
Abstract
The authors have previously shown that bilirubin-oxidation products (BOXes) are present in CSF of subarachnoid hemorrhage patients with vasospasm, and that BOXes cause vasoconstriction in vitro. This study determined whether BOXes cause vasospasm in vivo. Identical volumes of either lysed blood or standardized amounts of BOXes were injected into the cisterna magna of adult rats. BOX injections caused 6 of 10 rats to die within 10 minutes, whereas 12 of 12 rats survived for 24 hours after blood injections. The mechanism for this significant (P ⩽ 0.01) increase in mortality was unclear. To directly test whether BOXes produced vasospasm, a cranial window technique was used. Application of 20 μL of 10-μmol/L bilirubin had little effect on the vessels. However, application of BOXes produced marked, dose-dependent small artery and arteriole vasospasm that approached a 90% decrease in diameter by 40 minutes after application in some vessels, and persisted for at least 24 hours. To determine if BOX-mediated vasospasm led to cortical injury, histology and immunocytochemistry were performed on animals that survived for 24 hours. There was a BOX-related stress protein response for HSP25 and HSP32 (HO-1) without evidence of infarction. The finding that the BOXes produce vasospasm of cerebral vessels in vivo, in conjunction with BOXes being found in CSF of vasospasm patients, supports our hypothesis that BOXes contribute to or cause cerebral vasospasm after subarachnoid hemorrhage.
Blood products have been implicated in the pathogenesis of subarachnoid hemorrhage (SAH)–induced cerebral vasospasm (Findlay et al., 1991; Zhang et al., 1996; Mayberg, 1998; Weir et al., 1999). Extracts of CSF from SAH (CSFSAH) patients with vasospasm have been reported to be vasoactive, and molecules contained within the CSFSAH have been proposed to be the cause of cerebral vasospasm (Cadoux-Hudson et al., 1999, 2001; Kranc et al., 2000; Pyne et al., 2001a,b). Interestingly, the time course for cerebral vasospasm after SAH closely correlates with the appearance of bilirubin in the CSF of patients with SAH, although bilirubin alone does not cause vasospasm (Duff et al., 1988; Trost et al., 1993). Therefore, we proposed that a breakdown product of bilirubin might contribute to SAH-induced cerebral vasospasm.
We have discovered three molecules in the CSF of SAH patients with vasospasm, and shown them to be oxidized forms of bilirubin (Kranc et al., 2000). These Bilirubin OXidation products (BOXes) produce vasospasm in vitro, as assessed by measuring metabolic and contractile changes of isolated vascular smooth muscle (Kranc et al., 2000). In this, and previous studies, we have defined vasospasm as a pathologic vasoconstriction including a failure to relax (Cadoux-Hudson et al., 1999, 2001; Clark et al., 2001; Pyne et al., 2001a,b). We have suggested that these molecules may play a role in SAH-induced cerebral vasospasm because they were present in the CSF of these patients and caused vasospasm in vitro. However, until now, we had not demonstrated that BOXes could cause vasospasm in vivo.
The aim of this work was to demonstrate that BOXes can cause cerebral vasospasm in vivo using a rat model. The data demonstrate a potent vasoconstriction of rat cerebral vessels and therefore support the hypothesis that BOXes may contribute to the vasospasm.
MATERIALS AND METHODS
Chemicals
All chemicals were reagent grade.
Bilirubin and BOXes preparation
The peroxidation of bilirubin was used to produce BOXes as described previously (Kranc et al., 2000). We have chosen to use the combination of the BOXes (BOX A, BOX B, and methyl-vinyl-maleimide) as produced by peroxidation of bilirubin because we believe that this is likely to be the most clinically relevant mixture of compounds present in the patients causing vasospasm. For this study, we have used at least three different batches of BOXes. We standardized the doses applied to the rats according to our previous methods (Kranc et al., 2000) and used these doses in this study.
Animals
Male Sprague Dawley rats (350 to 400 g, Charles River, Wilmington, MA, U.S.A.) were used. All surgical procedures were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the University of Cincinnati Animal Care and Use Committee.
Surgical methods
The anesthetic was 2.5% isoflurane with 70% nitrous oxide and 27.5% oxygen. It was administered via a nose cone to ensure deep sedation as verified by an absence of hind limb and forelimb pain reflexes as well as the absence of corneal reflexes. Normal, nonlabored breathing was maintained throughout the surgery. Temperature was monitored with a rectal probe, and body temperature was maintained at 37°C with the animal on a thermal blanket.
