
Abstract
Changes in intracellular and extracellular pH may influence the vulnerability of brain tissue to anoxic or ischemic damage. In the present study, we investigated whether the increased vulnerability of aged brain tissue to anoxic damage is associated with age-related alterations in pH regulation. We obtained evidence for altered pH regulation by measuring concurrent changes in intracellular and extracellular pH before, during, and after anoxia in hippocampal slices from young adult (6–8 months old) and aged (24–27 months old) rats. We found indications of impaired pH regulation in aged hippocampal slices (a) before anoxia, as seen in a lower resting intracellular pH, (b) during anoxia, as seen in a slower decline in extracellular pH, and (c) during recovery after anoxia, as seen in a slower rate of recovery of intracellular pH. Age-related changes in pH regulation may contribute to the faster onset of anoxic depolarization in aged brain tissue during anoxia.
Keywords
Aging increases the susceptibility of brain tissue to anoxic damage. Evidence for this increased vulnerability includes an earlier onset of anoxic depolarization [the event that signals the massive redistribution of ions across brain cell membranes (see Hansen, 1985)] and diminished recovery of synaptic activity after anoxia in hippocampal slices from aging rats compared with slices from younger rats (Roberts et al., 1990; Roberts and Chih, 1995). Since the duration of anoxic depolarization is related to the severity of neuronal injury (Balestrino and Somjen, 1986; Roberts and Sick, 1987; Balestrino et al., 1989; Somjen et al., 1990; Gill et al., 1992), earlier anoxic depolarization may lead to greater anoxic damage in aging brain tissue compared with younger brain tissue if the durations of anoxia are the same in all age groups.
Age-related impairment of pH regulation (Roberts and Sick, 1996) may be involved in this greater vulnerability since any alterations in intracellular pH (pHi) and extracellular pH (pHo) during and after anoxia could affect the onset of anoxic depolarization and the recovery of brain function after anoxia. For example, mild to moderate extracellular acidosis (pHo of ∼6.5–7.3) delays the onset of anoxic depolarization (Tombaugh, 1994) and the rise in intracellular Ca2+ during simulated ischemia (Ebine et al., 1994) in hippocampal slices and reduces apparent neuronal injury due to oxygen-glucose deprivation (Giffard et al., 1990; Tombaugh and Sapolsky, 1990). Although reductions in pHo can lead to lower values of pHi (Pirttila and Kauppinen, 1993; Mellergard et al., 1994), whether mild extracellular acidosis influences recovery of brain tissue from anoxia or ischemia by reducing pHi remains unclear. In fact, intracellular acidosis has more often been associated with anoxic or ischemic damage (e.g., see Siesjo, 1981, 1992; Smith et al., 1986; Maruki et al., 1993; Katsura et al., 1994). Since anaerobic glycolysis is inhibited by low pHi (Trivedi and Danforth, 1966; Erecinska et al., 1995) and anoxic depolarization is closely associated with the onset of energy failure (Ekholm et al., 1993), lower pHi may hasten the occurrence of anoxic depolarization.
In this study, we investigated whether aging altered pH regulation before, during, and after anoxia. We obtained evidence for altered pH regulation by examining the relationship between pHi and pHo. We also examined the connection between changes in pH (both pHi and pHo) and the occurrence of anoxic depolarization, since this connection has never been characterized either in vivo or in vitro. In addition, we studied the relationship between pHi and pHo changes during and after anoxia. We carried out our studies in hippocampal slices from young adult (6–8 months old) and aged (24–27 months old) rats. We found evidence for impaired pH regulation in aged hippocampal slices in that aged slices had a lower resting pHi, a slower rate of decline in pHo during anoxia, and slower rates of recovery of pHi and of pHo following anoxia.
MATERIALS AND METHODS
Hippocampal slices were prepared as described previously (Roberts and Sick, 1987, 1988; Roberts et al., 1990) from male Fisher 344 rats purchased from the National Institute on Aging. Rats were classified as young adult (6–8 months of age) or aged (24–27 months of age). Four-hundred-micron-thick slices were initially stored at room temperature (22–24°C) in a holding chamber containing artificial CSF (ACSF) of formula (in mM) 126 NaCl, 3.5 KCl, 2.0 CaCl2, 2.0 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, and 10.0 glucose and were bubbled with a gas mixture containing 95% O2/5% CO2. The slices were then placed into ACSF containing 20 μM of the acetoxymethylester form of SNARF-1 (SNARF-1-AM) (Molecular Probes), 0.02% Pluronic 127 (Molecular Probes), and 0.5% dimethyl sulfoxide. The slices were incubated in this ACSF for at least 1 h to allow sufficient time for diffusion of SNARF-1-AM into the intracellular compartment, where the AM group was removed via hydrolysis, trapping SNARF-1 molecules intracellularly.
