
Abstract
Hypoxia leads to a rapid increase in vesicular release of glutamate. In addition, hypoxic glutamate release might be caused by reversed operation of neuronal glutamate transporters. An increase in extracellular glutamate concentration might be an important factor in generating anoxic depolarizations (AD) and subsequent neuronal damage. To study the AD and the vesicular release in hippocampal slices from CD1 wild-type mice and mice in which the neuronal glutamate transporter excitatory amino acid carrier 1 (EAAC1) had been knocked out, the authors performed recordings of field potentials and patch clamp recordings of CA1 pyramidal cells. Latency to anoxic depolarizations was enhanced in EAAC1−/− mice, whereas the hypoxia-induced increase in miniature excitatory postsynaptic current frequency occurred with similarly short latencies and to a similar extent in control and mutated animals. Additional block of glial glutamate uptake with TBOA (dl-threo-β-benzyloxyaspartate), a nontransportable and potent inhibitor, dramatically reduced the latency to onset of AD and abolished the difference between wild-type mice and EAAC1−/− mice. The authors conclude that the neuronal glutamate transporter greatly influences the latency to generation of AD. Because ADs are not prevented in EAAC1-deficient mice, vesicular release mechanisms also seem to be involved. They become prominent when glial glutamate transport is blocked.
It is generally accepted that extracellular accumulation of glutamate during hypoxia plays an important role in the generation of anoxic depolarizations (ADs) and in the subsequent cellular damage (Benveniste et al., 1984; Choi and Rothman, 1990). The mechanisms that are responsible for the increase in extracellular glutamate concentration before AD are still controversial. Previous studies revealed that oxygen deprivation might preferentially target the function of the presynaptic vesicular release machinery (Katchman and Hershkowitz, 1993; Kulik et al., 2000), and we have recently shown that, in neocortical neurons, the earliest consequence of acute hypoxia is a marked increase in spontaneous vesicular transmitter release (Fleidervish et al., 2001). Conversely, a number of studies proposed an impairment of glutamate uptake (Szatkowski et al., 1990; Roettger and Lipton, 1996; Jabaudon et al., 2000). Furthermore, Rossi et al. recently proposed that reversed operation of the neuronal glutamate transporter is predominately responsible for anoxic depolarization (Rossi et al., 2000).
In the hippocampus, three Na+-dependent glutamate transporters have been identified. Glutamate transporter 1 (GLT1) and
MATERIALS AND METHODS
Slice preparation and maintenance
Adult (>6 weeks old) EAAC1−/− and wild-type mice of either sex were deeply anesthetized with ether, decapitated, and the brain was removed. Animal procedures were approved by the local Berlin government authorities. Horizontal slices (400 μm) containing the ventral hippocampus, entorhinal, perirhinal, and temporal cortices were cut using a vibroslicer (Camp-den Instruments, Loughborough, U.K.). Slices were placed in a holding chamber containing artificial cerebrospinal fluid (in mmol/L: 129 NaCl, 21 NaHCO3, 3 KCl, 1.25 NaH2PO4, 1.6 CaCl2, 1.8 MgSO4, 10 glucose, saturated with 95% O2/5% CO2, pH 7.4 at room temperature). After more than 1 hour of incubation, slices were transferred to an interface-type recording chamber and perfused at a rate of 1.2 to 1.8 mL/minute with carbogenated artificial cerebrospinal fluid at 34°C.
Electrophysiologic recordings
Whole cell recordings were made from hippocampal CA1 neurons using the “blind” patch-clamp technique (Hamill et al., 1981; Blanton et al., 1989) with an EPC7 patch clamp amplifier (List, Darmstadt, Germany). Patch pipettes were pulled from borosilicate glass (Hilgenberg, Malsfeld, Germany) and had resistances of 6 to 8 MΩ when filled with pipette solution containing the following: 135 Cs-gluconate, 6 CsCl, 2 MgCl2, 10 N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.3 or 135 CsF, 6 CsCl, 2 MgCl2, 10 HEPES, pH 7.3. The software ISO2 (MFK Computer, Frankfurt, Germany) was used for the generation of pulses and recordings. Data were low-pass filtered at 3 kHz and digitized at 10 to 20 kHz. Care was taken to maintain series resistance as low as possible (<14 MΩ).
