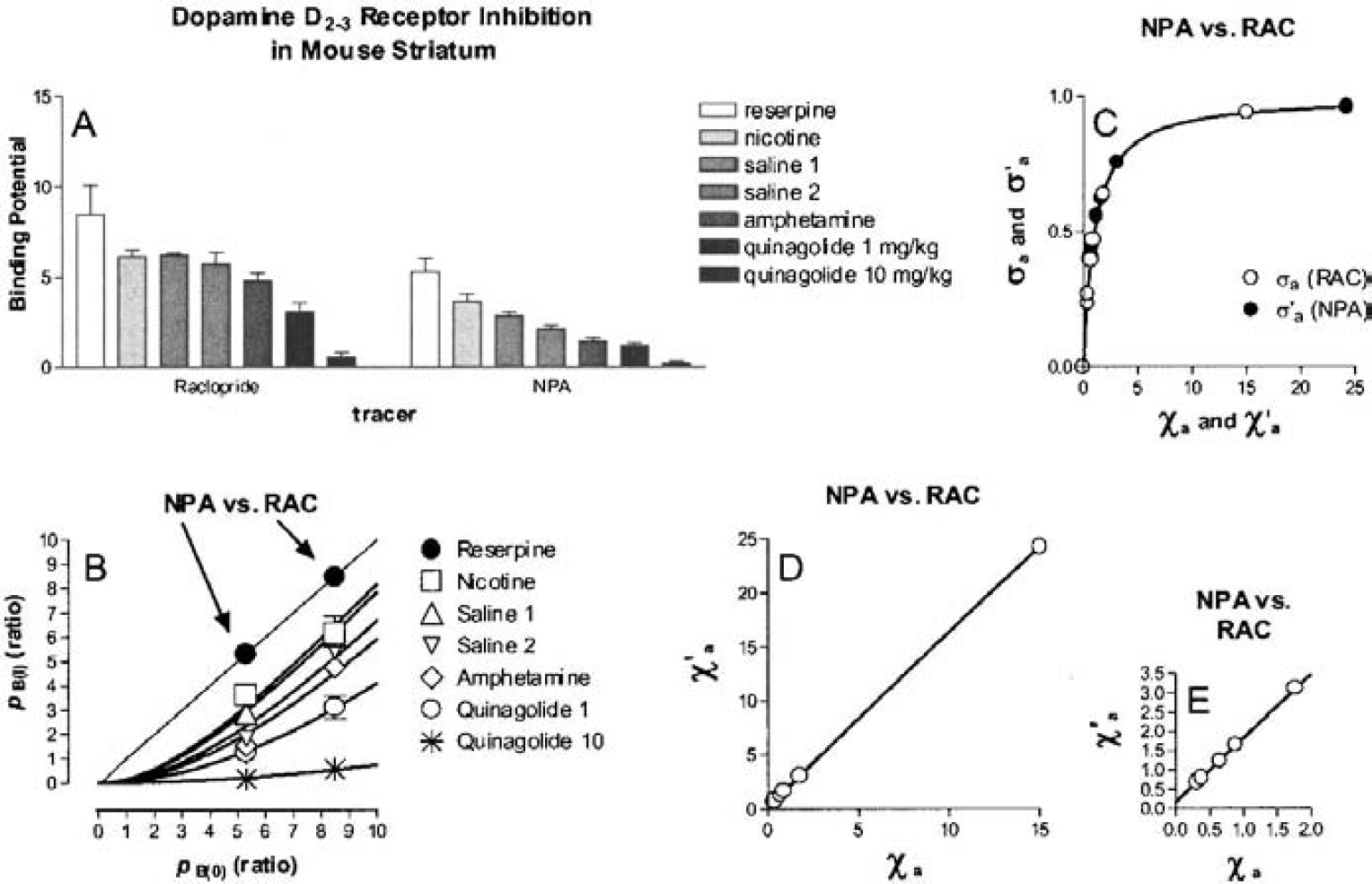
Binding potentials of [11C]raclopride and [3H]NPA in the striatum of mice after the administration of several pharmacologic treatments, and the estimation of occupancy by dopamine, a putative endogenous agonist inhibitor, or quinagolide, a known exogenous agonist inhibitor, under the same condition.