The slices were next transferred to an interface-type recording chamber where they were exposed to SNARF-free ACSF for 1 h at 36–37°C to wash out extracellular SNARF-1. Slices were considered free of extracellular SNARF-1 when their pH, remained unchanged for ≥10 min following their placement into the chamber. A stable pH, was usually reached in slices after ≤30 min. When data regarding pHi were needed, a slice was illuminated with 543-nm light from a helium-neon laser to excite SNARF-1 molecules into fluorescence. The laser beam was focused so that it illuminated a spot ∼400 μm in diameter. The laser spot was centered on the pyramidal cell layer of hippocampal subregion CA1. Spectra were obtained that contained single peaks for the protonated and unprotonated forms of SNARF-1, respectively. Details for obtaining pHi readings from SNARF-1 spectra are provided elsewhere (Roberts and Sick, 1996). Fluorescence was detected with a scanning array spectrophotometer (EG&G Princeton Applied Research Corp.).
As has been previously reported (Whittingham et al., 1992), SNARF-1 appeared more concentrated in the cell bodies of CA1 pyramidal cells. The pH-sensitive dye BCECF, which, like SNARF-1, is a fluorescein derivative, is also concentrated in pyramidal cell bodies (Fujiwara et al., 1992).
While extracellular SNARF-1 was being washed out, a single-barrel microelectrode filled with 150 mM NaCl was placed into a slice at stratum pyramidale of CA1. Synaptic transmission was then evaluated by stimulating the slice's Schaffer collaterals at 0.2 Hz with 0.5-ms-long constant-current pulses and recording the resulting extracellular postsynaptic field potentials (orthodromic population spikes). If the largest orthodromic population spikes that could be elicited were around ≥2 mV in magnitude, the single-barrel microelectrode was removed, and a double-barrel H+-sensitive microelectrode was placed into the slice (for construction and calibration of H+ microelectrodes, see Ammann, 1986). We used Hydrogen Ionophore II—Cocktail A (Fluka) as the H+-selective membrane in H+-sensitive microelectrodes. pHo and the extracellular DC potential (recorded from the reference barrel of the H+ microelectrode) were monitored continuously on a chart recorder. Orthodromic population spikes were recorded as needed from the reference barrel.
After sufficient time had passed for washing out extracellular SNARF-1 and for stabilization of extracellular H+ activity following electrode placement, the slice was stimulated at 0.1 Hz, and the magnitude of the orthodromic population spike was determined. Stimulus intensities were more than sufficient to produce the largest possible orthodromic population spike in CA, pyramidal cells. Also during this time, SNARF-1 spectra were obtained for determination of pH, during normoxia. The slice was than made anoxic by switching the gas mixture to 95% N2/5% CO2. Anoxic depolarization was identified by a sudden negative shift in the extracellular DC potential. The duration of anoxic depolarization was held to 1 min since this duration allows partial recovery of synaptic transmission following anoxia in both age groups used in this study (see Roberts et al., 1990; Roberts and Chih, 1995). After 1 min of anoxic depolarization, anoxic insults were terminated by the reintro-duction of 95% O2/5% CO2 to the chamber. Recovery of pH, and pHo was followed for ∼30 min while synaptic activity was followed for ∼1 h.
All statistical tests were carried out with the SAS statistical software package for microcomputers (SAS Institute, Cary, NC, U.S.A.).
