
Abstract
The widely prescribed drug desferrioxamine is a known activator of the hypoxia-inducible transcription factor 1 (HIF-1) and the subsequent transcription of erythropoietin. In the brain, HIF-1 is a master switch of the transcriptional response to hypoxia, whereas erythropoietin is a potent neuroprotectant. The authors show that desferrioxamine dose-dependently and time-dependently induces tolerance against focal cerebral ischemia in rats and mice, and against oxygen–glucose deprivation in purified cortical neurons. Desferrioxamine induced HIF-1 DNA binding and transcription of erythropoietin in vivo, the temporal kinetics of which were congruent with tolerance induction. Desferrioxamine is a promising drug for the induction of tolerance in humans when ischemia can be anticipated.
Tolerance to cerebral ischemia can be induced experimentally by a number of physical or pharmacologic strategies (i.e., ischemic preconditioning), such as hypoxia, hyperoxia, short ischemia, spreading depression, lipopolysaccharide, cytokines, respiratory chain inhibition (Kitagawa et al., 1990; Kawahara et al., 1997; Nawashiro et al., 1997; Riepe et al., 1997; Tasaki et al., 1997; Gidday et al., 1999; Ginis et al., 1999; Prass et al., 1999; Wiegand et al., 1999). Ischemic tolerance of the brain exists in humans (Weih et al., 1999; Moncayo et al., 2000). Because there are a number of clinical conditions under which cerebral ischemia can be anticipated (e.g., surgery of the heart or brain, subarachnoid hemorrhage), it is desirable to develop clinically applicable approaches to induce an ischemia-tolerant state of the brain.
However, few of the published experimental protocols for ischemic preconditioning can be applied to humans. Desferrioxamine, an iron chelator that is licensed to treat hemochromatosis caused by iron overload from repeated blood transfusion in patients with various forms of anemia (Brittenham et al., 1994), increases hematocrit through the induction of the hypoxia-inducible transcription factor 1 (HIF-1) and transcription of the major stimulator of erythropoiesis, erythropoietin (Wang and Semenza, 1993). HIF-1 is a master switch of the response of the brain to hypoxia (Semenza, 2000), and erythropoietin is a potent neuroprotectant in vivo and in vitro (Morishita et al., 1997; Sadamoto et al., 1998; Sakanaka et al., 1998; Bernaudin et al., 1999; Brines et al., 2000; Sinor and Greenberg, 2000; Siren et al., 2001). In neonatal rats, desferrioxamine induced HIF-1 and protected against hypoxia and ischemia applied 24 hours after the intraperitoneal injection of the drug (Bergeron et al., 2000; Jones and Bergeron, 2001). The neonatal brain has important differences to the adult CNS with respect to sensitivity to substrate deprivation. In addition, the mechanisms of damage after global hypoxia are morphologically and mechanistically different from those after focal cerebral ischemia (stroke). Whether desferrioxamine can induce tolerance in the adult brain and protect against stroke is unknown. Therefore, we investigate whether desferrioxamine also preconditions the adult rodent brain, and whether desferrioxamine-induced tolerance is dependent on protein synthesis and correlates to HIF-1 DNA binding and erythropoietin transcription. In addition, we investigated whether desferrioxamine-induced tolerance requires effects of the drug on CBF or glial–neuronal communication by testing its effectiveness in pure cortical neurons in culture.
MATERIALS AND METHODS
All experiments were performed and quantified in a randomized fashion by investigators blinded to the treatment groups. The local authorities approved all surgical procedures. Middle cerebral artery occlusion (MCAO) with reperfusion was induced in male Wistar rats (BGVV, Berlin, Germany; 280 to 320 g body weight) and male and female Sv129/J mice (BGVV; 18 to 22 g body weight). Induction of MCAO in rats was performed using halothane anesthesia by reversible tandem occlusion of the distal middle cerebral artery (under microscopic control, with a steel hook attached to a micromanipulator through a small temporoparietal burr hole) and the ipsilateral common carotid artery for 90 minutes according to the method of Brint et al. (1988). Physiologic parameters (arterial blood pressure and gases after femoral and artery and vein cannulation, rectal body temperature) were measured and controlled within physiologic limits. Then, MCAO was induced in mice using an 8/0 nylon monofilament as described by Hara et al. (1996). Reperfusion was induced after 45 minutes by removing the monofilament using brief anesthesia. During surgery and 2 hours after reperfusion, body temperature was measured and maintained between 37.0°C and 37.5°C with a heating pad.