Mortality study
Following the methods of Turner et al. (1999), 150 μL of lysed blood was injected into the cisterna magna while animals were under general anesthesia. Whole blood was collected from a single donor animal for lysing. The donor animal was deeply anesthetized and blood was removed by cardiac puncture. The blood was subjected to 5 freeze-thaw cycles to lyse red cell membranes, aliquoted, and stored at −20°C until use.
Bilirubin oxidation product injections
BOXes were synthesized as described by Kranc et al. (2000). Fifty microliters of BOXes were diluted in 100 μL of sterile normal saline. BOX injections were performed using the same volumes and the same methods as were used for the lysed blood injections.
Cranial window
Anesthetized rats were placed in a stereotaxic frame (Kopf Instruments Tujunga, CA, U.S.A.). After a dorsal scalp incision and blunt dissection to remove loose connective tissue, a cranial window was produced beginning 1 mm lateral to bregma using aseptic technique. A 3-mm-wide (extending from 1 mm to 4 mm lateral to bregma) by 5-mm (extending from 2.5 mm anterior to 2.5 mm posterior to bregma) window was created and the surface vessels visualized. A standardized volume and concentration of BOXes or bilirubin (20 μL of a 10-μmol/L solution) was applied to the surface of the dura, or under the dura in specific cases. Using the Axiovision software (Carl Zeiss Inc., Thornwood, NY, U.S.A.) images were acquired before the addition of the BOXes or bilirubin and then every 2 to 3 minutes for approximately 1 hour.
After the procedure, the muscle and skin were sutured closed. Animals were allowed to recover on a warming pad with constant monitoring until fully recovered from the anesthesia, after which they were returned to their home cages.
Dose dependence
The relative dose-dependent effects of BOXes on vessel diameter were assessed by taking a saturated stock solution and diluting it by up to 1,000-fold. The dilutions were as follows: 1:1, 1:8, 1:16, 1:32, 1:100, 1:500, and 1:1,000. The vessel diameter was measured immediately before the application of the BOXes and at 40 minutes after application of the BOXes.
Vessel diameters
MCID imaging hardware and software (Imaging Research, Inc., Ontario, Canada) was used to image and measure changes in vessel diameters. Images of the surface vessels were taken during the surgical procedure, and were imported into MCID for diameter measurements. Images were acquired before the application of bilirubin or BOXes and then at various times after application of bilirubin or BOXes. Measurements were made on the same three or four vessels that were considered clear and unobstructed in the surgical field from each animal during the course of the surgery. The diameter of cerebral vessels was determined by measuring the number of pixels across the vessel diameter. Pixel calibration was accomplished by calibrating the number of pixels per 1 mm.
Histology
Twenty-four hours after the surgery, the rats were anesthetized again with isoflurane and placed in the stereotaxic frame. The craniotomy was opened and the vessels were visualized, imaged, and measured in the same manner as during the original surgery. The animals were removed from the stereotaxic frame and given ketamine (100 mg/kg), xylazine (20 mg/kg), and heparin intraperitoneally. Once the animals were deeply anesthetized, they were perfused with 0.9% saline and then 4% paraformaldehyde in 0.1-mol/L phosphate buffer through the ascending aorta. After perfusion, the brains were removed and placed in 30% sucrose in 0.1-mol/L phosphate buffer until they were sectioned for immunohistochemistry.
Immunohistochemistry was performed using standard avidin–biotin techniques (Turner et al., 1998, 1999). A monoclonal antibody to HSP25 (StressGen, Victoria, British Columbia, Canada), a monoclonal antibody to HSP32/HO-1 (StressGen), and a polyclonal HSP 32/HO-1 antibody (Turner et al., 1998) were used for the immunocytochemistry. Briefly, 50-μm sections were cut on a sliding microtome. These sections were blocked with blocking buffer containing 2% goat serum, 0.1% bovine serum albumin, and 0.3% Triton X-100 in 0.1-mol/L phosphate buffer. They were then incubated with the antibodies (HSP25 at 1:2,000 and HO-1 at 1:8,000) overnight at room temperature. The next day they were washed with phosphate-buffered saline and incubated with a biotinylated goat anti-mouse second antibody (Vector, Burlingame, CA, U.S.A.) for 2 hours. After additional phosphate-buffered saline washes, they were incubated in Vectastain Elite ABC kit using the immunoperoxidase system (Vector) for 112 hours. Sigma Fast diaminobenzidine tablets (Sigma, St. Louis, MO, U.S.A.) were added after three more phosphate-buffered saline washes to visualize the protein. Sections were washed three times, mounted, dried, and coverslipped.