Extracellular DC-coupled recordings of field potentials were made with electrodes filled with artificial cerebrospinal fluid that were positioned in the stratum pyramidale of area CA1 and documented on a chart recorder (Dash 4).
Induction of hypoxia
Hypoxic episodes were produced by switching the gas flow over the slice from 95% O2/5% CO2 to 95% N2/5% CO2, as first described by Leblond and Krnjevic (1989). We have previously shown that, under these conditions, changes in neuronal behavior already occur 15 to 20 seconds after switching to O2-free gas mixture (Fleidervish et al., 2001). The hypoxic episode duration was maintained for up to 15 minutes in patch recordings and up to 30 minutes in field-potential recordings. Only one hypoxic episode per slice was induced. However, when an AD was observed, reoxygenation was started within 30 seconds after onset of AD. Figure 1 illustrates the experimental arrangement for patch clamp and field-potential recordings in hippocampal slices.
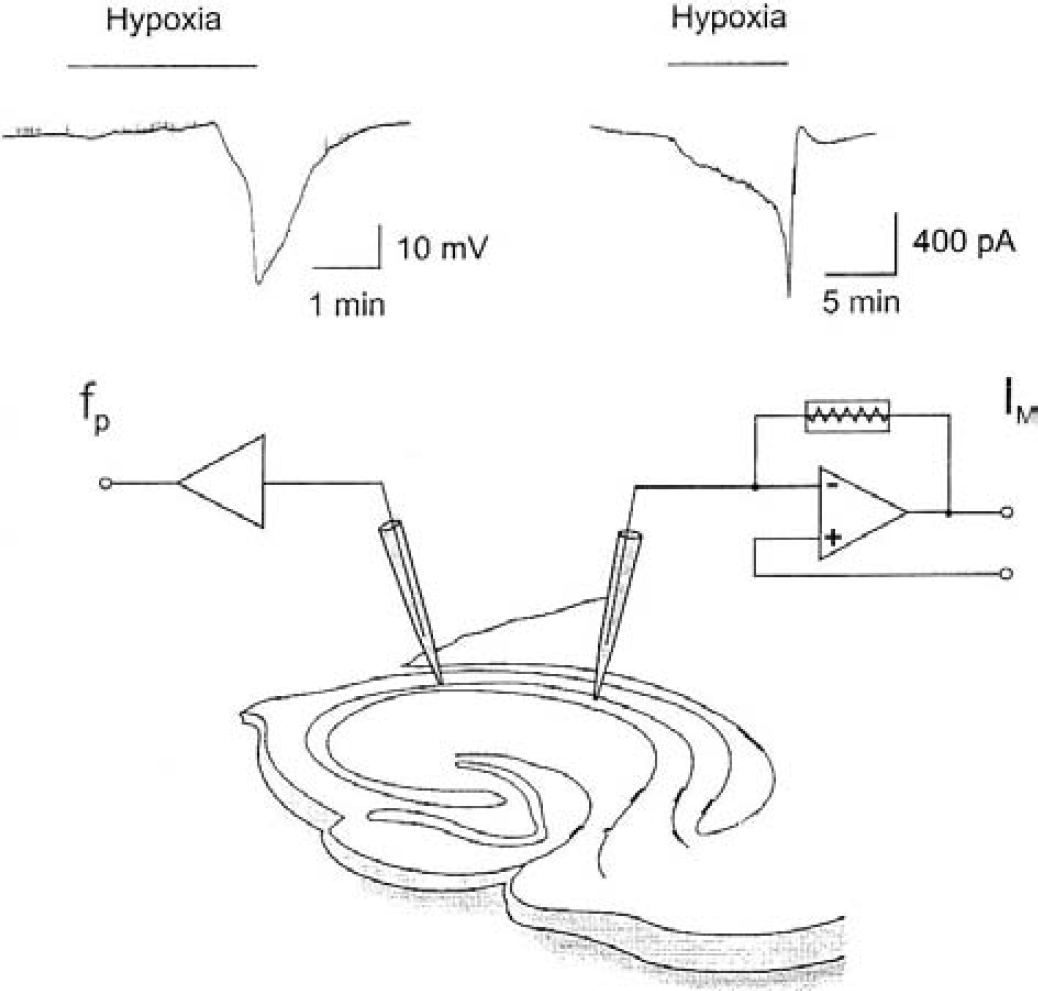
Experimental arrangement for whole cell patch clamp recordings in a voltage-clamped configuration (IM) and field potential (fp) recordings in hippocampal slices.
