
Abstract
Treatment after hypoxia—ischemia (HI) in immature rats with the N-methyl-
In the cerebral cortex of the immature rat, the early recovery period after hypoxia—ischemia (HI) is characterized by an incomplete restitution of ATP and phosphocreatine. There is a persistent increase in lactate levels and normal or low NAD+/NADH ratio. The cerebral metabolic rate of glucose (CMRglc) is increased in relation to a normal or low energy utilization, but the CBF is unimpaired until 48 hours after the insult (Mujsce et al., 1990; Palmer et al., 1990; Yager et al., 1991; Yager et al., 1992; Vannucci et al., 1994; Gilland and Hagberg, 1996; Gilland et al., 1997). This has been interpreted as signs of an impaired oxidative metabolism, and a reduction in the activity of cytochrome aa3 (cytochrome c oxidase), the final component of the electron transport chain, was observed in the hippocampus, but not in the cortex, early after HI in the immature rat (Nelson and Silverstein, 1994). In the newborn lamb, there is a reduction of ADP-stimulated mitochondrial respiration and of cerebral oxygen utilization after asphyxia (Rosenberg, 1986; Rosenberg et al., 1989), and an impairment of mitochondrial respiration has been seen after both focal and global ischemia in adult rats (Rehncrona et al., 1979; Hillered et al., 1984; Sims and Pulsinelli, 1987; Kuroda et al., 1996).
Treatment after HI with the N-methyl-
MATERIALS AND METHODS
Animals and chemicals
Inbred Wistar F rat pups of either sex were purchased from Møllegaard Breeding & Research Centre A/S (Skensved, Denmark) and fed by their dams until the start of the experiments at 7 days of age. All chemicals and enzymes for determining tissue metabolites were obtained from Sigma-Aldrich Sweden (Göteborg, Sweden) and Boehringer Mannheim Scandinavia AB (Bromma, Sweden).
Protocol
Animals in each litter were randomized to one of three groups: (1) the non-HI control group; (2) the HI group, which underwent HI and received saline injections; and (3) the HI plus MK-801 group, which underwent HI and received 0.5 mg/kg MK-801 intraperitoneally immediately after HI. In the rats used for measurement of energy metabolites and cerebral energy utilization 3 hours after HI, the non-HI controls were from separate litters to the HI and HI plus MK-801 groups. Cerebral energy metabolites at 8 hours after HI were measured in separate litters from the 3-hour time point. These rats were kept at 33°C ambient temperature, and were separated from the dams between 1 and 8 hours after HI (to eliminate the mild hypothermic effect of MK-801 that is evident from 4 hours after HI if the pups are kept in room temperature with their dams [Gilland and Hagberg, 1997]).
The rectal temperature was measured with a thermistor probe (BAT-12, Physitemp Instruments, Clifton, NJ, U.S.A.) immediately after removal from the dam. In rats used for measurement of energy utilization, the temperature was measured before decapitation at 3 hours after HI, whereas in the rats used for measurement of mitochondrial respiration, temperature was measured 1 hour after HI and rats were put back with the dam.
Induction of hypoxia—ischemia
Anesthesia was induced and maintained with halothane in oxygen—nitrous oxide on snout mask. The left common carotid artery was cut between ligatures of prolene suture (Rice et al., 1981). Animals were allowed to recover from anesthesia for at least 1 hour. The HI was induced by exposure to 7.7% ± 0.01 % oxygen in nitrogen for 70 minutes in a humidified chamber at 36°C. This induces a reversible reduction of CBF and infarction in the left (ipsilateral) hemisphere, whereas there is no reduction of CBF or any gross infarction in the contralateral hemisphere (Hagberg et al., 1997). After the insult, the pups were returned to their dams. In each litter used for measurement of pyruvate plus malate—supported mitochondrial respiration, one rat in the HI group was decapitated at 14 days after HI, and the hemispheres were dissected out and weighed to determine the degree of brain damage (Hagberg et al., 1994).
Measurement of cerebral energy utilization
Cerebral energy utilization was measured by the changes in energy metabolites during 1 minute after decapitation (Lowry et al., 1964; Gatfield et al., 1966). Shutting of the blood supply converts the brain into a closed biological system and limits the chemical events to those that can occur anaerobically (Lowry et al., 1964). Two rats were paired and received the same treatment. At 3 hours after HI, both rats were decapitated. The head of one was immediately frozen in liquid nitrogen, whereas the head of the other was left for 1 minute in room temperature before it was frozen. Portions (10 to 50 mg) of the parietal cortex were dissected out at −20°C, weighed, and solubilized in 3 mol/L HClO4 (Lowry and Passonneau, 1972; Gilland and Hagberg, 1996) at −10°C. After centrifugation at 5000 × g for 10 minutes, one volume of the supernatant was neutralized with 2.5 volumes of 2 mol/L KHCO3 and again centrifuged to remove precipitated KHCO3. The supernatant was used for enzymatic—fluorometric determination of ATP, glucose, and lactate (Lowry and Passonneau, 1972). Changes in metabolite levels were determined by subtracting the values of the second rat from the values of the first rat in each pair. Changes in high-energy phosphate bindings (Δ~P) were calculated according to the formula: Δ~P (μmol/min/g) = 2ΔATP + Δphosphocreatine + 2Δglucose + 1.45(Δlactate − 2Δglucose). Changes in ADP were neglected, since they are small in the immature brain during short time intervals (Duffy et al., 1975) and changes in glycogen were replaced by Δlactate − 2Δglucose (i.e., the excess lactate production over glucose disappearance [Gatfield et al., 1966]). All brains and specimens were stored at −80°C.