Statistics
A P value of ⩽ 0.05 was considered significant. Fisher's exact test was used for determining statistical difference for the mortality study. An analysis of variance was used to determine differences of diameters in vessels exposed to BOXes versus those exposed to bilirubin.
RESULTS
The initial experiments assessed the effects of subarachnoid injections of BOXes. A standardized amount of the BOXes was injected into the cisterna magna of anesthetized rats and compared with rats injected with lysed blood. Identical volumes were injected in both groups to control for volume effects. In the group that received BOX injections, 6 of the 10 rats died within 10 minutes. In the group that received hemolyzed blood injections, none of the 12 (0 of 12) rats died. This is a significant increase in mortality (P < 0.01) caused by BOXes as determined with the Fisher's exact test. In the animals that survived for 24 hours after the cisternal injections of BOXes, there was no evidence of focal or global ischemic injury as judged by Nissl staining, or HSP70 and HO-1 immunohistochemistry on sections from the brainstem and the forebrain. Postmortem examination of the animals that received cisternal BOX injections (data not shown) showed evidence of subarachnoid hemorrhage and marked venous dilation. We postulated but could not prove that the BOXes caused diffuse arterial vasospasm with arterial-venous shunting, venous hypertension, and venous dilation, which may have resulted in secondary subarachnoid hemorrhage (Figs. 1 and 2).
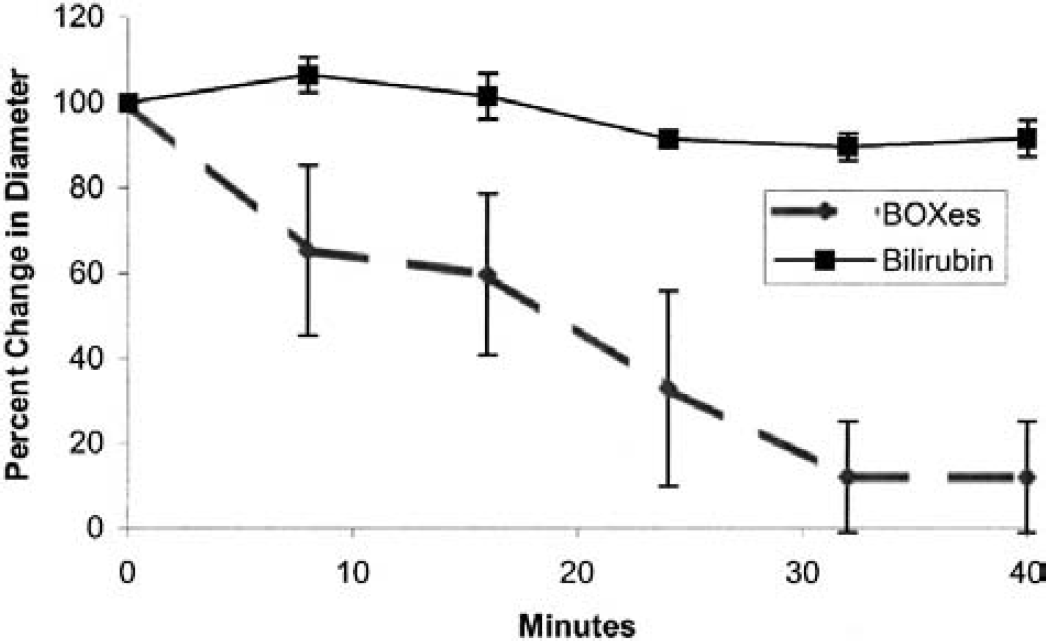
Bilirubin-oxidation products (BOXes) and bilirubin on arteries: the average percent change in vessel diameter caused by topical administration of BOXes and 10 μmol/L bilirubin to the surface of the brain. A significant vasoconstriction occurs after application of the BOXes. Eight vessels were examined from each BOXes- and bilirubin-treated rat with an average starting control diameter of 0.021 ± 0.0035 and 0.019 ± 0.0021 mm, respectively. (These diameters are indicative of surface arteries from seven and six rats, respectively). The data are normalized to the starting diameter of 100% to show the relative change in diameter.