Data analysis
Data averaging, fitting, and current peak detection were made using MFK software (MFK, Niedernhausen, Germany). In field-potential recordings, the latency to onset of anoxic depolarization is the time measured between onset of hypoxia and the beginning of the negative potential shift. The time measured between the beginning of the negative potential shift and 50% of the final amplitude of the negative potential shift is the time to reach half maximal amplitude of AD. If not otherwise noted, values are given as mean ± SD and a Student's t-test was performed for statistical analysis (P < 0.05 regarded as significant).
RESULTS
The effects of hypoxia were studied in a total of 35 hippocampal slices from 21 EAAC1−/− mice and 36 hippocampal slices from 19 wild-type mice.
Hypoxia-induced anoxic depolarizations in control and EAAC1-deficient mice
Patch recordings were performed on CA1 pyramidal neurons of wild-type mice and EAAC1-deficient mice. The cells were voltage-clamped at −70 mV and hypoxia-induced changes in holding currents were plotted as a function of time. The amplitude of the holding current before induction of hypoxia was 220 ± 80 pA (n = 8) in wild-type mice and 202 ± 75 pA (n = 7) in EAAC1−/− mice. During the initial phase of hypoxia, a slowly increasing inward current developed within 1 minute, which was later replaced by a secondary large and comparatively fast inward current (anoxic current) with amplitudes of up to 7 nA (Fig. 1). Because the neurons were dialyzed with Cs+, anoxia-induced outward potassium currents (Leblond and Krnjevic 1989; Mourre et al., 1989; Fujimura et al., 1997; Yamamoto et al., 1997; Zawar et al., 1999) could not be observed.
Reoxygenation was commenced within 30 seconds after onset of the secondary fast inward current. During reoxygenation, the holding current was diminished to levels decreased from those of control. Subsequently, the holding current recovered to control levels within 2 minutes. Consistent with previous reports (Hansen, 1985; Somjen et al., 1990; Kral et al., 1993) hypoxia frequently induced an initial slight positive shift in field potentials followed by some latency by a negative potential shift of approximately 20 mV. The slow field potentials slowly recovered to baseline on reoxygenation.
