
Abstract
The Ca2+ mobilization across the neuronal membrane is regarded as a crucial factor in the development of neuronal damage in ischemia. Because glucocorticoids have been reported to aggravate ischemic neuronal injury, the effects of dexamethasone on ischemia-induced membrane depolarization, histologic outcome, and changes in the intracellular Ca2+ concentration in the gerbil hippocampus were examined in vivo and in vitro. The effects of metyrapone, an inhibitor of glucocorticoid synthesis, were also evaluated. Changes in the direct-current potential shift in the hippocampal CA1 area produced by transient forebrain ischemia for 2.5 minutes were compared among animals pretreated with dexamethasone (3 μg, intracerebroventricularly), metyrapone (100 mg/kg, intraperitoneally), and saline. The histologic outcome was evaluated 7 days after ischemia by assessing the delayed neuronal death in the hippocampal CA1 pyramidal cells of these animals. A hypoxia-induced intracellular Ca2+ increase was evaluated by in vitro microfluorometry in gerbil hippocampal slices, and the effect of dexamethasone (120 μg/L in the medium) on the cytosolic Ca2+ accumulation was examined. The effect in a Ca2+-free ischemialike condition was also investigated. Preischemic administration of dexamethasone reduced the onset latency of ischemia-induced membrane depolarization by 22%, and aggravated neuronal damage in vivo. In contrast, pretreatment with metyrapone improved the histologic outcome. The onset time of the increase in the intracellular concentration of Ca2+ provoked by in vitro hypoxia was advanced in dexamethasone-treated slices. The Ca2+-free in vitro hypoxia reduced the elevation compared with that in the Ca2+-containing condition. Treatment with dexamethasone facilitated the increase on both the initiation and the extent in the Ca2+-free condition. Aggravation of ischemic neuronal injury by endogenous or exogenous glucocorticoids is thus thought to be caused by the advanced onset times of both the ischemia-induced direct-current potential shift and the increase in the intracellular Ca2+ concentration.
Keywords
In cerebral ischemia, an energy failure triggers depolarization of the neuronal membrane (Hansen, 1985), and various excitatory neurotransmitters such as glutamate and aspartate are released together (Benveniste et al., 1984; Phebus and Clemens, 1989; Adachi et al., 1991) from both neurons and glia (Mitani et al., 1994a). A marked influx of Ca2+ into postsynaptic neurons then occurs, which provokes the catastrophic enzymatic process leading to irreversible neuronal injury (Mitani et al., 1994b). Glucocorticoids have been demonstrated to exacerbate ischemia-induced neuronal damage in the brain (Koide et al., 1986). This action is speculated to be caused by the accumulation of excitatory amino acids in the extracellular space because of an inhibition of the uptake of these amino acids into astrocytes (Virgin et al., 1991; Chou et al., 1994). Further, these agents increase the basal free cytosolic Ca2+ concentrations in cultured hippocampal neurons in a nonischemic condition, and exacerbate the extent of Ca2+ mobilization induced by excitatory amino acids (Elliott and Sapolsky, 1992, 1993). Because excitatory neurotransmitter release and Ca2+ mobilization in ischemia are closely related to neuronal damage, we studied the in vivo effects of dexamethasone on ischemia-induced alterations in the direct-current (DC) potential shift (anoxic depolarization; AD) in the gerbil hippocampus, and the in vitro effects of dexamethasone on a hypoxia-induced increase in the intracellular Ca2+ concentration ([Ca2+]i). We also investigated the in vivo effects of metyrapone, a potent and rapid inhibitor of glucocorticoid production, to evaluate the effects of endogenous glucocorticoids in ischemia.
MATERIALS AND METHODS
This study was approved by the Committee on Animal Experimentation at Ehime University School of Medicine, Ehime, Japan. Male Mongolian gerbils weighing 60 to 80 g (Seiwa Experimental Animals, Fukuoka, Japan) were housed in groups in a room controlled at 23° ± 1°C and maintained in an alternating 12-hour light and 12-hour dark cycle (lights on at 6:00
Experiment 1: In vivo experiments on AD and the histologic outcome
In this experiment, 15 animals were prepared and then assigned to dexamethasone, metyrapone, and control groups (5 animals each). The animals were anesthetized and maintained with 2% halothane in balanced 50% O2 and 50% N2O. Through a ventral middle cervical incision, both common carotid arteries were exposed and separated carefully from adjacent nerves and tissues. Silk threads (4-0) were looped around these arteries. After the animal was placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, U.S.A.) in the prone position, the skull was exposed and a small burr hole was drilled in the left hemisphere at 2 mm anterior and 2 mm lateral to the bregma for the insertion of a thermocouple needle probe (TN-800; Unique Medical Corp., Tokyo, Japan). The tip of the thermocouple needle probe was positioned about 2 mm below the brain surface. An identical probe was inserted into the rectum. Brain and rectal temperatures were carefully maintained at 37° to 38°C with a heating lamp (Koehler type illumination lamp; Olympus, Tokyo, Japan). Two additional burr holes were drilled; one in the left hemisphere (0.5 mm posterior and 2.0 mm lateral to the bregma) for drug administration and the other in the right hemisphere (2.0 mm posterior and 2.0 mm lateral to the bregma) for the measurement of the DC potential shift.