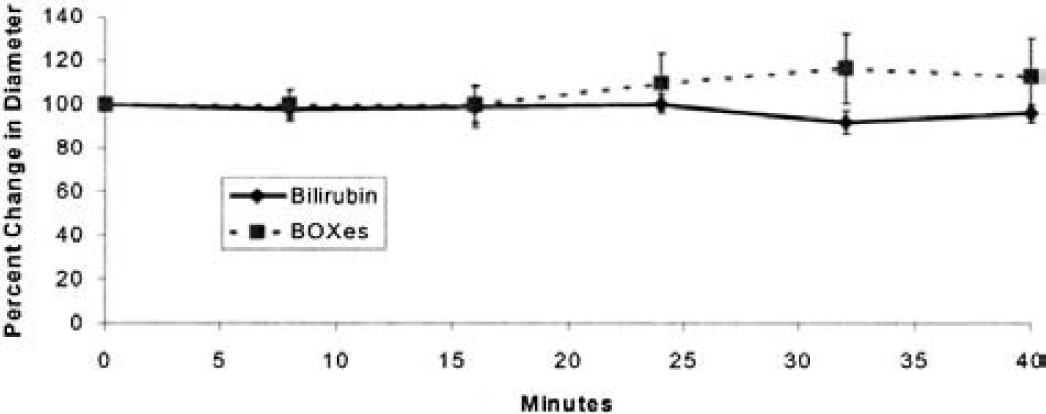
Bilirubin-oxidation products (BOXes) and bilirubin on veins: the average percent change in diameter caused by topical administration of BOXes compared with bilirubin to the surface veins of the brain. There is a small but statistically significant increase in diameter of the surface veins of the rats exposed to BOXes. Four veins were measured in each animal in each group, with an average starting control diameter of 0.095 ± 0.041 and 0.140 ± 0.098 mm for the bilirubin- and BOX-treated animals. These are the same rats that were analyzed in Fig. 1. The data are normalized to the starting diameter of 100% to show the relative change in diameter.
To directly test whether BOXes produced vasospasm, a cranial window technique was adopted. Application of bilirubin (10 μmol/L solution) to the surface of the dura had little effect on the vessel diameters as shown in Fig. 1. However, application of BOXes produced marked changes in the diameter of selected arteries (Fig. 1). There was a detectable decrease in vessel diameter by 7.5 minutes after BOXes application, and significantly more vasoconstriction occurring 32 to 40 minutes later (Fig. 1). Figure 3A shows the surface of the rat parietal cortex before any treatment. The largest vessels are likely veins, and most of the intermediate and smaller vessels are probably arteries. Figure 3B shows the same region 40 minutes after the addition of BOXes. The portions of the vessels shown with arrows in Fig. 3A can no longer be detected 40 minutes after application of the BOXes in Fig. 3B. Figure 3C demonstrates that these same vessels remain constricted 24 hours later. We assume that these vessels have remained in vasospasm for the 24-hour period and consider this to be prolonged vasospasm after a single application of the BOXes. Figure 3C also shows a decrease in the diameter of a majority of the vessels in the field (even after 24 hours), both in the larger arteries and in the small arteries seen in Fig. 3A. In contrast, the application of bilirubin produced little overall change in the caliber of the vessels in the cranial window (Figs. 4A and 4B). Note that there are some slightly smaller arteries after the application of bilirubin in Fig. 4B, and that there are some vessels that slightly increased their caliber (Fig. 4B) compared with the control field (Fig. 4A). Quantification of artery diameters after the application of bilirubin over time showed a small but nonsignificant decrease in vessel diameters (Fig. 1).
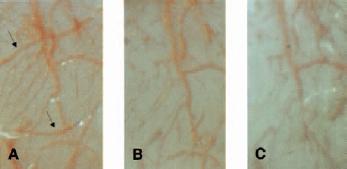
Micrographs of bilirubin-oxidation products (BOXes) causing vasospasm. BOXes application to surface vessels of brain in one representative rat.