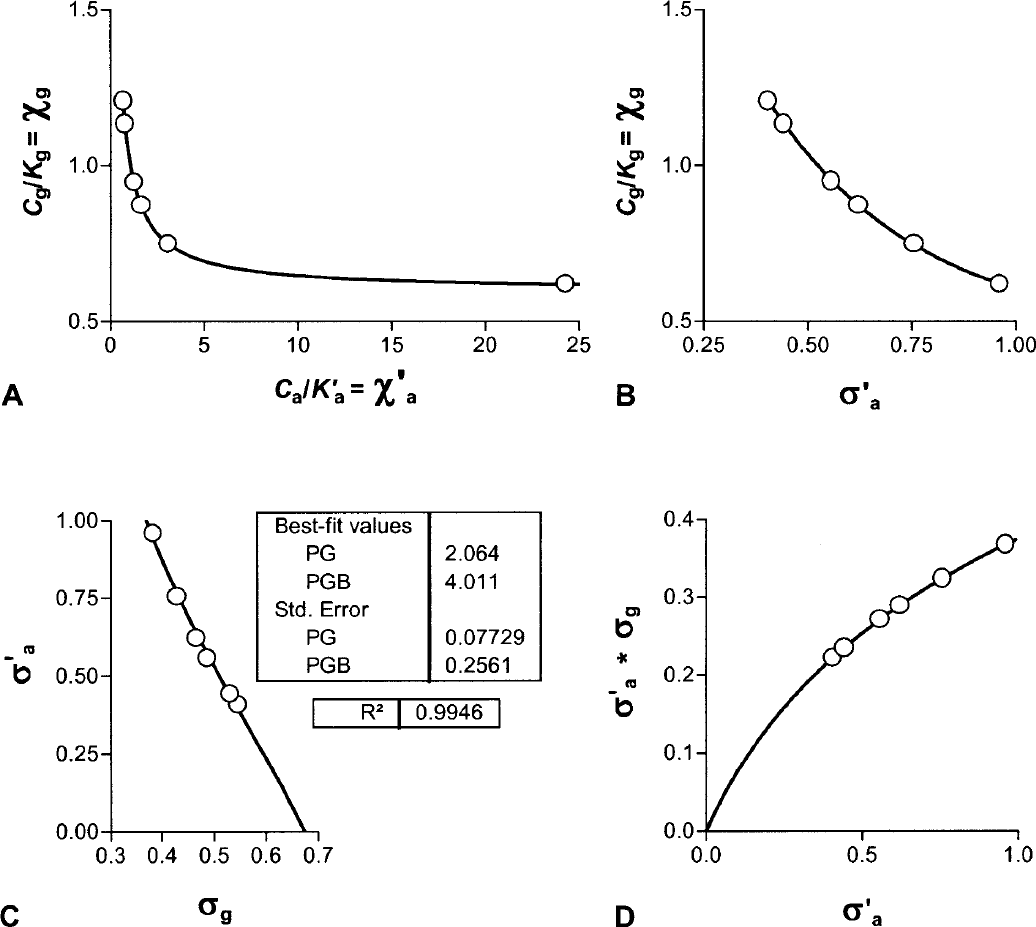
From these changes of the binding potential and relative to the those measured in the agonist depleted condition, the receptor occupancies (σa) were calculated according to Eq. 6 and plotted as a function of the concentration of dopamine relative to its affinity, whereas χa (Fig. 2C) was calculated from Eq. 7. By definition the relation obeyed the Michaelis-Menten formulation for both ligands. As predicted, the occupancy was higher for [3H]NPA than for [11C]raclopride in each pharmacologic condition. The resulting pairs of σa versus χa were plotted in Fig. 2C, and values of χ′a were higher than values of χa. Figures 2D and 2E show the ratio between the antagonist and agonist affinities fitted to a second-order polynomial, which approaches a slope of 1.66 ± 0.11 (standard error) at the highest values of χ′a. The slope indicates that the affinity of dopamine receptors toward an agonist approaches a value 1.66-fold that of the receptors toward an antagonist, assuming that the total concentration of available binding sites is the same for agonists and antagonist ligands. Another way of expressing the same result is to state that the Cg/Kg ratio (χg) declines to a ratio close to 0.66 for large values of χ′a.