One group was given 10 μL of saline intracerebroventricularly through the burr hole via a 27-gauge needle. Another group was administered dexamethasone-water soluble (3 μg dissolved in saline) intracerebroventricularly in a volume of 10 μL. The remaining group was injected with metyrapone (100 mg/kg) intraperitoneally, and with 10 μL of saline intracerebroventricularly.
The electrode consisted of a glass micropipette with a tip diameter of about 4 μm, which was filled with 2 mol/L NaCl with an Ag/AgCl electrode in the barrel. This local electrode was implanted in the right hippocampal CA1 field (2.0 mm posterior and 2.0 mm lateral to the bregma, and 2.0 mm below the brain surface) through the burr hole described above 30 minutes after the drug or saline administration. The remote electrode (Ag/AgCl) was inserted subcutaneously at the back of the neck. The DC potential shift was monitored between these electrodes with a model AB-621G DC amplifier (Nihon Konden, Tokyo, Japan).
After a stabilization period of 30 minutes, transient forebrain ischemia for 2.5 minutes was achieved by pulling the threads around the bilateral common carotid arteries with 8-g weights, while maintaining the brain and rectal temperatures at 37.5° ± 0.2°C (Mitani et al., 1991). After the 2.5 minutes' ischemia, the threads were cut to restore the blood flow. Rectal and brain temperatures were maintained at 37° to 38°C under halothane anesthesia for 30 minutes after the reflow. The difference in the DC potential shift was compared by analyzing its onset latency, amplitude, and recovery time of depolarization to half-maximal amplitude. After the measurement, the thermocouple probes and the electrodes were gently removed. All surgical incisions were sutured, and the animal was removed from the stereotaxic apparatus. The animal was brought to its cage in a room maintained at constant temperature and allowed access to food and water ad libitum.
Seven days after this transient forebrain ischemia, the animals were anesthetized with an intraperitoneal injection of sodium pentobarbital (50 mg/kg). The brains were perfused with heparinized saline and fixed with 10% buffered formalin. The brains were removed after perfusion. After dehydration with graded concentrations of alcohol solutions, the brains were embedded in paraffin. Brain slices, 5 μm thick, were stained with hematoxylin and eosin. The numbers of preserved pyramidal cells in the hippocampal CA1 field per 1 mm length of stratum pyramidale in each hemisphere were counted in the same level of coronal section (1.5 mm posterior to the bregma). The average of the values on both sides was then obtained for each animal.
Another set of 12 gerbils was prepared to determine the physiologic variables that may exert an influence on the extent of neuronal damage in ischemia. These animals were anesthetized with 2% halothane in balanced 50% O2 and 50% N2O. Four animals were then given saline intracerebroventricularly, and another four animals were given dexamethasone (3 μg) according to the procedure described above. The remaining four animals were injected with metyrapone (100 mg/kg) intraperitoneally and saline intracerebroventricularly. The rectal temperature was maintained at 37° to 38°C. The abdominal aorta was exposed after an incision was made in the abdomen with the animal in the supine position. Sixty minutes after the drug or saline administration, 1 mL of arterial blood was collected through the abdominal aorta to analyze the serum glucose, electrolytes, hemoglobin, hematocrit, and arterial blood gas levels according to routine laboratory procedures (blood glucose testing system by electrode, MPG01; Daikin, Osaka, Japan; ABL505, Radiometer, Copenhagen, Denmark).
Experiment 2: In vitro measurement of [Ca2+]i
Gerbils were anesthetized with ether and decapitated. The hippocampi were rapidly removed and placed in an ice-cold physiologic medium containing (in mmol/L) NaCl, 124; KCl, 5; CaCl2, 2; MgCl2, 2; NaH2PO4, 1.25; NaHCO3, 26; and glucose, 10. Hippocampal transverse slices approximately 300-μm thick were cut with a vibrating slicer (DTK-1000; Dosaka Co., Kyoto, Japan); two to three slices were obtained from each hippocampus. Slices were incubated in physiologic medium equilibrated with a 95% O2, 5% CO2 gas mixture for 60 minutes at 26°C. Slices were preloaded with a fluorescent indicator, rhod-2 acetoxy-methyl ester (Dojin, Kumamoto, Japan), which was diluted to 20 μmol/L in the physiologic medium and equilibrated with a 95% O2, 5% CO2 gas mixture for 45 minutes at 26°C. After the loading incubation, slices in the dexamethasone group were incubated in physiologic medium containing 120 μg/L dexamethasone for 60 minutes at 26°C. Control slices were incubated further in physiologic medium for 60 minutes.
The [Ca2+]i levels were measured using an inverted fluorescence microscope, a high-performance video camera, and an image-processor setup. A low-magnification objective lens (×4) and a side illumination system were used to visualize the fluorescent image of the slice. The slice was transferred to a flow-through chamber (volume ~0.2 mL) mounted on the fluorescence microscope that was equipped with a heat plate stage (IMT2; Olympus) and superfused at 3 mL/min with the appropriate medium at 36.5°C. The temperature of the medium in the chamber was monitored using a thermocouple needle probe (0.4-mm diameter; TN-800; Unique Medical Corp.) and a thermocouple meter (TME-300; Unique Medical Corp.). The slice was excited with 550-nm light produced by an ultraviolet (UV) lamp (100 W; Osram, Munich, Germany), filtered by an interference filter (550 nm, band width < 16 nm) and conducted to the slice through an optic fiber (5-mm diameter). The fluorescence signals (>580 nm) were captured on an SIT (silicon-intensified target) camera (C2400-8; Hamamatsu Photonics, Hamamatsu, Japan) and processed using an image processor (Argus-100; Hamamatsu Photonics).