Both field potential and patch recordings revealed that the latency to onset of anoxic depolarization or anoxic current in wild-type mice was significantly (P < 0.05) shorter than in EAAC1-deficient mice. Typical experiments are displayed in Fig. 2Aa for field potential recordings of anoxic depolarizations and in Fig. 2Ab for anoxic current recordings. The latency to anoxic depolarizations was 4.2 ± 1.1 minutes (n = 8) in wild-type mice and 13.7 ± 7 minutes (n = 9, P < 0.05) in EAAC1-deficient mice. The amplitudes of ADs were 18.4 ± 8 mV in wild-type mice and 20 ± 4 mV in EAAC−/− mice, which is not significantly different (P > 0.05). The time to reach half maximal amplitude of ADs was 15 ± 5 seconds in EAAC1−/− mice and 32 ± 7 seconds in wild-type mice (P < 0.01).
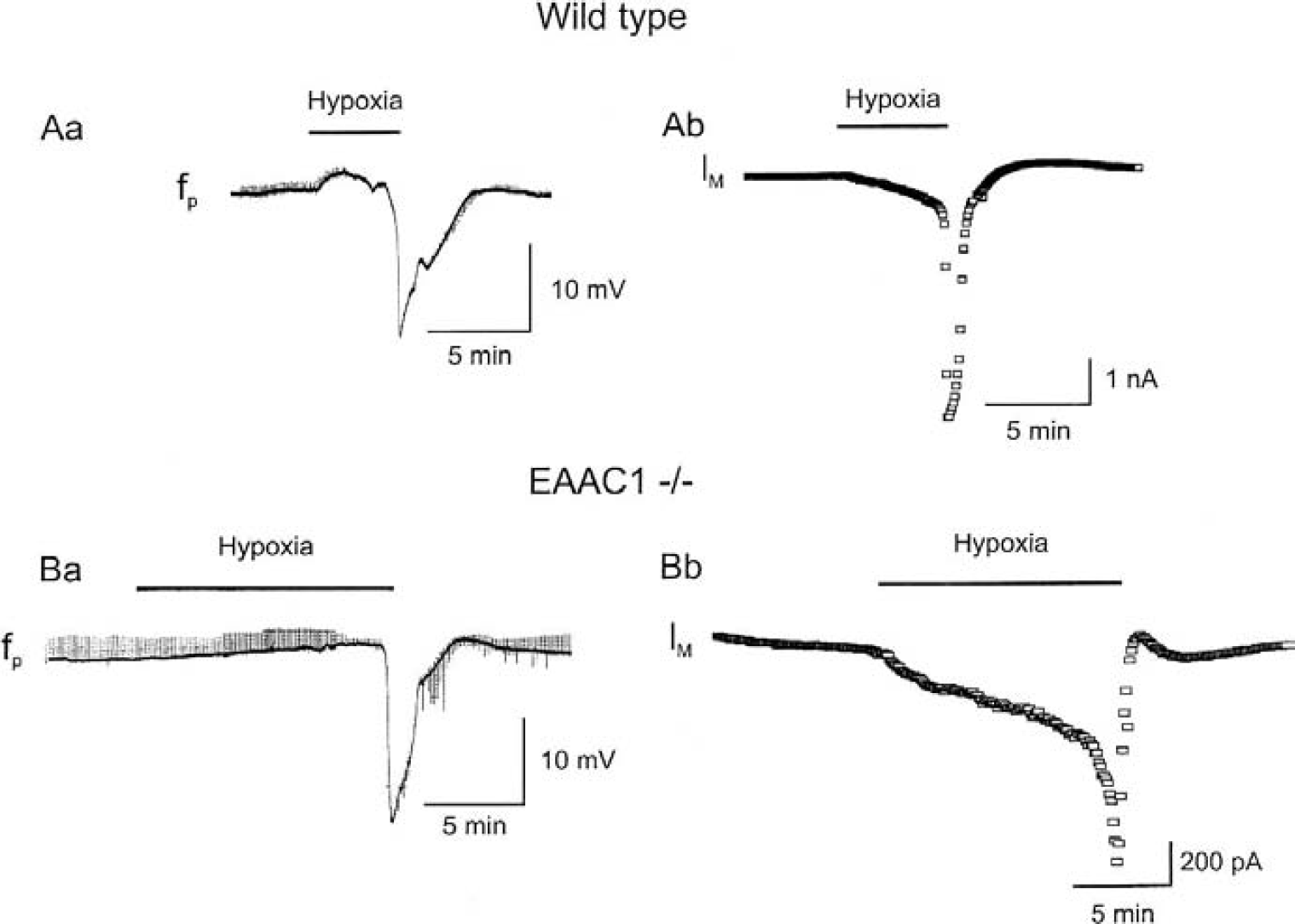
Hypoxia-induced anoxic depolarization is significantly later in each EAAC1−/− mouse than in wild-type mouse.
In patch recordings, the hypoxic episode duration was limited to 15 minutes. Therefore, a secondary large anoxic current was observed in only four of seven cells of EAAC1-deficient mice, whereas this anoxic current was seen in eight cells from eight different slices in control wild-type mice. The latency to onset of anoxic current was 5.7 ± 2 minutes (n = 8) in wild-type mice and 11.2 ± 3.8 minutes (n = 4) in EAAC1−/− mice. The difference in latency to onset of AD and anoxic current was not statistically significant. The peak amplitudes of 2,380 ± 720 pA in wild-type mice and 1,860 ± 532 pA in EAAC−/− mice showed no significant differences.
Effects of hypoxia on primary inward currents
A slow inward current was noted before onset of the large anoxic current. The amplitude of this primary inward current at 3 minutes after onset of hypoxia was significantly smaller in EAAC1−/− mice (78 ± 48 pA, n = 11) as compared with wild type (155 ± 61 pA, n = 8). This slow primary inward current might represent the reduced efficacy of the hyperpolarizing electrogenic Na-K-pump, as recently suggested by Kulik et al. (2000) but also might be contaminated by accumulation of extracellular glutamate or γ-aminobutyric acid) and subsequent increases in membrane conductance. We therefore studied the effects of APV (2-amino-5-phosphonopentanoic acid), NBQX (6-nitro-7-sulphamoylbenzo[f]quinoxa-line-2,3-dione disodium), and bicuculline on these currents. However, the initial inward current induced by hypoxia was not significantly different from that of control in the presence of 30-μmol/L APV + 25-μmol/L NBQX (wild type n = 5, EAAC1−/− n = 7) or 25-μmol/L bicuculline (wild type n = 8, EAAC1−/− n = 4), respectively.