Infarct sizes were determined in rats 7 days after MCAO and in mice 2 days after MCAO using digital imaging processing of frozen brain sections stained with vanadium acid fuchsin according to the protocol of Victorov et al. (2000), or with hematoxylin staining, respectively. In the mouse experiments edema correction (calculating the indirect infarct volume as the volume of the contralateral hemisphere minus the noninfarcted volume of the ipsilateral hemisphere) did not yield different results from those of raw infarct volumes, which are reported here.
The HIF-1 DNA binding activity was quantified in rat brain tissue as described previously (Ruscher et al., 1998, 2000). Briefly, nuclear extracts were prepared and 1 pmol Cy5-labeled specific probe (5′-Cy5-AGTTGAGGGGACTTTCCCAGGC-3′) was incubated with 25 μg nuclear extract protein in binding buffer (20 mmol/L HEPES, 50 mmol/L potassium chloride, 1 mmol/L ethylene diamine tetraacetic acid, 1 mmol/L dithiothreitol, 25 ng poly(dI)·poly(dC), 10% glycerol) for 15 minutes at room temperature. Specificity was confirmed by the addition of a 50-fold excess of unlabeled specific competitor (specific probes without Cy5 label) or unlabeled nonspecific competitor (5′-GACGTATGAGTCAGTCCA-3′). The HIF-1 supershifts were performed using a polyclonal antibody against HIF-1α (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.). The mixture was separated on a nondenaturing polyacrylamide gel at 4°C using an external temperature-regulated ALFexpress (Amersham Pharmacia Biotech, Freiburg, Germany) DNA sequencer. Gels were analyzed directly by ALFwin and Allelix software (Amersham Pharmacia Biotech) as described previously (Ruscher et al., 2000).
Transcriptional regulation of erythropoietin was measured in rat brain tissue with a semiquantitative competitive reverse transcription polymerase chain reaction (PCR) approach using the housekeeping gene β-actin as internal control, and deletion standards as external controls (Freyer et al., 1999). Briefly, total cellular RNA was isolated using trizol reagent (Life Technology, Karlsruhe, Germany), contaminating DNA was removed with DNase (Promega, Madison, WI, U.S.A.), and extracted RNA was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Promega). Deletion fragments of rat β-actin and erythropoietin were used as external standards for quantification of PCR products. A deletion fragment of rat β-actin was constructed and competitive β-actin PCR was performed as described previously (Freyer et al., 1999). A deletion fragment of rat erythropoietin was constructed by PCR using the primer 5′-AGCTCTGGGAAGTTCTTCTGG-3′ and the 42mer primer 5′-AGCTCTGGGAAGTTCTTCTGGAGTGTCCGTAGGACAGGCCGG-3′ (bases 3410-3478) with an internal gap of 21 nucleotides. Subsequently, the erythropoietin deletion fragment was cloned in pCRII-vector (Invitrogen, Karlsruhe, Germany) and confirmed by a cycle-sequencing procedure (Amersham Pharmacia Biotech). Competitive erythropoietin PCR was performed with forward primer 5′-Cy5-AGCTCTGGGAAGTTCTTCTGG-3′ and reverse primer 5′-TACGTAGCCTCACTTCACTGC-3′ spanning one intron and yielding a PCR product of 371 bp. The respective deletion fragment was 350-bp long. Competitive PCR was performed by amplifying a constant amount of probe complimentary DNA and a serial-diluted DNA of the respective deletion standard. The PCR reaction mixture contained 5 μl 10-fold PCR buffer, 1.5 mmol/L magnesium chloride, 0.25 mmol/L dNTPs (2′-deoxynucleoside-5′-triphospates), 0.5 μmol/L of each primer, and 2.5 U Taq DNA-polymerase (Perkin-Elmer Applied Biosystems, Weiterstadt, Germany) in a final 50-μL volume. Amplification for erythropoietin was performed by running 40 cycles under the following conditions: denaturation at 95°C for 5 minutes, annealing at 65°C for 60 seconds, and extension at 72°C for 60 seconds. The forward primer was Cy5-labeled to allow for a direct quantification of PCR products. The resulting two Cy5-labeled PCR products, one of the amplified deletion standard and one of the amplified target complimentary DNA, were analyzed and quantified by fluorescence detection using the ALFexpress DNA sequencer. The initial amount of target cDNA was calculated as described (Freyer et al., 1999).