Figures 3A and 3B show the relation between the normalized concentration of the GTP-free G protein and the concentration and occupancy of the endogenous agonist assumed to be dopamine, showing the decline of the estimated GTP-free G-protein concentration at elevated concentrations (Fig. 3A) and occupancies (Fig. 3B) of the endogenous agonist competitor. Figure 3C yields estimates for χG and p“B(0) listed in Table 3, which show that the inherent affinities of the receptors toward G protein and agonist are similar (4.0 vs. 5.3) and that the relative mass of available G protein is only half of the mass of receptors (2.1 vs. 4.0). The product of the occupancies of G protein and agonist defines the fraction of the total number of receptors in the ternary configuration. Figure 3D shows that this fraction reaches only 40% of the receptors at total agonist occupancy, indicating that no more than 40% of the receptors can be fully activated.
Baseline variables
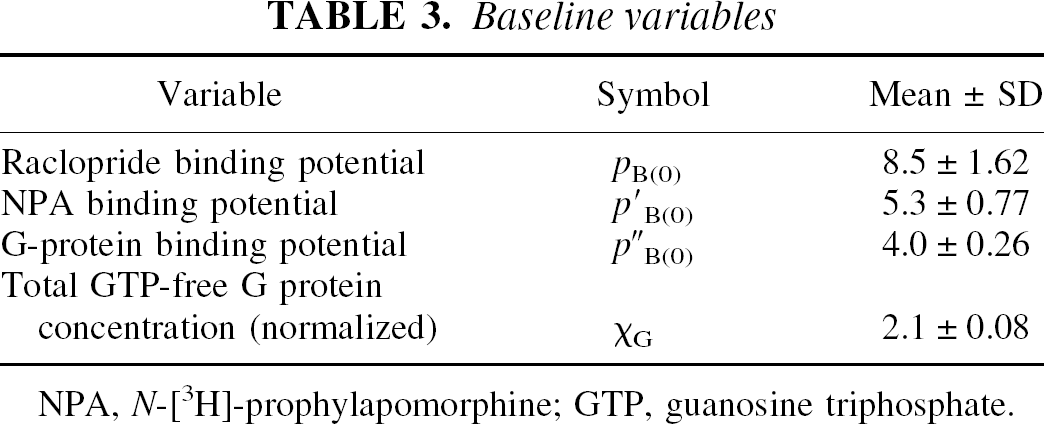
NPA, N-[3H]-prophylapomorphine; GTP, guanosine triphosphate.
Binding potentials of G protein versus binding potentials of endogenous agonist inhibitor.
DISCUSSION
Partial depletion of dopamine increases the availability of binding sites for [11C]raclopride and other benzamide radioligands in the human brain. The extent of this increase is greater in patients with schizophrenia than in healthy volunteers (Abi-Dargham et al., 2000), a finding that was interpreted to reveal the presence of elevated basal occupancy in schizophrenia. However, the partial dopamine depletion increased the availability of D2 antagonist binding sites by less than 20%, even in schizophrenic subjects. In contrast, the present study and earlier results of experimental animals (Ross and Jackson, 1989a,b; Ginovart et al., 1987) indicate that basal occupancy of antagonist bindings sites in the mouse is approximately 40%. If complete dopamine depletions are not obtained in the human brain, differences in pB could be attributed to differential availability of the extracellular dopamine in addition to differences in occupancy.
The present results show that a radiolabeled dopamine agonist is highly sensitive to changes in extracellular dopamine concentrations, and may thus be more suited than antagonists for detecting pathophysiologic changes in occupancy of human brain receptors. Several radiolabeled agonist ligands have been tested as in vivo tracers for dopamine receptors (Closse et al., 1988; Nobrega and Seeman, 1994; Mukherjee et al., 2000). The present findings with [3H]NPA in living mouse suggest that 11C-labeled NPA ([11C]NPA; Hwang et al., 2000; Gillings and Cumming, 2000) is a good candidate for PET investigations of basal occupancy of dopamine receptors by endogenous dopamine in living human subjects.