Effect of hypoxia on miniature excitatory postsynaptic currents in EAAC1−/− and wild-type mice
Because spontaneous transmitter release also increases rapidly after onset of mild hypoxia (Katchman and Hershkowitz, 1993; Fleidervish et al., 2001), we were interested in hypoxia-induced changes of miniature excitatory postsynaptic current (mEPSC) frequency in EAAC1−/− and wild-type mice. To restrict the measured spontaneous events to mEPSCs, the experiments were done in the presence of tetrodotoxin (1 μmol/L), which prevents generation of sodium-dependent action potentials and action potential–dependent EPSCs. GABAergic inhibitory postsynaptic currents were not visible at the holding potential of −70 mV, which was close to the reversal potential of Cl−.
In both mouse strains, the frequency of mEPSCs started to increase during the first minute of hypoxia. Sample recordings of hypoxia-induced changes in mEPSC frequency are illustrated in Figs. 3Aa and 3Ba. Averaged mEPSCs (Figs. 3Ab and 3Bb) showed no difference in both rise time and decay time between wild-type and EAAC1−/− mice under control conditions as well as during the first 2 minutes of hypoxia. In Figs. 3Ac and 3Bc, the effect of hypoxia on the amplitudes of mEPSCs is illustrated. The cumulative amplitude histograms were based on measurement of the amplitudes of 500 mEPSCs under normoxic conditions (open circles) and under hypoxia (solid triangles). Events were included in the analysis only if the rise phase was smooth and if the decay returned completely to the base time. By these selections, superimposed events were excluded.

Hypoxia causes a rapid increase in miniature excitatory postsynaptic current (mEPSC) frequency in EAAC1−/− and in wild-type mice without any changes in amplitude and decay time of mEPSC.
The measurements were done before significant numbers of mEPSC became superimposed on each other, which occurred after 3 to 4 minutes of hypoxia. For both animal groups, hypoxia caused no alterations in amplitudes of mEPSCs (Kolmogoroff-Smirnov test).
Figs. 3Ad and 3Bd show the time courses of hypoxia-induced increase in mEPSC frequency from representative neurons of wild-type and EAAC1−/− mice. The statistical analysis of the frequency during the first 3 minutes of hypoxia is displayed in Figs. 3Ae and 3Be. Unexpectedly, the increase in mEPSC frequency at 3 minutes after onset of hypoxia was significantly (P < 0.05) higher in EAAC1−/− mice (621 ± 180%, n = 11) than in the wild type (439 ± 160%, n = 8).
Effects of ld -threo-β-benzyloxyaspartate on anoxic depolarizations in EAAC1−/− mice and wild-type mice
The results presented herein reveal that EAAC1−/− mice also are able to develop anoxic depolarizations. This might suggest that accumulation of glutamate by spontaneous vesicular transmitter release elicits ADs, rather than nonvesicular release through “reversed uptake.” Consequently, a blockade of glial glutamate uptake might accelerate the generation of ADs, suppressing the clearance of glutamate from the extracellular space. To test this hypothesis, we applied the glial glutamate uptake blocker TBOA (150 μmol/L) 10 minutes before induction of hypoxia. Application of TBOA dramatically reduced the latency to ADs. Sample recordings are shown in Fig. 4A. The AD started in less than 1 minute in both groups of animals (0.85 ± 0.4 minutes for EAAC1−/− mice (n = 5) and 0.9 ± 0.3 minutes in wild-type mice, n = 3) without any significant difference. Amplitude of ADs were 14.8 ± 0.8 mV in EAAC1−/− mice (n = 5) and 17 ± 0.9 mV (n = 3) in wild-type mice (see Fig. 4B). Thus, the amplitude of ADs in the presence of TBOA was smaller than under control conditions (Fig. 4B); however, this difference reached significance only in EAAC1−/− mice (P < 0.05). The rise times for ADs were significantly increased by TBOA, reaching half-maximal amplitudes in 92 ± 19 seconds (n = 5) in EAAC1−/− mice and 77 ± 30 seconds (n = 3) in wild-type mice.