RESULTS
Anoxia caused a rapid decline in both pHi and pHo in young adult and aged hippocampal slices (Figs. 1 and 2). In young adult slices before anoxia, pHi was 7.23 ± 0.05 (mean ± SD; n = 5), while pHo was 7.10 ± 0.07 (n = 6). These findings were not significantly different from pHi (7.16 ± 0.08) and pHo (7.01 ± 0.08) for young adult slices in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)–buffered (nonbicarbonate) ACSF (Roberts and Sick, 1996). At anoxic depolarization, pHi in young adult slices had decreased to 6.91 ± 0.13 (n = 5), while pHo dropped to 6.71 ± 0.11 (n = 6). Both pHi and pHo decreased further during anoxic depolarization, reaching their lowest values either after 1 min of anoxic depolarization or early during recovery from anoxia. The maximum decreases in pHi and pHo from their controls before anoxia averaged 0.43 ± 0.04 and 0.52 ± 0.10 unit, respectively.
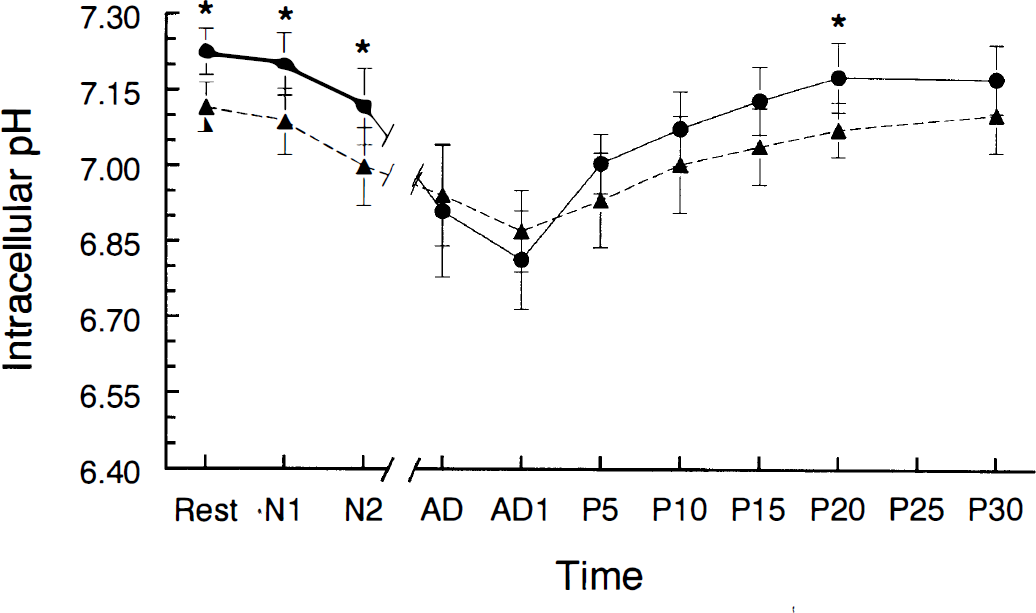
Changes in intracellular pH before, during, and after anoxia in young adult (filled circles) and aged (filled triangles) hippocampal slices. The different stages of the experiment were defined as (a) “Rest” (pHi before anoxia), (b) “N1” and “N2” (1 and 2 min of anoxia, respectively, without the appearance of anoxic depolarization), (c) “AD” (anoxic depolarization) and “AD1” (∼1 min after AD during anoxia), and (d) “P5-30” (5–30 min after reoxygenation). The breaks in the x-axis represent the interval between N2 and anoxic depolarization. *Young adult and aged values were significantly different from each other (p < 0.05, Student t test). Error bars are standard deviations. For number of observations at each time point, see Table 1. Note: Since time to anoxic depolarization varied from 2 to 7 min, the elapsed time between N2 and anoxic depolarization varied with each slice.