The differences between the properties of the radioligands [11C]raclopride and [3H]NPA are difficult to unravel because their effects are additive. However, the present study revealed two important differences. First, the maximally obtainable binding potential of the radioligands (i.e., the potential measured in the absence of competition from any competitor other than the tracer itself) is greater for raclopride than for NPA. This difference is presumed to reflect different volumes of distribution, receptor affinities, or maximum binding capacities of the two ligands. Second, the decline of the binding potential in the presumed presence of an agonist competitor is relatively greater for NPA than for raclopride. Therefore, the greater binding potential of one ligand in the baseline will be magnified during competition from an agonist by the greater responsiveness to competition of the other ligand. This difference is shown in Fig. 4A, in which the binding potentials are plotted against the normalized concentrations of the agonist, as calculated from the agonist-derived binding potentials in the present study.
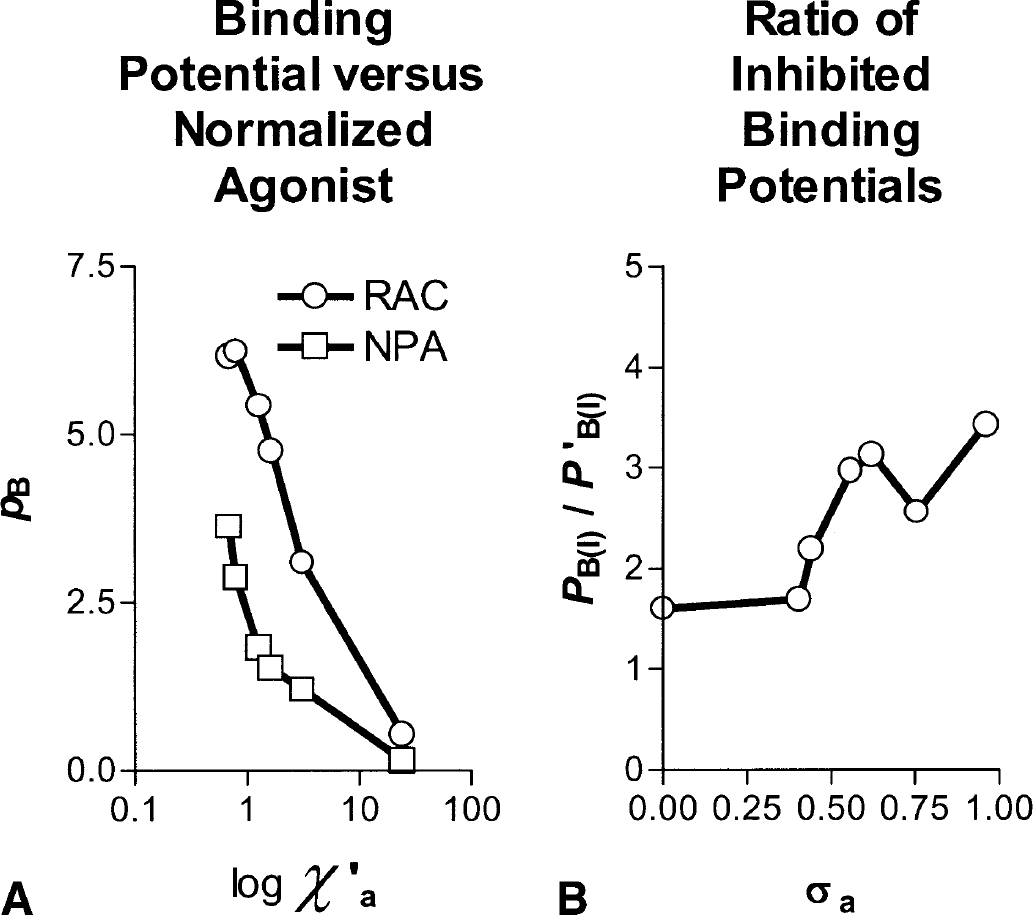
Binding potentials
Baseline ratio of binding potentials
In the striatum of living mouse, the apparent Bmax and Kd of [3H]NPA (Ross and Jackson, 1989a) and [3H]raclopride (Ross and Jackson, 1989b) are similar. For both radioligands, dopamine depletion with reserpine decreased and psychostimulants increased the magnitude of the apparent affinity without altering Bmax. The pB of [11C]raclopride in the striatum of reserpine-treated or saline-treated mice was close to the ratio of radioactivity in the striatum to that in cerebellum (< 1) 30 minutes after [3H]raclopride injection (Ross and Jackson, 1989b). Likewise, the estimates of pB for [3H]NPA in the mouse striatum were consistent with earlier reports of [3H]NPA binding in the rodent striatum during 1-hour tracer circulation (Hall et al., 1983; van der Werf et al., 1983; Benedetti et al., 1990).