Before the measurement of [Ca2+]i, the slice loaded with rhod-2 was excited with 550-nm light, and the picture (on a TV monitor) was examined to confirm that the dye was uniformly distributed throughout the slice. After placement of the slice in the chamber, the slice was perfused with normoxic medium (physiologic medium equilibrated with a 95% O2, 5% CO2 gas mixture) for 15 minutes, and the [Ca2+]i in the preischemic state was measured. In vitro hypoxia was induced by switching the normoxic medium to a glucose-free ischemialike medium equilibrated with a 95% N2, 5% CO2 gas mixture. The fluorescence intensity was measured in the image after the induction of in vitro hypoxia. The numerical values (pixels) were then divided by the value of the corresponding element that had been taken before the measurement. Thus, the ratio of [Ca2+]i was obtained every 10 seconds. To investigate the effect of dexamethasone, the slice pretreated with dexamethasone was perfused in the chamber with normoxic medium containing 120 μg/L dexamethasone. In vitro hypoxia was induced by switching the normoxic medium to a glucose-free ischemialike medium containing dexamethasone.
The effect of in vitro hypoxia in a condition free from extracellular Ca2+ was examined. First, the slice was perfused with Ca2+-containing normoxic medium for 15 minutes after placement of the slice in the chamber, and then the medium was changed to Ca2+-free normoxic medium that had been prepared by replacing CaCl2 with MgCl2. After 10 minutes, the medium was switched to Ca2+-free ischemialike medium. To investigate the effect of dexamethasone in a Ca2+-free condition, the slice pretreated with dexamethasone was perfused in the chamber with Ca2+-containing normoxic medium with dexamethasone, and then the medium was changed to Ca2+-free normoxic medium containing dexamethasone. After 10 minutes, the medium was switched to Ca2+-free ischemialike medium with dexamethasone.
Drugs and chemicals
Dexamethasone-water soluble and metyrapone were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Halothane was obtained from Takeda Chemical Industries (Osaka, Japan). Sodium pentobarbital was purchased from Abbott Laboratories (North Chicago, IL, U.S.A.). Other chemicals were all of reagent grade. The dose of dexamethasone-water soluble is expressed as the weight of the free base.
Statistical analysis
The data obtained from measuring the DC potential and the data on physiologic variables were analyzed by analysis of variance followed by Dunnett's test. The data from the histology were evaluated with the Kruskal-Wallis test followed by the Mann-Whitney test. The data from the fluorometry were analyzed using repeated measures two-way analysis of variance to detect differences among groups. When differences were found, Scheffé's test was used post hoc to compare each value with that in the control group.
RESULTS
Table 1 shows the physiologic variables from the in vivo experiment. There were no differences in any of the physiologic variables between the controls and the gerbils given dexamethasone or metyrapone.
Physiologic variables in control, dexamethasone-, and metyrapone-treated gerbils
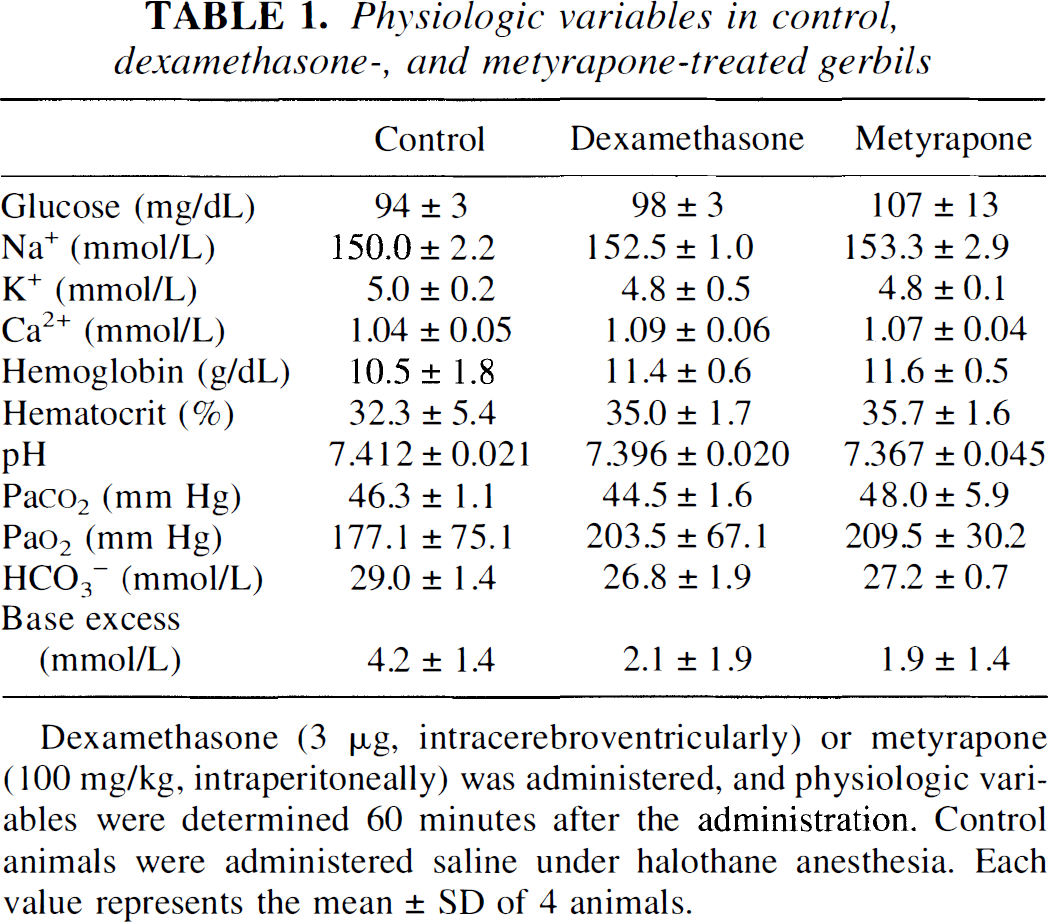
Dexamethasone (3 μg, intracerebroventricularly) or metyrapone (100 mg/kg, intraperitoneally) was administered, and physiologic variables were determined 60 minutes after the administration. Control animals were administered saline under halothane anesthesia. Each value represents the mean ± SD of 4 animals.