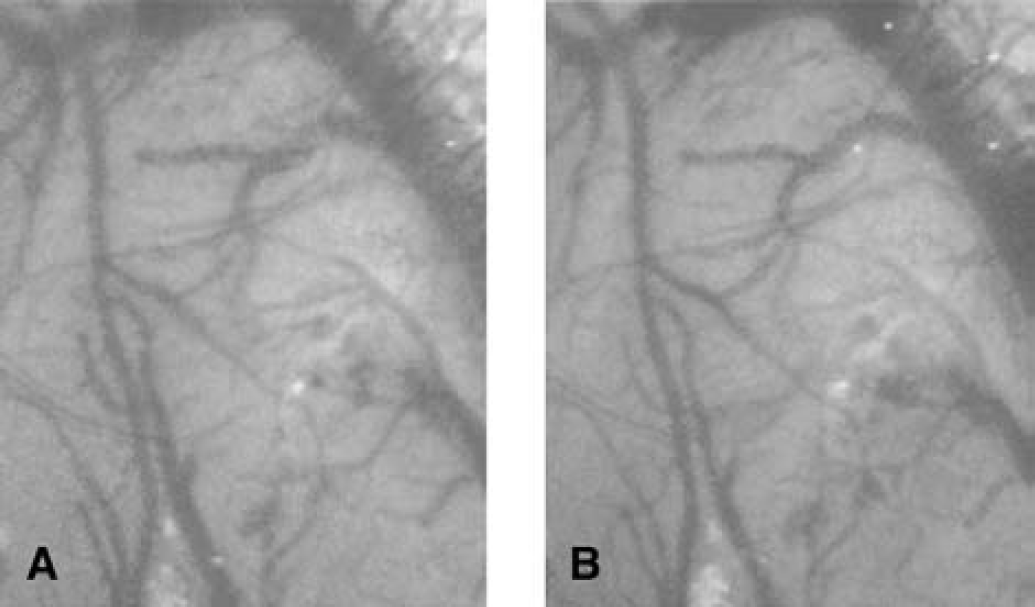
Bilirubin application to surface vessels of brain in a representative rat.
The largest vessels in Figs. 3A to 3C are mainly veins. Although it appeared that there was little visible effect of the BOXes on the veins (Figs. 3A and 3B), measurement of vein diameters demonstrated a modest but significant venodilation after the BOXes were applied compared with when bilirubin was applied, particularly by 30 to 40 minutes later (Fig. 2).
To determine whether the BOX-mediated vasospasm led to cortical injury, the animals were allowed to survive for 24 hours. Immunocytochemistry demonstrated that the HSP25 heat shock protein (Fig. 5, BOXes application) was induced mainly in astrocytes in the cortex at the site of BOXes application and often in the hippocampus (Figs. 5A and 5B) compared with induction of HSP25 in astrocytes at the cortical surface after bilirubin application (Fig. 5A, bilirubin application). Figure 5B shows hippocampal staining of astrocytes after BOXes but not bilirubin. In addition, it shows that both the BOXes and bilirubin induced Hsp25 in vessels, probably in capillary endothelial cells (Fig. 5B). Both BOXes and bilirubin induced HO-1/HSP32 in cortex under the application sites, although there were no significant differences between BOXes and bilirubin animals (not shown). Nissl histologic staining and NeuN immunohistochemistry showed no evidence of infarction in any of the animals.
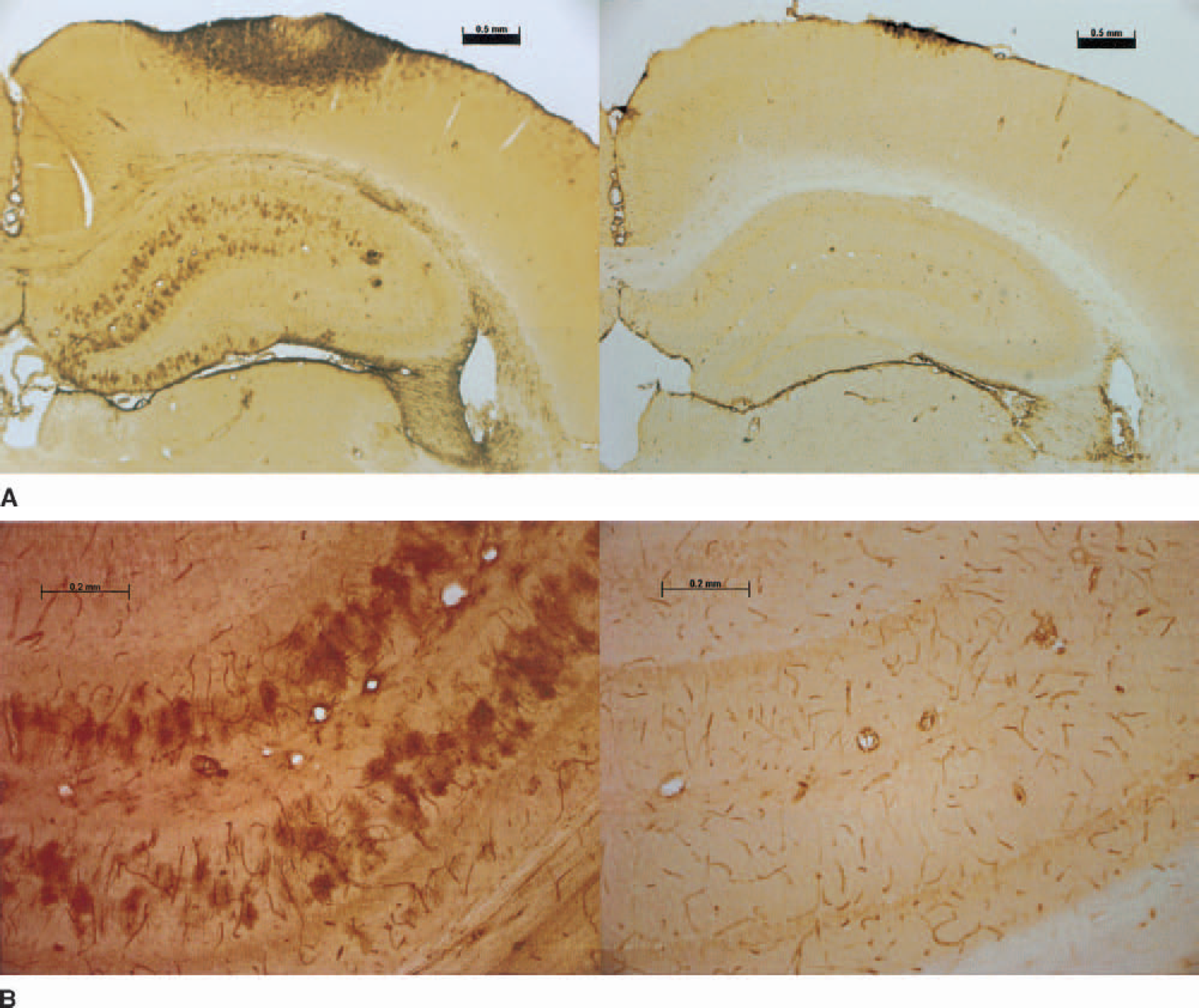
Hsp25 expression.
Figure 6 demonstrates the relative change in vessel diameter caused by the application of increasing doses of BOXes. There is a dose-dependent decrease in vessel diameter (consistent with vasoconstriction), although there is no evidence of constriction at the highest (1:1) dose. The lowest dose of BOXes (dilution of 1:1,000) produced moderate vasodilation. This “U-shaped” dose–response curve is discussed later in this article. Each point represents a minimum of six vessels averaged from three animals. The data from the saturated solution (1:1 dilution) is from two animals and five vessels. Therefore, 20 animals were used to generate these data.
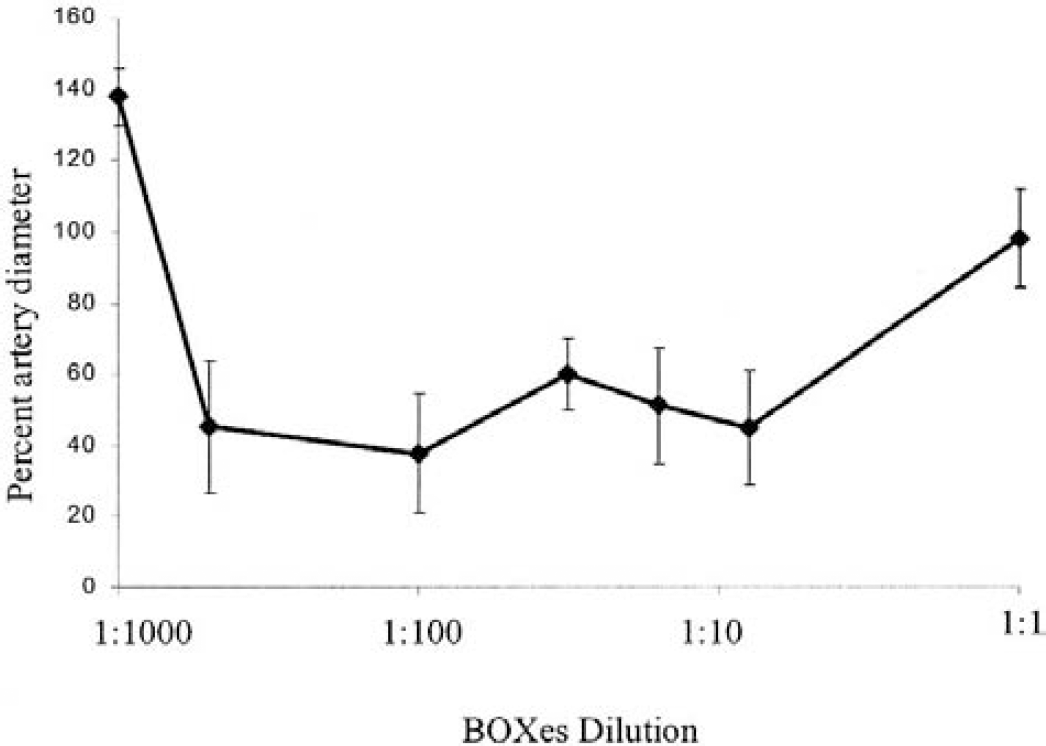