Primary neuronal cultures of cerebral cortex were obtained from embryos (E16–E18) of Wistar rats (Bundesinstitut für gesundheitlichen Verbraucherschutz und Veterinärmedizin, Berlin, Germany). Cultures were prepared by a modified protocol (Katchanov et al., 2001) according to Brewer (1995). The cultures comprising less than 10% astroglial cells were used for experiments after 8 days in vitro. A 120-minute deprivation of oxygen and glucose (OGD) was induced in an anaerobic (P
RESULTS
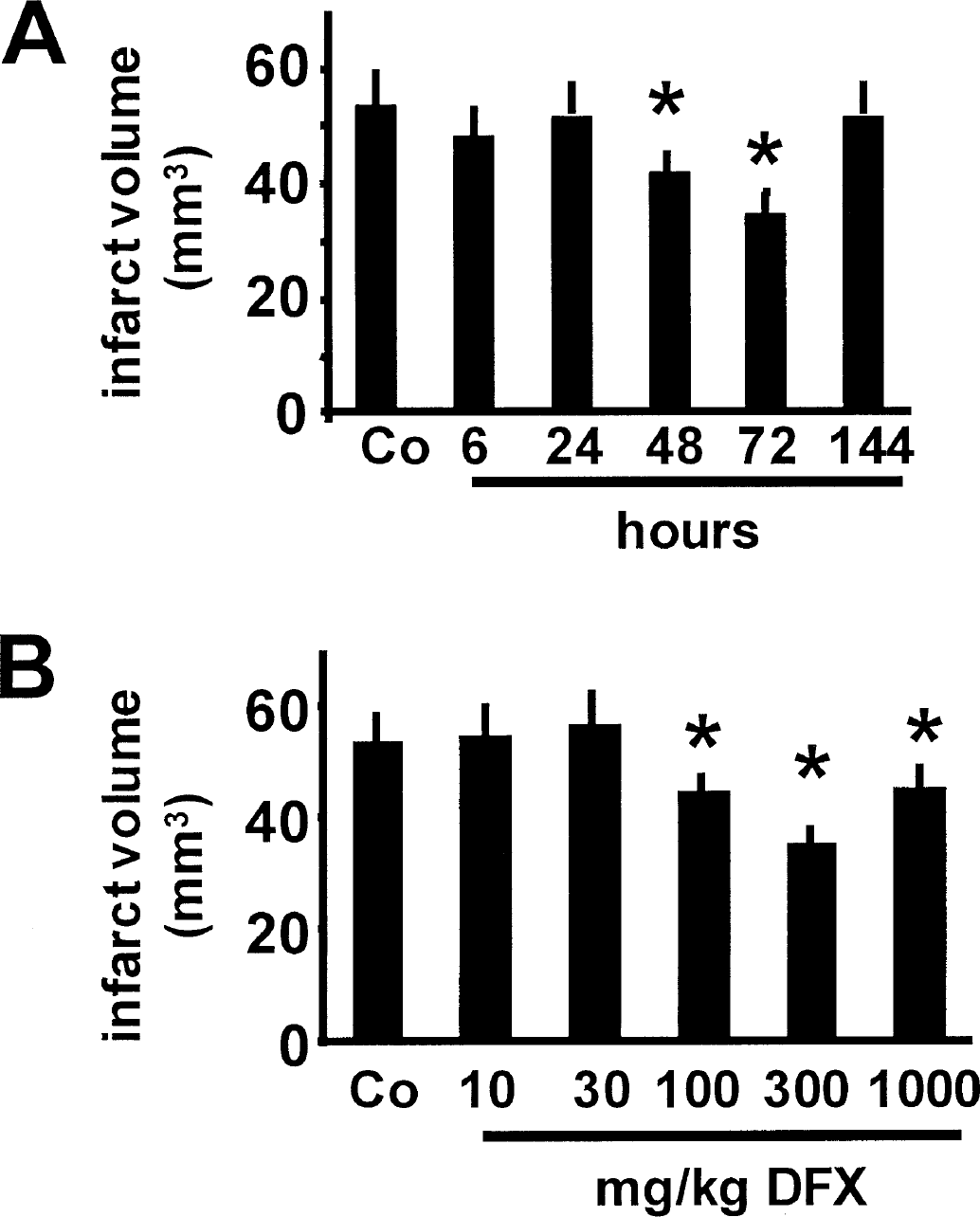
In a murine model of mild focal cerebral ischemia, intraperitoneal injection of 300 mg/kg body weight desferrioxamine (Sigma Chemical, Deisenhofen, Germany) induced statistically significant protection against 45-minute MCAO when desferrioxamine was given 48 or 72 hours before ischemic induction (Fig. 1A; infarct volume reduction, −22% and −35%, respectively). Tolerance was effectively induced by 100, 300, or 1,000 mg/kg desferrioxamine given intraperitoneally 72 hours before induction of MCAO (Fig. 1B; infarct volume reduction, −17%, −35%, and −16%, respectively).