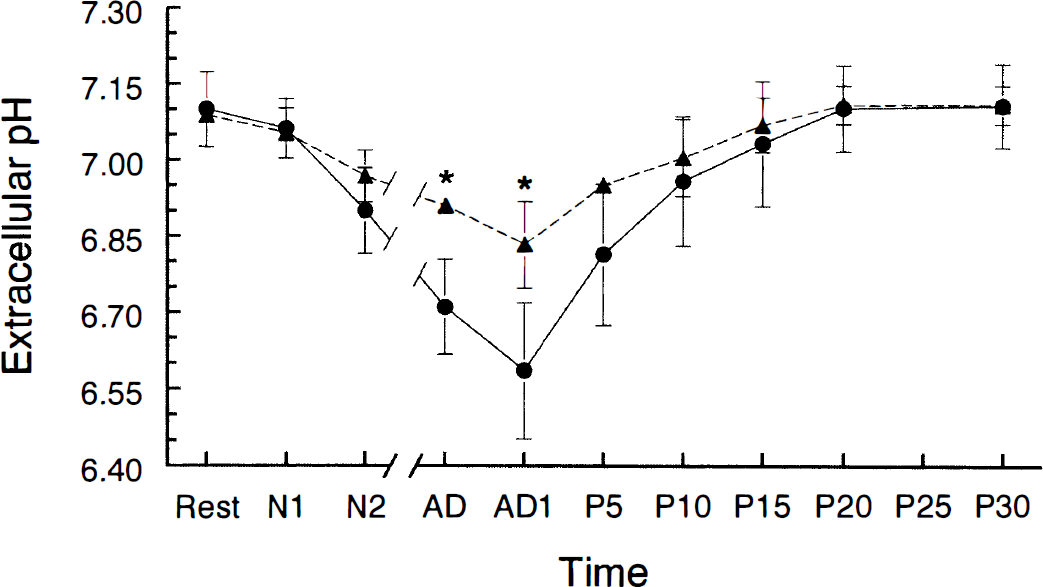
Changes in extracellular pH before, during, and after anoxia in young adult (filled circles) and aged (filled triangles) hippocampal slices. *Young adult and aged values were significantly different from each other (p < 0.05, Student t test). The abbreviations and breaks in the x-axis are explained in Fig. 1.
The resting pHi for aged slices (7.12 ± 0.05, n = 5) was significantly lower than that for young adult slices (p < 0.05) (Fig. 1). This resting pHi did not differ significantly from the resting pHi for aged slices in HEPES-buffered (nonbicarbonate) ACSF (7.06 ± 0.05) (Roberts and Sick, 1996). The difference in pHi between the two age groups persisted for the first 2 min of anoxia but disappeared at anoxic depolarization. pHi in aged slices was 6.94 ± 0.10 at anoxic depolarization. The maximum decline of pHi during anoxia in aged slices was 0.28 ± 0.11 unit and was significantly less than that seen in young adult slices (p < 0.05).
The resting pHo in aged slices (7.09 ± 0.02, n = 4) did not differ from resting pHo in young adult slices or from its value in HEPES-buffered (nonbicarbonate) ACSF (7.02 ± 0.12) (Roberts and Sick, 1996). pHo declined more slowly during the first 2 min of anoxia in aged slices compared with young adult slices (p < 0.05) (Table 1). Although the rate of decline in pHo during the period of time following the first 2 min of anoxia and concluding with onset of anoxic depolarization (N2-AD in Table 1 and Figs. 1 and 2) was not different between the two age groups, any possible age-related differences in this rate may have been obscured by the fact that anoxic depolarization occurred shortly after 2 min of anoxia in most aged slices (see below). pHo was higher in aged slices at anoxic depolarization (p < 0.05) (Fig. 2). Also, the total decrease in pHo at anoxic depolarization was smaller in aged slices (p < 0.05). pHo and pHi generally decreased at the same time during anoxia in both age groups (Fig. 3), but the magnitudes of these decreases were not correlated.