Dose–response curve for vasoconstriction produced by the bilirubin-oxidation products (BOXes). Different concentrations of the BOXes, ranging from 1:1,000 up to a 1:1 dilution, were applied to the surface of the rat brain and vessel diameters measured before and again 40 minutes after the application. The maximum dose given represents a saturated solution of BOXes and is assigned a concentration of 1 (1:1), and the lowest concentration given represented a 1:1,000 dilution of the maximal dose. Note that the 1:1,000 dose produced moderate vasodilation, the 1:1 dose had no significant effect on vessel diameters, and intermediate concentrations of 1:500 up to 1:8 produced marked vasoconstriction of the vessels. The error bars represent the standard deviation of the measurements. The values for the percent artery diameter at 1:500, 1:100, 1:32, 1:16, and 1:8 were significantly different than the values measured at 1:1 or 1:1000 (P < 0.01 using an unpaired t-test).
DISCUSSION
The results show that application of oxidized bilirubin (BOXes) to the surface of the brain produces profound arterial vasospasm, and that this cerebral vasospasm persists for at least 24 hours in the intact animal. These data provide strong support for our hypothesis that oxidized forms of bilirubin contribute to the vasospasm that occurs in patients after subarachnoid hemorrhage (Cadoux-Hudson et al., 1999, 2001; Kranc et al., 2000; Pyne et al., 2000, 2001a). Our previous data demonstrate that CSF from patients with vasospasm caused by subarachnoid hemorrhage produces contraction and increased metabolism of carotid rings in vitro (Pyne et al., 2000, 2001a). Moreover, the substances that lead to this vasospasm are small-molecular-weight molecules that are light sensitive, act as phosphatase inhibitors, and can be extracted with chloroform (Pyne et al., 2000). Based on these characteristics, we reasoned that the responsible molecules might be oxidized forms of bilirubin. To test this, bilirubin was oxidized with hydrogen peroxide, and three molecules were identified from this reaction including 4-methyl-3-vinylmaleimide and molecules we named as BOX A and BOX B (Kranc et al., 2000). Most