As shown in Fig. 1, forebrain ischemia provoked a sudden shift in the extracellular membrane potential in the hippocampal CA1 area. In control animals, the DC potential shift was observed at an onset latency of 70 ± 11 seconds (mean ± SD, n = 5), and the amplitude of the potential attained was 21.2 ± 3.2 mV. The control group recovery time as measured by the duration between the start of the reflow and the point at half-maximal amplitude was 68 ± 11 seconds. Administration of dexamethasone produced a 22% reduction of the onset latency of the DC potential shift. In contrast, pretreatment with metyrapone tended to prolong the onset latency, but the effect was not significant. The maximal amplitude of the DC potential shift and the recovery time of the depolarization revealed no significant differences between the three groups.
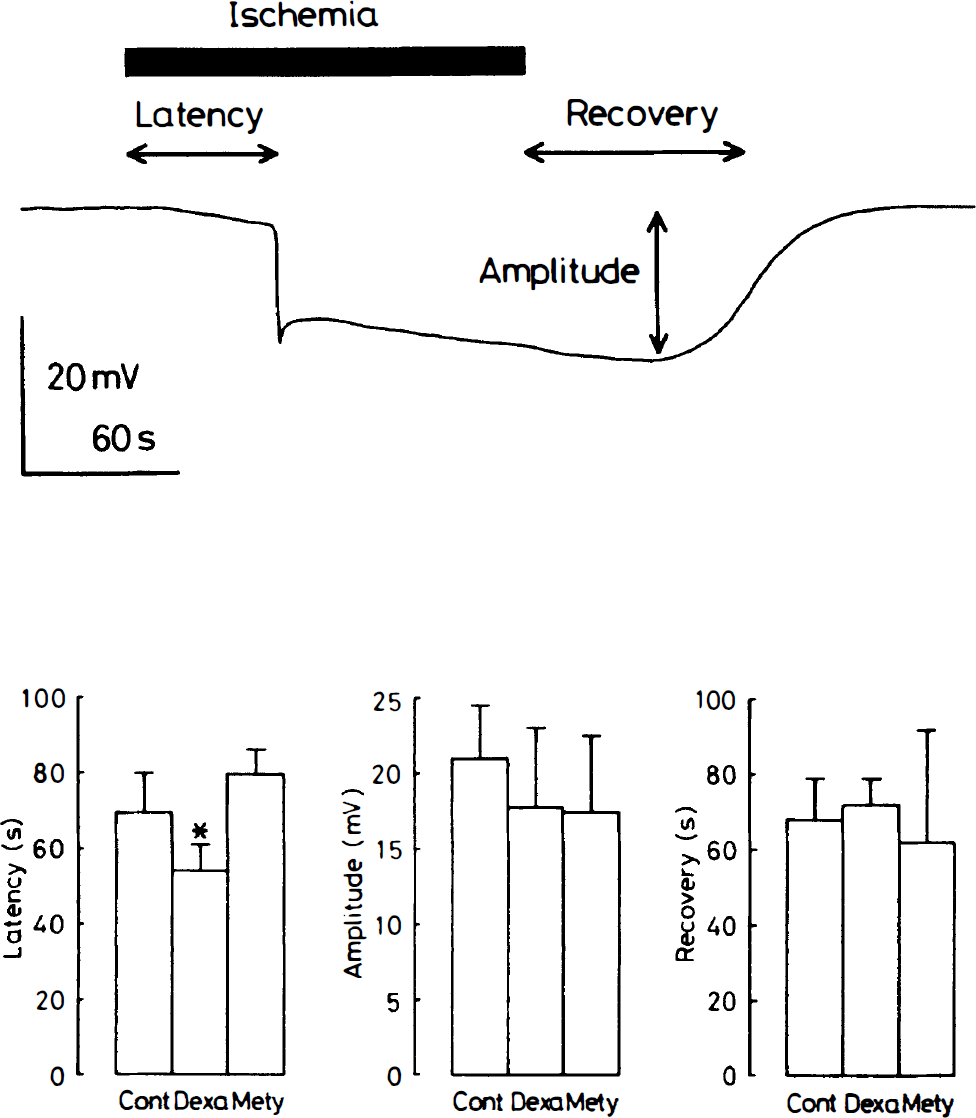
Effects of dexamethasone (3 μg, intracerebroventricularly) or metyrapone (100 mg/kg, intraperitoneally) administered 60 minutes before ischemia on anoxic depolarization (AD). The AD was analyzed regarding its onset latency, amplitude, and recovery time. Each column represents the mean ± SD of 5 animals. Cont, saline-injected control group; Dexa, dexamethasone-injected group; Mety, metyrapone-injected group. *P < 0.05 compared with the respective values in the control group.