Rates of changes in pH during and after anoxia
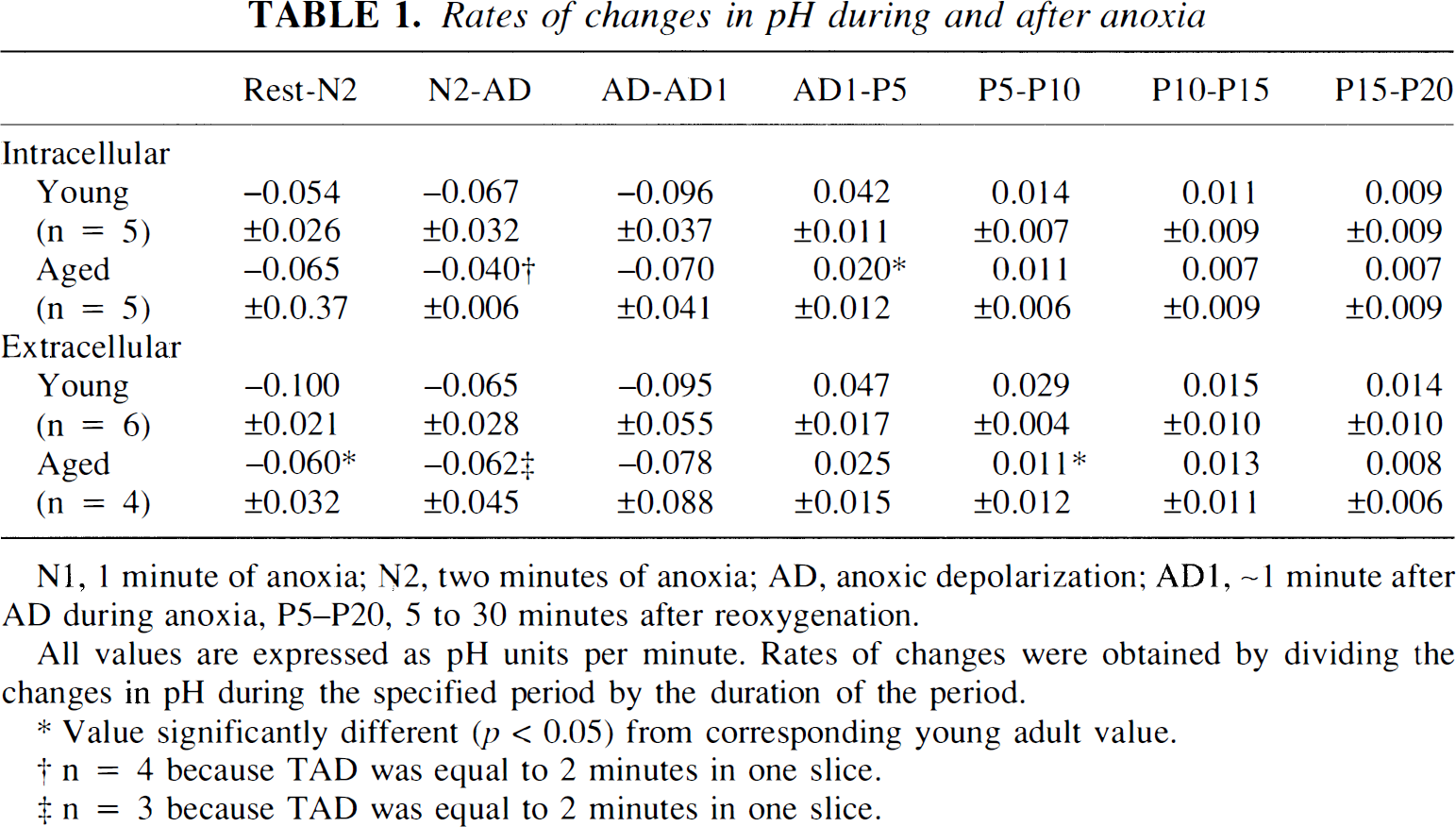
N1, 1 minute of anoxia; N2, two minutes of anoxia; AD, anoxic depolarization; AD1, ∼1 minute after AD during anoxia, P5–P20, 5 to 30 minutes after reoxygenation.
All values are expressed as pH units per minute. Rates of changes were obtained by dividing the changes in pH during the specified period by the duration of the period.
Value significantly different (p < 0.05) from corresponding young adult value.
n = 4 because TAD was equal to 2 minutes in one slice.
n = 3 because TAD was equal to 2 minutes in one slice.