The somewhat lower pB reported by Zijlstra et al. (1992) for (N-[11C]methyl)-norapomorphine (pB = 1.5) may have been due to lower affinity of that tracer for dopamine D2,3-receptors. The differential sensitivity of NPA and raclopride to competition from dopamine is not evident from the earlier analyses, which were performed in separate groups of mice. In the present study, we used the dynamic time–radioactivity curves measured in the brain during 1-hour dual-tracer circulation to calculate the values of pB in the striatum at equilibrium.
The apomorphines, like raclopride, do not distinguish between D2 and D3 receptors (Levant, 1997). In the present study, the pB(0) of [11C]raclopride exceeded that of [3H]NPA after reserpine treatment and in other conditions (Fig. 2A). A single class of binding sites for [11C]raclopride and [3H]NPA and no endogenous competition would yield the following ratio of the baseline binding potentials at equilibrium:
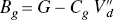
The 6-hydroxybenzoquinoline compound [3H]quinagolide is an agonist that binds to dopamine D2 receptors in rat brain sections with an apparent affinity of 0.6 nmol/L (Charuchinda et al., 1987). In the present study, the low dose of quinagolide displaced [3H]NPA slightly more effectively than [11C]raclopride, but the two ligands were not discriminated by the high quinagolide dose. Likewise, it was noted previously that nanomolar concentrations of NPA completely inhibited dopamine D2 antagonist binding in the rodent brain (Andersen and Nielsen, 1986). Partial agonism of dopamine D2 sites may underlie the neuroleptic property of NPA (Campbell et al., 1991; Tarazi et al., 1997). Like the apomorphine derivatives, quinagolide may also be a partial agonist, accounting for the ability of a low dose of quinagolide to differentiate only partially between the two radioligands.
Interaction with G protein
The findings are consistent with the hypothesis that dopamine or exogenous agonist competitors must appear to occupy a greater fraction of dopamine receptors when the occupancy is measured with an agonist radioligand than when it measured with an antagonist radioligand. We based the hypothesis on the claim that an agonist radioligand undergoes the same GTP-shift of affinity exhibited by dopamine and exogenous agonist competitors, and hence exhibits binding that reflects accurately the ratio of bound-to-free agonist. The antagonist radioligand undergoes no GTP-shift of affinity, and hence underestimates the bound-to-free ratio at all concentrations of dopamine or an exogenous agonist competitor. The ratio between the binding potentials determined with an antagonist and an agonist radioligand, as defined previously, has the simple solution
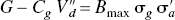
The long-term activation of G-protein–linked receptors can result in the phenomena of homologous and heterologous sensitization in the brain (Morelli and Di Chiara, 1987; Watts and Neve, 1996; Zhang et al., 2000). The several sensitization phenomena can be attributed to diverse mechanisms, including receptor recruitment (Holtback et al., 1999) and enhanced coupling of receptors to signal transduction pathways (Tenn and Niles, 1997). In addition, altered expression of G proteins has been specifically implicated in the sensitization of dopamine receptors by chronic reserpine treatment (Butkerait and Friedman, 1993) or morphine (Van Vliet et al., 1993). The present finding that G-protein availability limits the binding and, hence, the efficacy of dopamine D2 3 agonists in the mouse striatum suggests that altering the expression of Gi/o in the striatum should modulate the efficacy of dopamine receptor agonists.
Footnotes
Acknowledgments:
The authors thank Bisma El-Humadi and Paige Rauseo for expert technical assistance, and acknowledge the generous gift of quinagolide from Novartis.