Animals in the control group regained consciousness and the righting reflex within 30 minutes after halothane anesthesia was stopped. Similar to the control animals, animals treated with dexamethasone or metyrapone recovered from anesthesia within 30 minutes. No seizures were noted in the three groups in the 7-day period between ischemia and sacrifice.
Figure 2 shows the extent of neuronal damage in the hippocampal CA1 region in each group. In most of the animals of the control group subjected to forebrain ischemia for 2.5 minutes, the pyramidal cells were slightly degenerated after 7 days. In contrast, intracerebroventricular administration of dexamethasone 60 minutes before ischemia significantly aggravated the damage. Most of the neurons were preserved by preischemic treatment with metyrapone.
Figure 3 comprises photographs showing the elevation of [Ca2+]i in hippocampal slices induced by the in vitro ischemialike conditions.
As shown in Fig. 4, when the hippocampal slices were perfused with in vitro ischemialike medium, almost no increase in the ratio of [Ca2+]i was observed in the CA1 field within 250 seconds after the beginning of in vitro ischemia. Subsequently, an acute and large increase in [Ca2+]i spread throughout the CA1 field, and the ratio reached a plateau. The latency of the onset was 311 ± 25 seconds (n = 8). Then, 600 seconds later, the [Ca2+]i ratio reached 3.2 times the value observed at the beginning of in vitro ischemialike condition. When slices treated with dexamethasone (120 μg/L) were examined, the latency of onset of the increase in [Ca2+]i was significantly reduced (P < 0.001), the value being 249 ± 23 seconds (n = 8). The [Ca2+]i ratio after 600 seconds attained the same level as that in the control slices.
When slices were perfused with Ca2+-free in vitro ischemialike medium, an increase in [Ca2+]i was still observed in the CA1 field, although it was more gradual and occurred to a lesser extent compared with the increase observed in the Ca2+-containing condition. In contrast, in dexamethasone-treated slices, perfusion with Ca2+-free ischemialike medium produced an increase in [Ca2+]i earlier than in Ca2+-free control slices, and the ratio of fluorescence intensity after 600 seconds was larger than that in Ca2+-free controls.
DISCUSSION
In the present study, we observed a deleterious effect of dexamethasone on hippocampal neurons in vivo ischemia, and an improvement of ischemic neuronal damage by the blockade of endogenous glucocorticoids with metyrapone. Further, we observed advanced initiation of a hypoxia-induced increase in [Ca2+]i in the in vitro experiment.
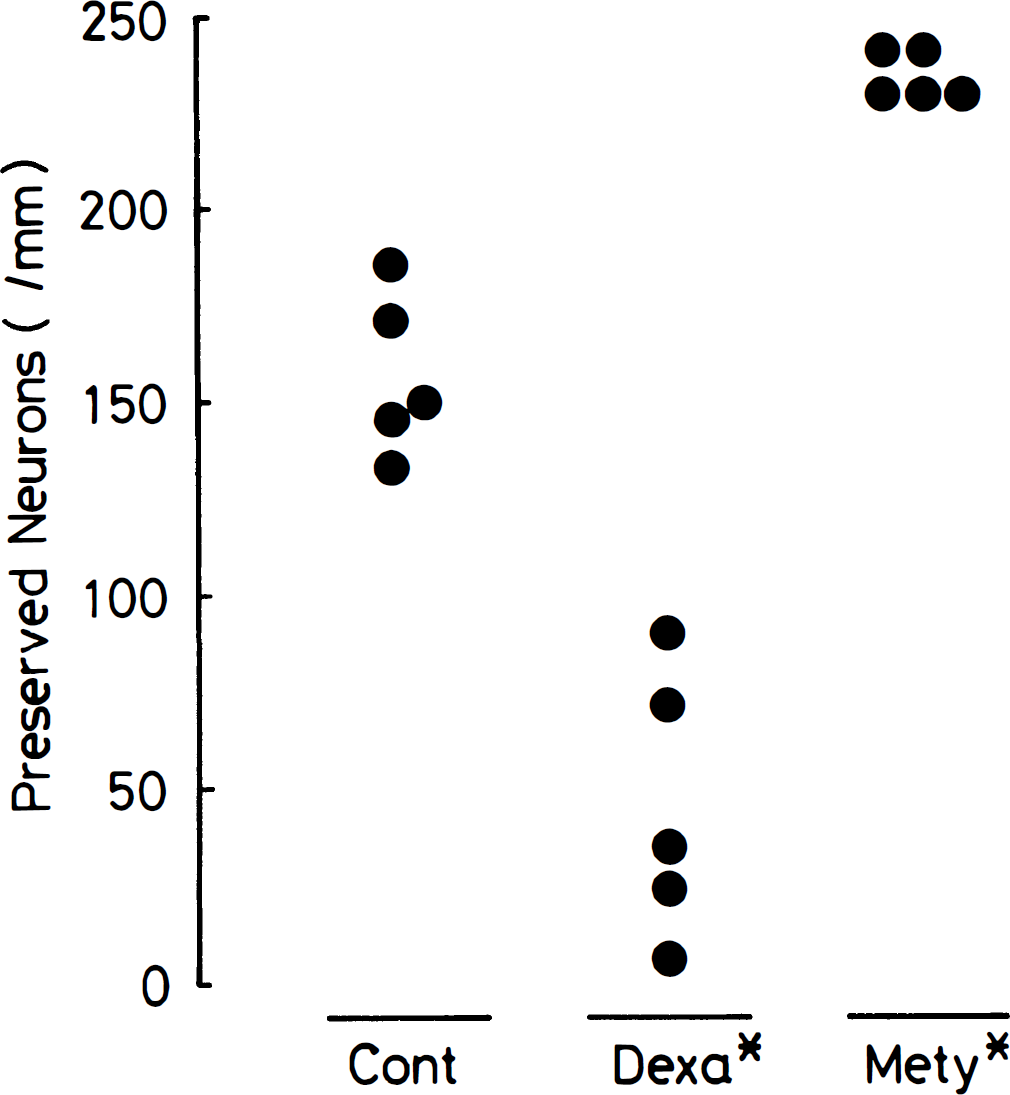
Effects of dexamethasone (3 μg, intracerebroventricularly) or metyrapone (100 mg/kg, intraperitoneally) administered 60 minutes before ischemia on delayed neuronal death of CA1 pyramidal cells. CA1 pyramidal cells were examined 7 days after 2.5-minutes' ischemia, and the number of pyramidal cells (ordinate) was determined. Values obtained from individual animals are shown. Cont, saline-injected control group; Dexa, dexamethasone-injected group; Mety, metyrapone-injected group. *P < 0.01 compared with the control group.