(
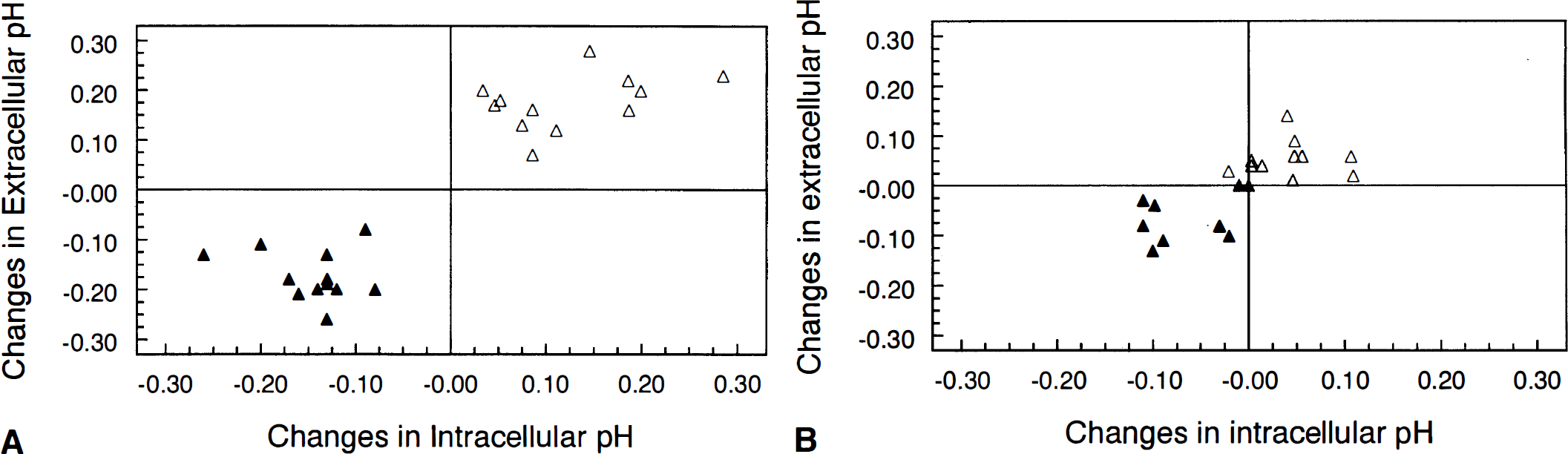
The duration of anoxia required for anoxic depolarization to take place (TAD) varied between 3.8 and 7 min with an average of 5.26 ± 1.02 min in young hippocampal slices. TAD in aged slices varied between 2 and 4 min and averaged 3.17 ± 0.80 min, which was significantly shorter (p < 0.05) than TAD in young adult slices. TAD was correlated with the magnitudes of the decreases in pHi (r = −0.74, p < 0.05) (Fig. 4) and in pHo (r = −0.62, p < 0.05) (Fig. 4B) at anoxic depolarization.
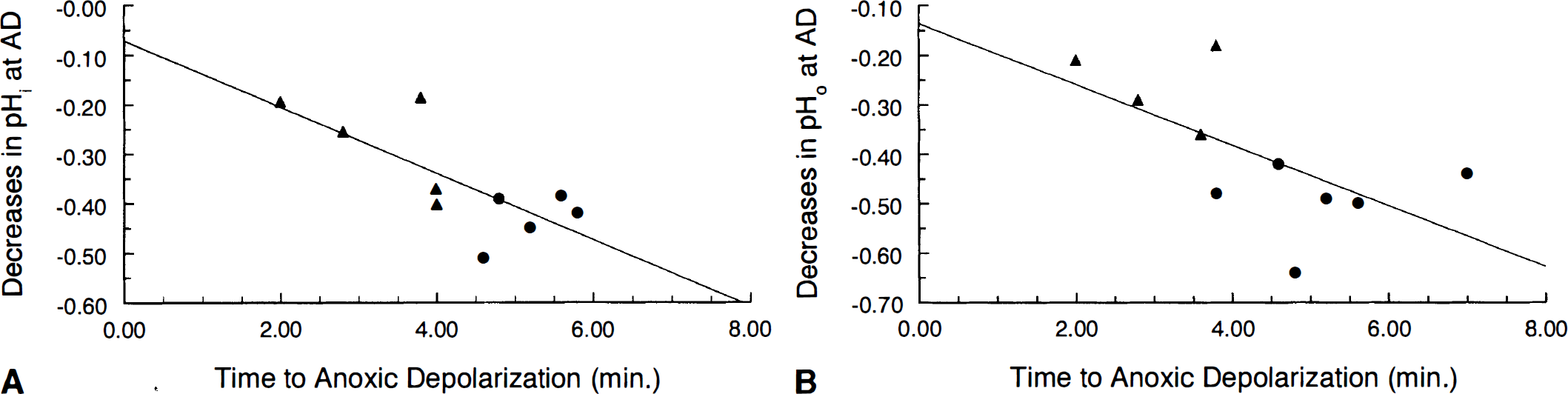
(
Both pHi and pHo returned to their resting levels between 15 and 20 min after reoxygenation in both young adult and aged hippocampal slices. In both age groups, pHi increased fastest during the first 5 min of reoxygenation (Table 1). Recovery of pHi during the initial 5 min was significantly slower in aged slices. Also, recovery of pHo was significantly slower in aged slices during the second 5-min period of recovery (P5–P10, Table 1). As was true during anoxia, increases in pHo were accompanied by increases in pHi in both age groups during recovery (Fig. 4), but the magnitudes of these increases were not correlated.