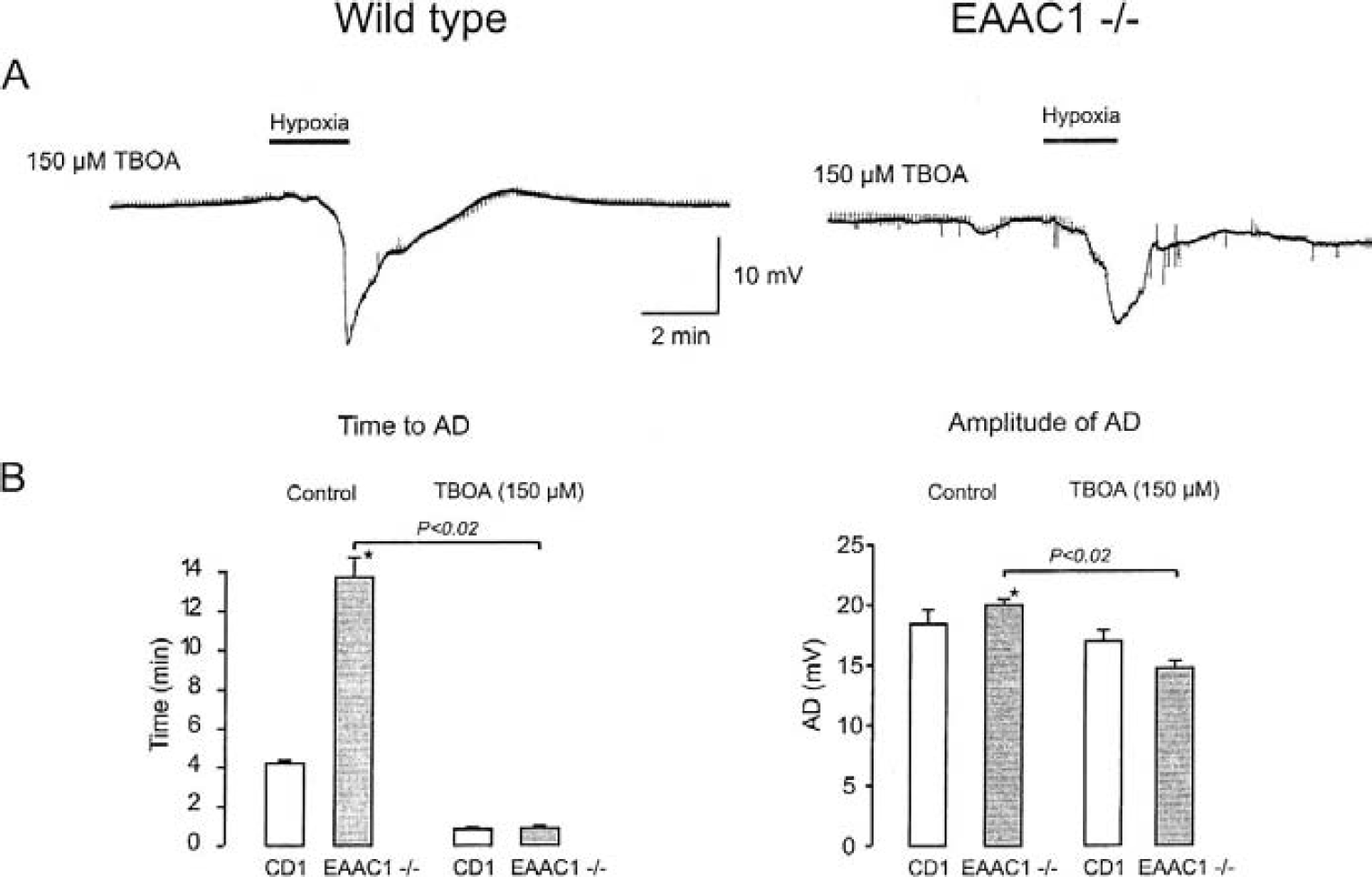
Blocking of glial glutamate uptake with
DISCUSSION
We used transgenic mice to analyze the role of glutamate transporters in the induction of anoxic depolarizations. Our data show that the latency to onset of ADs is significantly increased in EAAC1−/− mice in comparison to wild-type mice. This suggests that reversed transport through the neuronal EAAC1 transporter strongly contributes to the induction of ADs. However, because ADs were still elicitable in both mouse strains, additional mechanisms might influence the generation of ADs. Because hypoxia induced an increase in mEPSC frequency in both animal groups, we speculate that this can eventually lead to toxic accumulation of glutamate in the extracellular space. This speculation is supported by the observation that TBOA, a nontransportable glial glutamate uptake inhibitor (Shimamoto et al., 1998), dramatically shortens latency to AD in both animal groups. Our findings thereby also show that the reversal of glial glutamate transport is not critically involved in induction of ADs, although glial glutamate transport might also reverse during anoxia.