importantly, these molecules produced vasospasm in vitro (Kranc et al., 2000). We have recently obtained evidence that at least one of these molecules (BOX A) is present in the CSF of patients with vasospasm caused by subarachnoid hemorrhage, and is not detectable in CSF of patients who do not have vasospasm after subarachnoid hemorrhage (Kranc et al., 2000). The current study provides the first in vivo evidence that oxidized products of bilirubin can produce a potent and prolonged vasospasm.
The data support the following mechanism for the pathogenesis of vasospasm after subarachnoid hemorrhage. After the formation of a clot around cerebral blood vessels at the base of the brain, red blood cells lyse and release hemoglobin. The heme is released and metabolized, in and around the vessels, to biliverdin by heme oxygenase-1 and heme oxygenase-2 (Matz et al., 1996b; Turner et al., 1998, 1999). Bilirubin reductase then metabolizes the biliverdin to bilirubin. The bilirubin is then oxidized because of free radicals associated with iron release, inflammatory cells, or other prooxidant mechanisms (Macdonald and Weir, 1994). It is notable that bilirubin itself can act as an antioxidant, but does not produce vasospasm by itself, as confirmed by the present study and others (Morooka, 1978; Stocker et al., 1987; Macdonald et al., 1991; Trost et al., 1993).
The production of vasoactive oxidation products of bilirubin after subarachnoid hemorrhage can explain the fact that the cerebral vasospasm that occurs after a subarachnoid hemorrhage is often delayed many days to a week after the hemorrhage (Macdonald and Weir, 1991; Weir, 1995; Weir et al., 1999). It takes time for heme to be metabolized to bilirubin, with CSF bilirubin levels typically peaking at 3 to 5 days after subarachnoid hemorrhage (Macdonald and Weir, 1991; Mayberg, 1998; Weir et al., 1999). Others have noted that bilirubin concentrations in the CSF are closely related to the time course of vasospasm (Duff et al, 1988; Morooka, 1978). Presumably, it would take additional time for the bilirubin to be oxidized to vasoactive BOXes, thus explaining the onset of vasospasm many days after the rupture of an aneurysm.