As in previous experiments from this laboratory (Roberts et al., 1990; Roberts and Chih, 1995), there was no significant difference in the amplitude of orthodromic population spikes between the two age groups before anoxia. The percent recovery of the amplitude of the orthodromic population spike for each slice 60 min after reoxygenation averaged 47 ± 16% (n = 7) in young slices and 34 ± 19% (n = 6) in aged slices. The difference in recovery of the orthodromic population spike was not statistically significant between the two age groups.
DISCUSSION
In this study, we present evidence for impaired pH regulation in the aged brain (a) before anoxia, as seen in a lower resting pHi, (b) during anoxia, as seen in a decreased rate of decline in pHo, and (c) during recovery from anoxia, as seen in a slower rate of recovery in pHi. This impairment may affect normal brain function as well as the susceptibility of the aged brain to anoxic damage.
Before anoxia
The pHi before anoxia was lower by ∼0.1 unit in aged slices compared with young adult slices whether bicarbonate buffer was present (this study) or absent (Roberts and Sick, 1996). Since pHo did not differ between the two age groups, the transmembrane gradient for H+ was less steep in aged hippocampal cells. Potential explanations for this difference include an increased leakage of H+ through aged membranes or a decreased efficiency of acid extrusion in aged slices. A diminished pH gradient means that aged hippocampal cells expend less metabolic energy maintaining this gradient than they would if they maintained the same gradient present in young adult slices. However, a lower pHi could decrease glycolysis by inhibiting phosphofructokinase (Trivedi and Danforth, 1966; Erecinska et al., 1995). Depending upon the levels of glycolytic substrates and modulators in cells, a 0.1-unit decrease in pHi can significantly affect phosphofructokinase activity (e.g., see Trivedi and Danforth, 1966). A decrease in pHi can also slow signal transduction through interactions with second messenger systems (Conner and Hockberger, 1984) and decrease the capacity of the aged brain to buffer pH changes (Roberts and Sick, 1996).
Our results demonstrate that pHi can be maintained in the absence of bicarbonate-dependent pH regulation since pHi within each age group was not significantly different in the presence (this study) or nominal absence (Roberts and Sick, 1996) of HCO3/CO2 buffering. Furthermore, the fact that the difference in pHi between the two age groups remained the same whether bicarbonate was present or absent suggests that bicarbonate-dependent pH regulation contributes little to regulation of resting pHi in hippocampal cells. The reason for this is that the presence of bicarbonate-dependent pH regulation might have allowed aged hippocampal cells to make up any deficit of bicarbonate-independent pH regulation and to minimize any difference in pHi between the two age groups. This finding agrees with the suggestion that Na+-H+ exchange is the principal mechanism for regulating resting pHi in cultured hippocampal neurons (Raley-Susman et al., 1991). Na+-H+ exchange activity has been demonstrated in hippocampal slices (LaManna et al., 1992; Lin et al., 1996) and may then be involved in pH regulation in our experiments.
During anoxia
Before the onset of anoxic depolarization, pHi was lower in aged hippocampal slices. However, this lower pHi provided no protection from anoxic depolarization in that anoxic depolarization occurred sooner in aged hippocampal slices. This finding argues against the possibility that lower pHo exerted its protective effect by reducing pHi. In fact, a lower pHi prior to anoxic depolarization may have been detrimental to aged hippocampal slices because anaerobic glycolysis can be suppressed by low pHi (see previous section). Indeed, aged hippocampal slices appear less able to facilitate anaerobic glycolysis to support ion transport during anoxia (Roberts and Chih, 1995). Since anoxic depolarization is closely associated with the onset of energy failure (Ekholm et al., 1993), inhibition of glycolysis is likely to contribute to the earlier occurrence of anoxic depolarization in the aged brain.