Two isoforms of glutamate transporters are mainly expressed in hippocampal area CA1, where our measurements were done (Rothstein et al., 1994; Chaudhry et al., 1995; Lehre et al., 1995; Coco et al., 1997). Available evidence suggests that EAAC1 is expressed only in neurons (Kanai and Hediger, 1992; Rothstein et al., 1994; Kanai et al., 1995; Velaz Faircloth et al., 1996), whereas GLT1 is primarily expressed in the plasma membrane of astrocytes (Chaudhry et al., 1995; Lehre et al., 1995; Schmitt et al., 1996). GLT1 plays a pivotal role in control of glutamatergic transmission and in neuronal degeneration, whereas the physiologic and pathophysiologic relevance of EAAC1 has not been clarified in detail; for review see Masson et al. (1999). The EAAC1-deficient mouse shows no neurodegeneration or elevation of extracellular glutamate concentration to toxic levels (Peghini et al., 1997).
The observed differences in AD latency between EAAC1−/− mice and wild-type mice suggests that reversed transport from neuronal compartments is an important step in induction of ADs irrespective of the neuronal compartment from which glutamate is released. This is in line with recent findings by Rossi et al. (2000) and by Jabaudon et al. (2000). Rossi et al. showed a delay in generation of ADs when slices were preloaded with the transportable glutamate analogue PDC (
However, despite the increase in latency, ADs could still be generated in EAAC1−/− mice. We have previously shown that increases in spontaneous release of γ-aminobutyric acid and glutamate are probably the first event during hypoxia in mouse neocortical neurons (Fleidervish et al., 2001). This latent period was strongly shortened when glial glutamate uptake was blocked in EAAC1−/− and wild-type mice, suggesting that under conditions of reduced glutamate uptake into glia spontaneous transmitter release might contribute to generation of ADs. In accordance with our data, Jabaudon et al. found that the increased spontaneous vesicular transmitter release caused by energy depletion only caused extracellular glutamate accumulation when glial glutamate uptake was blocked (Jabaudon et al., 2000). This suggests that glial glutamate transport does not readily reverse and that it remains functional for prolonged periods of hypoxia preceding ADs in EAAC−/− mice. The reversal of glutamate transport is only expected during a strong depolarization to values near −20 mV unless the ionic transmembrane gradients are strongly altered. In the period preceding ADs, extracellular potassium concentrations slightly increase (Hansen, 1985; Muller and Somjen, 2000), whereas extracellular concentrations of Na+, Ca +, and Cl− ions drop only during ADs (Hansen and Zeuthen, 1981; Hansen, 1985; Muller and Somjen, 2000). Consequently, if glutamate transport is reversed preceding AD, the particular release compartment must be strongly depolarized. Because neither glial cells nor neuronal somata reveal large depolarizations before AD, the likely compartment where glutamate transport is reversed is the presynaptic terminal. In fact, at least 20% of ATP consumption in brain is used for maintaining the presynaptic release machinery functional (Ames, 2000), and synaptic terminals may be very sensitive to hypoxia. We therefore suggest that reversed transport of glutamate before induction of ADs is caused by depolarizations of synaptic terminals. Such depolarization should also increase spontaneous transmitter release. In a previous study, we have shown that the frequency of mEPSCs and miniature inhibitory postsynaptic currents increases within 30 seconds after onset of hypoxia, a result that could be confirmed in the present study for both EAAC1−/− and wild-type mice.
The massive synaptic release of glutamate at early stages of hypoxia will cause an increased extracellular concentration of the transmitter, depending on the efficacy of glutamate uptake. This implies that blockade of glial glutamate transport should accelerate the generation of AD.
In contrast to our results, a previous in vivo study (Obrenovitch, 1998) showed that an inhibition of glutamate uptake by
In summary, our data provide evidence for an active role of the neuronal glutamate transporter EAAC1 in the generation of ADs. Conversely, glial glutamate uptake protects hippocampal tissue from this pathologic pattern of activity. It is feasible that selective block of neuronal glutamate uptake could contribute to the prevention of adverse effects of hypoxia.