The discovery that a single application of the BOXes produced an extremely prolonged vasospasm was unexpected, but would be consistent with the clinical features of vasospasm. Once subarachnoid hemorrhage–induced vasospasm occurs in patients, it typically persists for a week or more (Heros et al., 1983; Newell et al., 1990; Weir et al., 1999). The prolonged vasospasm produced by the BOXes is consistent with the prolonged in vitro vasospasm produced by application of CSF from patients with vasospasm to carotid rings (Cadoux-Hudson et al., 2001; Pluta et al., 2000; Pyne et al., 2001a; Thomas and Rosenwasser, 1999; Wolf et al., 1998). Most compounds that produce physiologic constriction of cerebral blood vessels would not produce constriction that persists for 1 day, as observed here (McDaniel et al., 1991; Zhang and Paul, 1994; Zubkov et al., 2000). The prolonged vasospasm produced by the BOXes suggests a pathologic vasospasm and not a physiologic contraction of the vessels (Chyatte and Sundt, 1984; Mayberg, 1998; Weir et al., 1999). Although it is unclear how BOXes might maintain such a prolonged vasoconstriction, Kranc et al. (2000) suggested that BOXes could behave as alkylating agents and thereby irreversibly attack proteins involved in the contractile cycle. This as well as other putative mechanisms will require further study.
The “U-shaped” dose dependence of BOXes-induced vasoconstriction is similar to the results observed with phosphatase inhibition in smooth muscle. Phosphatase inhibition of smooth muscle can produce vasoconstriction or vasodilation, depending on which phosphatases are being inhibited (Bialojan et al., 1988; Takai, 1988; Ishihara et al., 1989; Siegman et al., 1989; Kimura et al., 1993). If phosphatase inhibition leads to an increase of myosin phosphorylation then constriction occurs, whereas if phosphatase inhibition leads to increased myosin light chain kinase phosphorylation then dilation occurs. Okadaic acid, a phosphatase inhibitor, can produce constriction or dilation, depending on the dose and the relative inhibition of protein phosphatase 2A (PP2A) as well as protein phosphatase 1 (PP1) (Pyne et al., 2000). Although it is unclear whether BOXes are working in the same way, this could explain the dose–response curve for the BOXes seen in Fig. 6. More work is needed to characterize the mechanisms of action of the BOXes on the vascular smooth-muscle function contributing to vasospasm.
The most important consequence of vasospasm is the strokes that occur in many, although not all, patients who develop this complication of SAH (Pickard et al., 1989; Kassel et al., 1996; Dorsch, 1998; Findlay and Deagle, 1998). Although the application of BOXes produced severe vasospasm, no evidence of infarction was observed in these experiments. This negative result prompted us to inject the BOXes under the dura. Although not described in detail, this produced equally severe vasospasm without producing infarction. The fact that the BOXes induced HSP25 in astrocytes and in the blood vessels suggests that the compounds stressed the tissue either directly or that the HSP25 was induced because of BOX-induced ischemia. Indeed, the injection of lysed blood into the subarachnoid space produces focal areas of heat shock protein induction that are consistent with areas of focal ischemia/vasospasm (Matz et al., 1996a,b; Turner et al., 1998). The finding that the BOXes produce vasospasm but do not produce stroke/infarction could mean that additional molecules, such as hemoglobin and endothelin, are also important for producing the strokes associated with vasospasm because of SAH (Macdonald and Weir, 1991; Mayberg, 1998; Weir et al., 1999). It is notable that cisternal/subarachnoid injections of the BOXes led to rapid death in a significant number of animals. However, this was probably not caused by infarction because the rapidity of this response would suggest either direct toxicity or rapid vasospasm perhaps affecting cardiorespiratory functions. Future experiments are required to determine whether the BOXes are directly neurotoxic and the role of BOX-induced vasospasm contributing to the HSP expression in these animals.
In conclusion, the data demonstrate that BOXes produce a severe and prolonged vasospasm of dural and cerebral vessels. This finding supports the possibility that the oxidized products of bilirubin found in the CSF of patients with vasospasm after SAH (Kranc et al., 2000; Pyne et al., 2000, 2001a) directly contribute to and/or are the primary cause of SAH-induced cerebral vasospasm.