A lower pHi prior to anoxic depolarization in aged hippocampal slices did not lead to greater acidosis at anoxic depolarization. In fact, depolarization occurred at about the same pHi (6.91 in young adult and 6.94 in aged rats). This observation supports the hypothesis that there may be a common threshold pHi for the two age groups at which anoxic depolarization occurs. This hypothesis is consistent with earlier suggestions that acidosis becomes damaging to the brain when it reaches a threshold level (Siesjo et al., 1990). Further experimentation will be needed to determine the extent to which pHi is involved in changing the latency of anoxic depolarization.
Another observation was that pHi and pHo generally changed in the same direction in both age groups in that a decline in pHi was accompanied by a decline in pHo (Fig. 3). This link between pHi and pHo implies that H+ was extruded during anoxia. Acid extrusion may have been impaired in the aged group during anoxia, as seen in the slower decline of pHo in the aged group during the initial 2 min of anoxia (Table 1). Also, although there were no differences in values of resting pHo between the two age groups, pHo was lower in the young adult group at anoxic depolarization. This smaller total decrease in pHo at anoxic depolarization in aged slices may have also been due to impaired acid extrusion, as well as to the fact that anoxic depolarization occurred sooner in the aged group.
The greater extracellular acidosis found during anoxia in young adult slices compared with aged slices may have helped delay the onset of anoxic depolarization in young adult slices since mild acidosis has been found to delay anoxic depolarization in hippocampal slices (Tombaugh, 1994). Further evidence for a delay of anoxic depolarization with extracellular acidosis includes our finding that decreases in pHo at anoxic depolarization were positively correlated with increases in TAD (Fig. 4B). Lower pHo may delay anoxic depolarization by reducing N-methyl-
During postanoxic recovery
Intracellular H+ concentrations increased more during anoxia in young adult slices (171%) than in aged slices (95%). However, pHi recovered to preanoxic levels at approximately the same time in both age groups. Thus, pH regulation may be more efficient in young adult slices. Providing further evidence for impaired acid extrusion in aged slices was our finding that the rate of recovery of pHi was faster in the young adult slices during the first 5 min after anoxia (Table 1). Differences in the recovery of pHi during reperfusion following ischemia have also been found in the brains of young and middle-aged gerbils (Funahashi et al., 1994).
Both pHi and pHo recovered more slowly than extracellular K+ following anoxia in hippocampal slices. A previous study has shown that, after reoxygenation, young adult slices need ≤3 min for extracellular K+ to return to preanoxic levels (Roberts et al., 1990), while both pHi and pHo needed between 15 and 20 min to recover after reoxygenation in this study. Aging slows recovery of extracellular K+ following anoxia (Roberts et al., 1990; Roberts and Chih, 1995), which probably means that extracellular Na+ also recovered more slowly. This could in turn slow Na+-dependent pHi regulation. Thus, slower recovery of the K+ gradient across cell membranes may have also contributed to the reduced rate of pHi recovery in aged slices.
The diminished ability of aged hippocampal slices to regain extracellular K+ homeostasis promptly has also been associated with diminished recovery of synaptic activity after anoxia (Roberts et al., 1990; Roberts, 1993). In this study, synaptic activity recovered less in aged slices, but the difference was not statistically significant. However, this lack of significance is probably due to small sample size, as larger samples have shown that postanoxic recovery of synaptic transmission is less in aged slices ((Roberts et al., 1990; Roberts and Chih, 1995).
It remains unclear whether recovery of synaptic activity following anoxia benefits from lower pH levels. For example, delayed recovery of pHi during reperfusion by hypercapnia reduced evoked potential recovery independent of recovery of high-energy phosphate levels in vivo (Maruki et al., 1993). However, mild preischemic extracellular acidosis was found to improve recovery of synaptic activity in hippocampal slices subject to a fixed duration of anoxic depolarization (Tombaugh, 1994). In this study, we found no correlation between the rate of recovery of pHi or pHo and the recovery of synaptic transmission.
In summary, our results suggest that aging impairs pH regulation before, during, and after anoxia in hippocampal slices. This impairment may contribute to the faster onset of anoxic depolarization in aged hippocampal slices and thus increase the vulnerability of the aged brain to anoxic damage.
Footnotes
Acknowledgment:
We thank Patricia Mumford and Joe Feng (both of the Department of Neurology) for their technical assistance with our experiments. We also thank Dr. Brant Watson (Department of Neurology) for lending us a helium-neon laser for our experiments.