The most effective dose (300 mg/kg body weight) was used for further experiments in rats. In a more severe model of ischemia and reperfusion, pretreating rats with desferrioxamine 72 hours before 90-minute MCAO reduced infarct volumes significantly by 40% (Fig. 2A). Coapplication of the protein synthesis inhibitor cycloheximide (1 mg/kg body weight, Sigma Chemicals) together with desferrioxamine, completely abrogated the preconditioning effect of desferrioxamine. Cycloheximide alone (1 mg/kg body weight) 72 hours before MCAO did not affect infarct volumes (Fig. 2A).
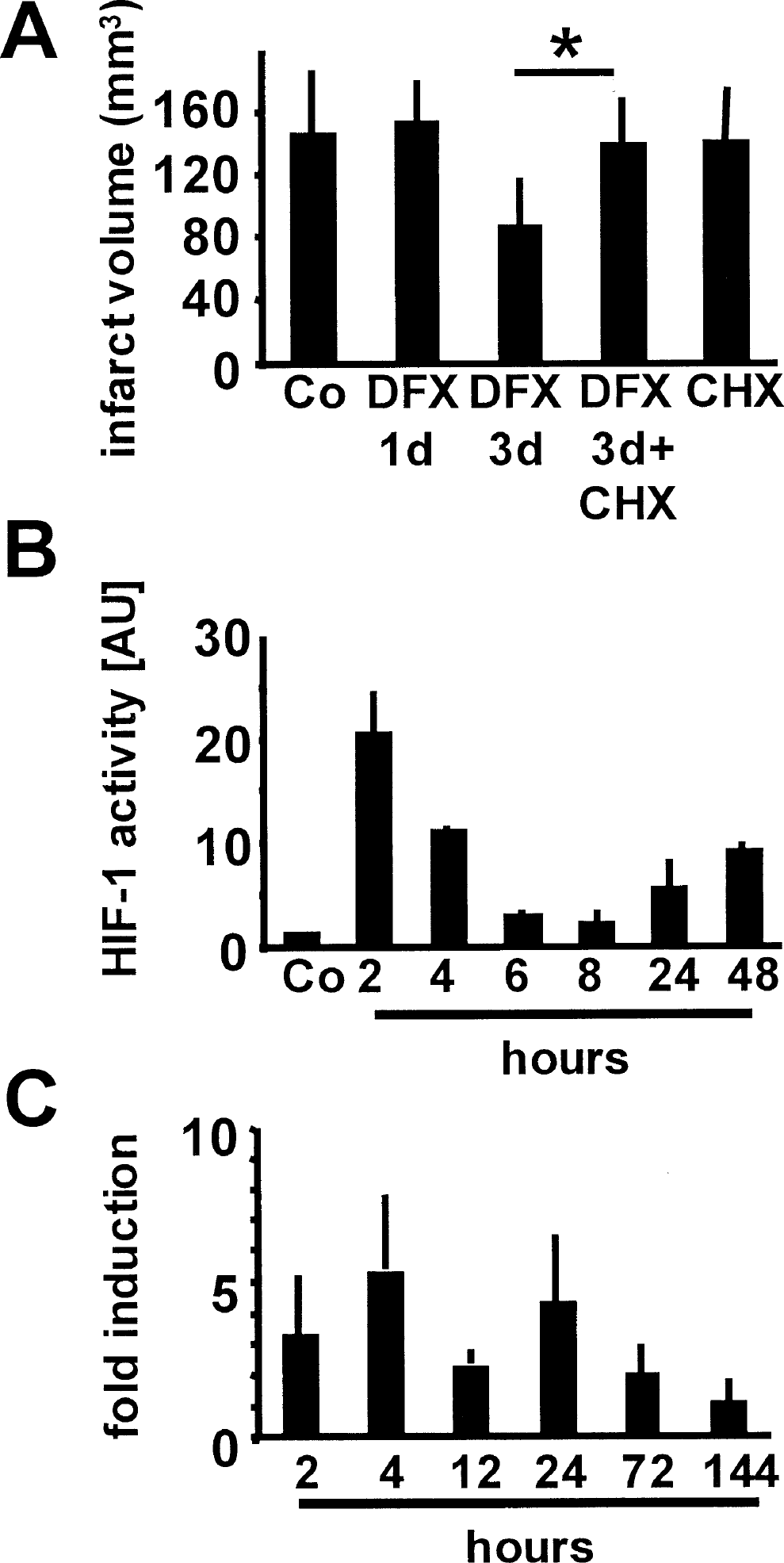
Time kinetics of intraperitoneal desferrioxamine-induced (300 mg/kg body weight) tolerance in the mouse. Numbers on the x-axis represent hours between desferrioxamine injection and the induction of middle cerebral artery occlusion (MCAO; n = 6–9), whereas the y-axis numbers represent infarct volume (mm3). *Significantly different from control MCAO.
Desferrioxamine (DFX)-induced tolerance in the rat. Interval between desferrioxamine injection and induction of MCAO was 72 hours. Intraperitoneal cycloheximide (CHX; 1 mg/kg body weight) given with intraperitoneal desferrioxamine (300 mg/kg body weight) completely abrogates tolerance induction. Intraperitoneal cycloheximide alone (1 mg/kg body weight) given 72 hours before MCAO induction had no effect on infarct volumes (n = 8–10). The y-axis represents infarct volume (mm3).
Fluorescent gel shift assays revealed that the tolerance-inducing dose of desferrioxamine rapidly induces DNA binding of HIF-1. Two hours after desferrioxamine application, HIF-1 DNA binding was increased 20-fold in the rat (Fig. 2B). HIF-1 is implicated in the transactivation of more than 40 genes, most of which are concerned with tissue protection and anaerobic metabolism. In the brain, erythropoietin appears to be of major importance in protective HIF-1 signaling (Ruscher et al. 1998; Zaman et al., 1999). Using a semiquantitative competitive PCR approach, 300 mg/kg body weight intraperitoneal desferrioxamine in the rat robustly induced transcription of erythropoietin (Fig. 2C).