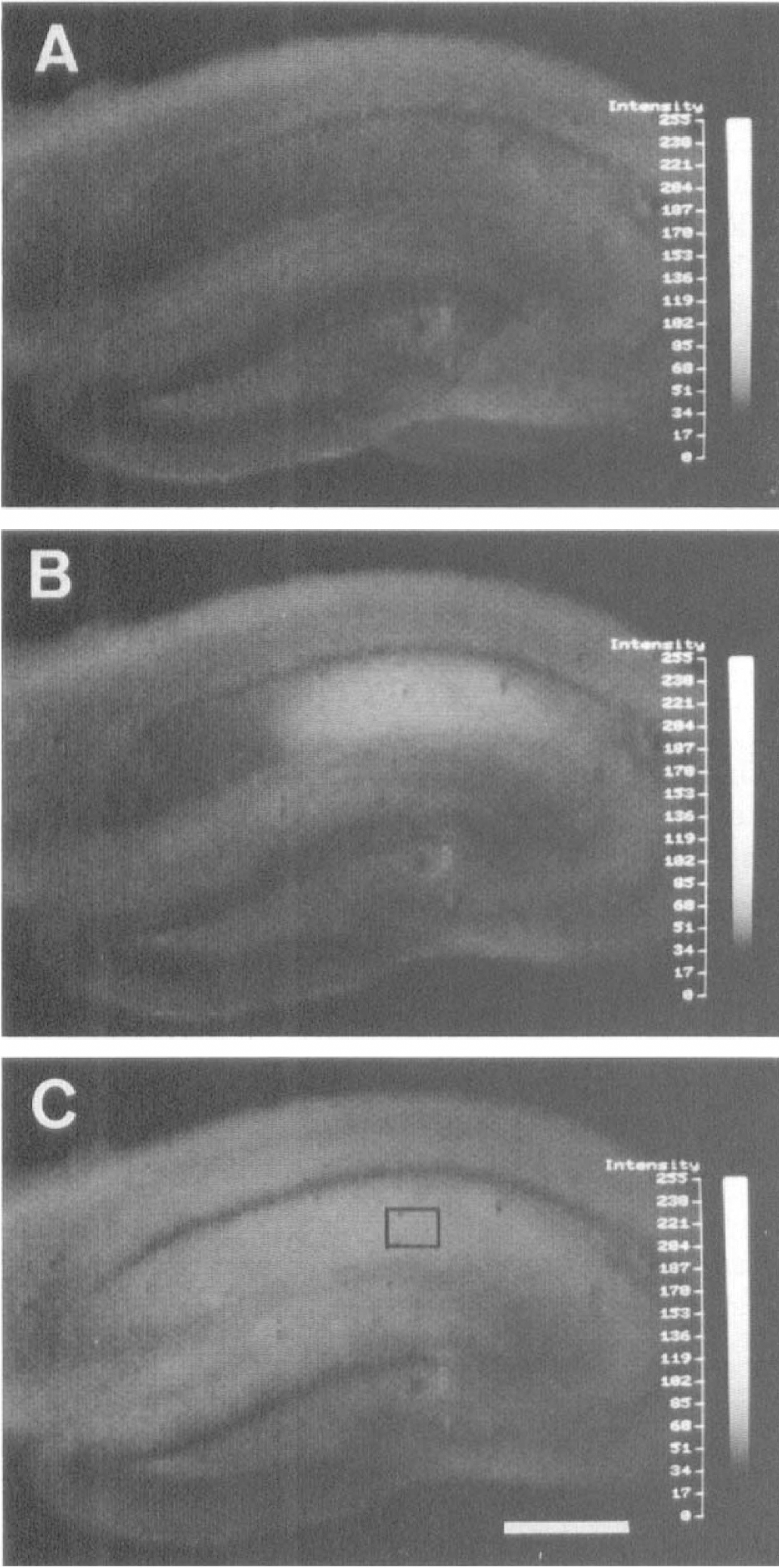
Photographs showing the [Ca2+]i elevation induced by in vitro hypoxia in a control hippocampal slice 200 seconds (
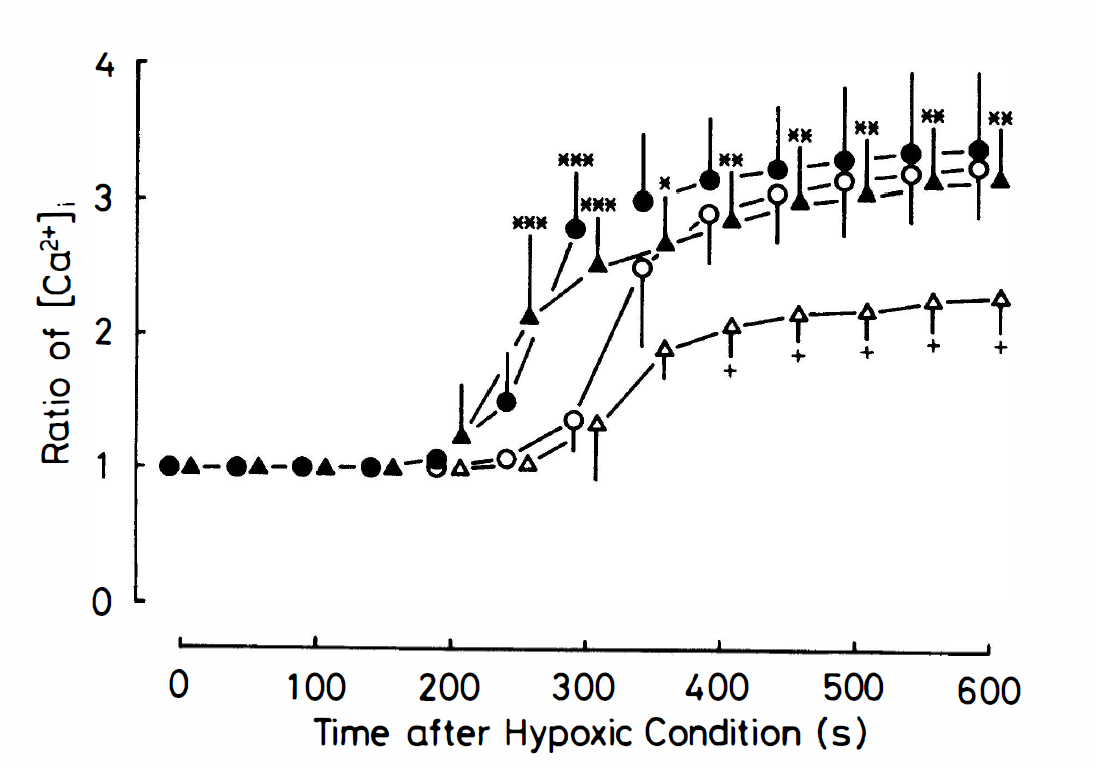
Changes in the ratio of [Ca2+]i in slices of the gerbil hippocampal CA1 field that underwent in vitro ischemialike conditions. Each value represents the mean ± SD of 8 slices for control group (○), dexamethasone (120 μg/L)-treated group (•), Ca2+-free control group (△), and Ca2+-free dexamethasone (120 μg/L)-treated group (▴). *P < 0.05, **P < 0.01, ***P < 0.001 compared with the respective values in each corresponding control group. +P < 0.01 compared with the respective values in the Ca2+-containing control group.
The extracellular DC potential shift closely reflects the movement of Na+, K+, Cl−, and Ca2+ across the membrane (Hansen, 1985). Because depletion of ATP during ischemia causes the sudden depolarization of the neuronal membrane as a result of insufficient ion transport, the latency from the start of ischemia to the sudden depolarization represents the duration of ATP consumption in the CNS. Therefore, the reduction of the onset latency by dexamethasone observed in this study indicates the possibility of an accelerated consumption of ATP or a decrease in the ATP store in the CNS. The latter mechanism is likely, because glucocorticoids have been shown to inhibit glucose uptake into hippocampal neurons and glia both in vitro and in vivo by ~30% in a nonischemic state (Kadekaro et al., 1988; Horner et al., 1990; Virgin et al., 1991).
In the histologic observation after 7 days, transient forebrain ischemia for 2.5 minutes at a brain temperature of 37.5°C produced mild damage in the hippocampal CA1 region in control animals. This result is in good agreement with a previous report (Mitani et al., 1991). Intracerebroventricular administration of dexamethasone 60 minutes before ischemia markedly aggravated this injury, whereas pretreatment with metyrapone ameliorated the damage. The CA1 area is innervated by glutamatergic fibers (Storm-Mathisen, 1981), which cause neuronal death by their excess release of glutamate during ischemia (Jørgensen and Diemer, 1982). Because a large amount of glutamate is released in the abrupt phase of AD, the facilitation of the beginning of this phase by dexamethasone seems to indicate an enhanced glutamate release. Further, glucocorticoids have been reported to exacerbate neuronal injury produced by kainic acid, which damages neurons by its structural similarity to glutamate (Stein-Behrens et al., 1992). In any case, because neuronal death is caused by a depletion of energy substrates or excessive demands for energy, glucocorticoids may make neurons relatively energy-depleted, and thus more vulnerable to excitotoxins.