The pathobiology of focal cerebral ischemia in vivo is complex and involves the interaction of destructive and protective signaling cascades (Dirnagl et al., 1999). The ability to model selected aspects of these cascades in vitro reduces complexity to reveal basic principles (Bruer et al., 1997; Grabb and Choi, 1999; Gonzalez-Zulueta et al., 2000). To show that the effect of desferrioxamine does not necessarily involve induction of changes in systemic circulatory parameters, vascular reactivity, or glia-neuron interactions, we tested whether desferrioxamine is also effective in inducing tolerance in neurons in culture. Similar to the in vivo condition, desferrioxamine time-dependently induced tolerance against OGD. The maximum protection (−47% LDH release) was obtained when desferrioxamine was applied to the culture medium 48 hours before OGD (Fig. 3A). The 72-hour interval was not tested because purified primary cortical neurons can only be kept in culture for a limited time. The most effective desferrioxamine concentration in the 48-hour interval was 150 μmol/L (Fig. 3B), and induction of protection was completely abrogated by co-application of the protein synthesis inhibitor cycloheximide (Fig. 3C). Thus, the in vitro effects of desferrioxamine were in complete agreement with the in vivo effects, indicating that desferrioxamine-induced ischemic tolerance might be explained, at least in part, by a neuronal effect of the compound.
DISCUSSION
The main findings of our study were that (1) desferrioxamine induced robust tolerance against reversible focal cerebral ischemia in two experimental stroke models in mice and rats; (2) desferrioxamine induced tolerance that is time-dependent and dose-dependent, with a maximum effect when given 3 days before ischemia induction at a dose of 300 mg/kg body weight; (3) desferrioxamine-induced protection involves de novo protein synthesis; (4) desferrioxamine induced a rapid and dramatic increase in the DNA binding of the transcription factor HIF-1, which is followed by the transactivation of erythropoietin; and (5) desferrioxamine-induced ischemic tolerance can be modeled in vitro in purified cortical neurons.
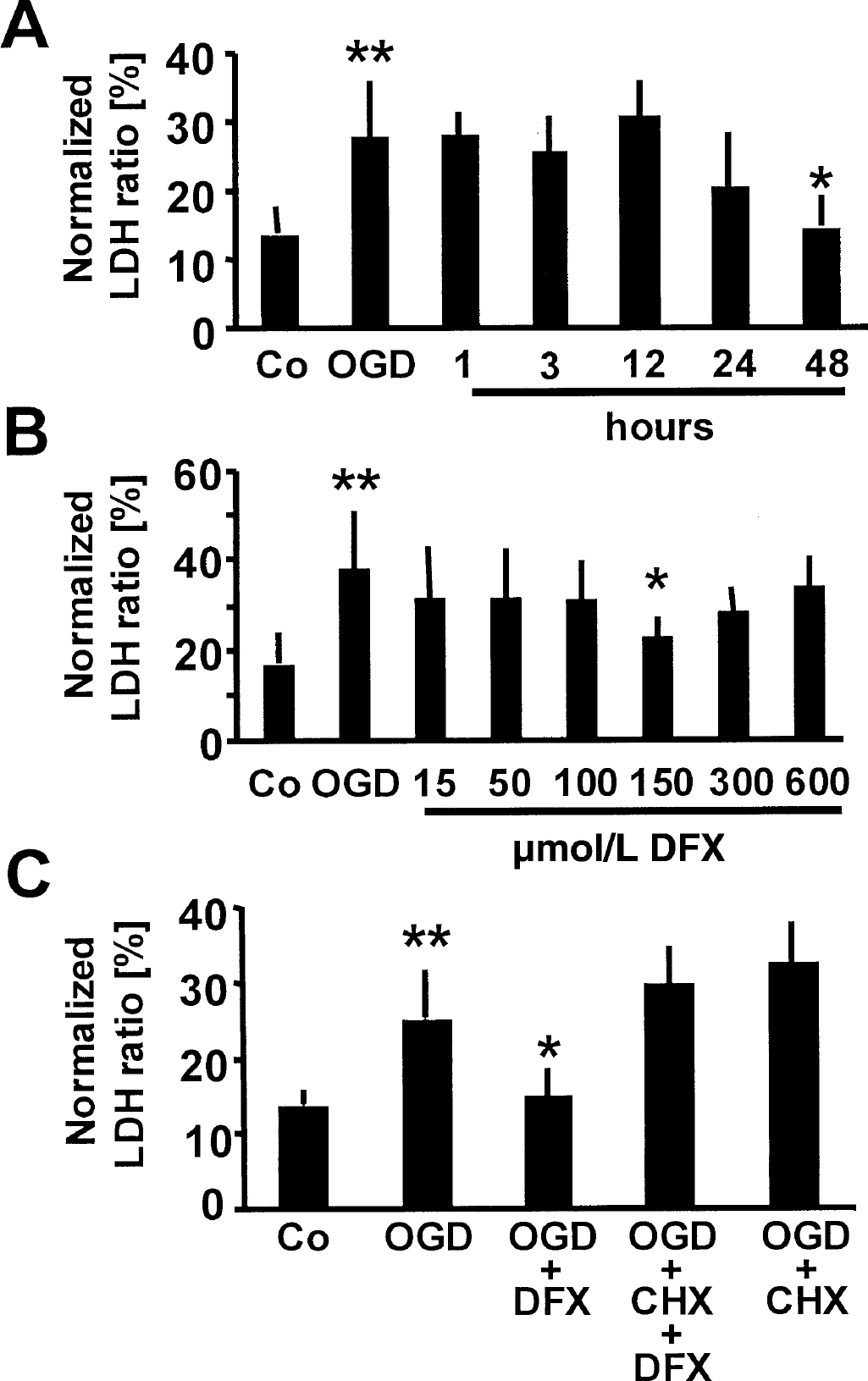
Desferrioxamine (150 μmol/L) time-dependently induces tolerance against oxygen–glucose deprivation (OGD; conditions pooled from three different experiments) in rat primary cortical neurons. The x-axis represents nonhypoxic controls, OGD indicates no desferrioxamine pretreatment, and numbers represent hours of desferrioxamine pretreatment before OGD.
How does desferrioxamine protect the brain? Our data are in agreement with the recent literature, which points at a sequence involving HIF-1 and erythropoietin. However, the evidence we present is correlational and does not functionally prove the relevance of this pathway, nor does it rule out other possible protective gene products. HIF-1 is involved in the regulation of glucose transporters and glycolytic enzymes, and regulates basic cellular proliferation and survival (Semenza, 2000). Of particular relevance is that HIF-1 induces receptors and cytokines needed for the growth and regulation of the vasculature, which was one of the reasons why we sought to confirm our in vivo data in the absence of vasculature and blood flow. However, one has to consider other possibilities by which desferrioxamine may promote cell survival. For example, as an iron chelator, desferrioxamine may block the formation of reactive oxygen species. Furthermore, desferrioxamine can block the cell cycle, and may thereby prevent neuronal cell death (Farinelli and Greene, 1996; Katchanov et al., 2001).
Recently, a number of investigators have used various in vivo and in vitro models to show that erythropoietin and its receptor are expressed in the brain and act as endogenous neuroprotectants (Masuda et al., 1994; Digicaylioglu et al., 1995; Marti et al., 1996; Morishita et al., 1997; Sadamoto et al., 1998; Sakanaka et al., 1998; Bernaudin et al., 1999, 2000), whereas the exogenous delivery of erythropoietin is a promising strategy for the management of stroke (Brines et al., 2000; Calapai et al., 2000; Siren et al., 2001). The effector proteins of erythropoietin-induced neuroprotection are currently under intense investigation, but a common motive seems to be a transcriptional or posttranslational antiapoptotic effect (Siren et al., 2001; Digicaylioglu and Lipton, 2001).
In humans, desferrioxamine is given in doses of up to several grams per day (Brittenham et al., 1994). In our rodent experiments, the most effective dose was 300 mg/kg body weight, which is approximately 10-fold higher that administered to humans. Because of the higher metabolism and excretion in rodents, it is not uncommon that equivalent doses (and unwanted side effects) are at least 10 times higher than in humans. Because desferrioxamine is a safe drug that does not affect systemic hematocrit after a single dose, it may be an interesting candidate for the induction of an ischemia-tolerant state of the brain in humans.