Concerning the histologic improvement by metyrapone, the finding in the current study is consistent with previous reports that metyrapone reduced the brain injury induced by ischemia or kainic acid in rats (Smith-Swintosky et al., 1996; Stein and Sapolsky, 1988). Because metyrapone is a potent and rapid inhibitor of glucocorticoid synthesis and secretion, endogenous glucocorticoids seem to take part in the development of ischemic neuronal damage. Further, because the plasma concentration of corticosterone is markedly increased by cerebral ischemia (Fassbender et al., 1994), blockade of the increased action of corticosterone in the postischemic period as well as in an ischemic event may contribute to the improvement.
Glucocorticoids increase the plasma concentration of glucose (Dearden et al., 1986; Hall et al., 1994), and hyperglycemia is known to exacerbate ischemic neuronal injury (Ginsberg et al., 1980; D'Alecy et al., 1986; Pulsinelli et al., 1982; Woo et al., 1988). This aggravation is speculated to be a result of intraneuronal lactic acidosis associated with enhanced anaerobic glucose metabolism (Nedergaard et al., 1991). However, in the current study, the plasma concentration of glucose did not differ between the control group and the dexamethasone-treated group. Therefore, it is not likely that a difference in the plasma concentration of glucose affected the histologic outcome, although the current value represents a preischemic state. Another element specific to dexamethasone besides hyperglycemia seems to have provided the deleterious effect. There is a report that systemic administration of dexamethasone (2 mg/kg) three times before ischemia aggravated the ischemic damage provoked by transient forebrain ischemia in rats in both functional and histologic aspects (Koide et al., 1986). Although neuronal damage in that animal model was reversed by achieving normoglycemia with preischemic insulin treatment, insulin treatment did not completely improve the histologic outcome (Wass et al., 1996). These findings also appear to show another deleterious factor besides hyperglycemia. Inhibitions of glucose uptake, protein synthesis, and cell division are conceivable as mechanisms of the acute effect of dexamethasone (Munck et al., 1984), whereas chronic administration may harm neurons by modifying nucleic acids through steroid receptors in the nucleus.
A sudden and large increase in [Ca2+]i in the CA1 area was observed in the in vitro experiment. This finding reflects the selective vulnerability in this area, because the elevation of [Ca2+]i in postsynaptic neurons after ischemia is regarded as a crucial factor in the development of neuronal damage (Rothman and Olney, 1987). The reduction of the onset latency of the increase in [Ca2+]i by dexamethasone treatment is in good agreement with the facilitation of AD observed in the measurement of the DC potential shift. In another study, glucocorticoids were shown to increase basal [Ca2+]i in a nonischemic state, and this increase has been thought to be caused by impairment of the efflux of Ca2+ from the cytosol, rather than facilitation of its influx (Elliott and Sapolsky, 1993). These findings imply that aggravation of ischemic neuronal damage by dexamethasone in the in vivo ischemia is a result of an increased [Ca2+]i because of the disruption of Ca2+ transport to the extracellular space owing to energy depletion. The neurodegenerative action of dexamethasone in ischemia in the in vivo experiment seems to be a result of these effects.
The elevation of cytosolic concentration of Ca2+ in ischemia is induced by both the influx of Ca2+ from the extracellular space and the release of Ca2+ from intracellular stores such as the endoplasmic reticulum and mitochondria. In this study, the increase in [Ca2+]i was still observed in the Ca2+-free hypoxic condition, although the extent was smaller than that in the Ca2+-containing condition. Therefore, it is speculated that Ca2+ released from intracellular stores, as well as Ca2+ from the extracellular space, plays an important role in the elevation of [Ca2+]i in ischemia. Because dexamethasone treatment reduced the onset latency of the increase and increased [Ca2+]i after 600 seconds, dexamethasone may contribute to the increase in [Ca2+]i by facilitating release of Ca2+ from intracellular stores.
Two mechanisms for release of Ca2+ from the endoplasmic reticulum to the cytosol have been clarified (Tsien and Tsien, 1990; Henzi and MacDermott, 1992). One is release through ryanodine receptors, which exist on the membrane of the endoplasmic reticulum. When Ca2+ flows into the cytosol from the extracellular space through some types of Ca2+ channels during an ischemic event, the increase in cytosolic Ca2+ stimulates ryanodine receptors, thereby releasing Ca2+ from intracellular Ca2+ stores (Ca2+-induced Ca2+ release). The other mechanism involves the inositol 1,4,5-triphosphate (IP3) receptors on the endoplasmic reticulum. Stimulation of postsynaptic metabotropic glutamate receptors enhances the activation of phospholipase C, resulting in facilitation of phosphatidylinositol (PI) turnover. Thus, IP3 receptor-linked Ca2+ channels open in response to the increase in intracellular concentration of IP3, which causes the release of Ca2+ (IP3-induced Ca2+ release). In the current study, the mechanism by which dexamethasone reduced the onset latency of the release of Ca2+ from intracellular stores was not made clear. However, because glutamate-induced PI hydrolysis has been shown to be inhibited by glucocorticoids (Kolasa et al., 1992), the mechanism of release from intracellular Ca2+ stores by Ca2+-induced Ca2+ release is more conceivable than that by IP3-induced Ca2+ release.
In this study, we observed a facilitating effect of glucocorticoids on the onset time of AD and the increase in [Ca2+]i in ischemia. Because the increase in [Ca2+]i in the early stage of ischemia plays an important role in the cascade leading to neuronal damage, the current findings contribute to our understanding of the neurodegenerative action of both endogenous and exogenous glucocorticoids on ischemic neuronal damage.