Measurement of mitochondrial respiration
Mitochondrial respiration in brain tissue homogenates were measured according to Sims and Blass (1986). Since each measurement took 30 minutes to perform, six rats from each litter that underwent HI (the HI and HI plus MK-801 groups) were decapitated successively 1 to 4 hours after HI and the results pooled, irrespective of time after HI. The cortex of the left hemisphere was dissected out and immersed in ice-cold 10 mmol/L Tris buffer (pH 7.4) containing 0.32 mol/L sucrose and 1 mmol/L ethylendiammine tetraacetic acid (EDTA; potassium salt). The tissue was rinsed and the surface cleared from meninges and pial vessels. The anterior and posterior poles were cut away, and a central portion (weighing 60 ± 12 mg) was homogenized in 1-mL buffer by hand (four up-and-down strokes with a total clearance of 0.12 mm and eight up-and-down strokes with a total clearance of 0.05 mm) in a 2-mL glass homogenizer (Kontes, Vineland, NJ, U.S.A.).
Respiratory activity was measured at 28°C in a closed and magnetically stirred glass chamber (volume 0.54 mL), using a Clark-type oxygen electrode (L. Escheweiler & Co., Kiel, Germany). A total of 90 μL of the homogenate was added to a reaction buffer (pH 7.4), which consisted of 100 mmol/L KCl, 75 mmol/L mannitol, 25 mmol/L sucrose, 5 mmol/L Trisphosphate, 0.05 mmol/L EDTA, 10 mmol/L Tris-HCl, and 0.1% bovine serum albumin. After 3 minutes, the chamber was closed, and, after baseline measurement, 10 μL of 0.28 mol/L pyruvate and 0.14 mol/L malate was added (final concentrations 5 mmol/L and 2.5 mmol/L, respectively). State 3 (+ADP) respiration was measured after addition of 1.5 μL of 50 mmol/L ADP, and state 4 (–ADP) respiration was determined after the ADP was depleted. Uncoupling was initiated by adding 1 to 2 μL of 0.9 mmol/L carbonyl cyanide m-chlorophenylhydrazone in 32% ethanol (final concentration 1.7 to 3.3 μmol/L). In separate rats randomized only to the non-HI control and the HI groups, glutamate and succinate—supported respiration was measured (after baseline measurement) by adding 10 μL of 0.55 mol/L glutamate and 0.14 mol/L malate (final concentrations 10 mmol/L and 2.5 mmol/L, respectively), whereupon state 4 respiration was measured, and 2 μL of 50 mmol/L ADP, whereupon state 3 respiration was measured. Addition of 1 μL of 1 mmol/L rotenone completely inhibited oxygen uptake until 10 μL of 0.55 mol/L succinate was added. State 3 and state 4 respiration was measured as described earlier. Between samples, the chamber was rinsed with ethanol, distilled water, and reaction buffer to avoid contamination of the next sample by rotenone. All substrates and EDTA were neutralized with KOH before use.
Statistics
Analysis of variance was used for comparisons between groups with Scheffe's post hoc test. Paired Student's t test was used for comparisons between hemispheres. Differences with P < 0.05 were considered statistically significant. Values are given as mean ± SD.
RESULTS
Physiologic variables
There was no difference in rectal temperature between the HI and HI plus MK-801 groups, either at 1 hour or 3 hours after HI and drug administration (Table 1). Blood lactate levels at 3 hours after HI were 0.80 ± 0.06 mmol/L (n = 5) in the HI plus MK-801 group, and 0.86 ± 0.05 mmol/L (n = 3) in the HI group. The brain damage in the HI group, expressed as weight deficit of the ipsilateral hemisphere compared with the contralateral hemisphere, was 38% ± 16% (n = 4) at 14 days after HI. There was no mortality during or after HI.
Physiologic variables after hypoxia-ischemia (HI) and in non-HI controls

Values represent mean ± SD.
P < 0.05 versus the non-HI group.
P < 0.05 versus the HI group.
Cerebral energy metabolites
In both the HI and HI plus MK-801 groups at 3 and 8 hours after HI, the ATP, phosphocreatine, and lactate levels in the ipsilateral parietal cortex were altered compared with their respective contralateral cortex and compared with the ipsilateral cortex of the non-HI controls (Table 2 and Table 3). Glucose levels were significantly higher in the contralateral cortex of the HI plus MK-801 group. There were no significant differences in the ATP, phosphocreatine, and lactate levels between the HI and HI plus MK-801 groups at 3 hours after HI, but at 8 hours after HI, the HI plus MK-801 group had higher concentrations of ATP, phosphocreatine, and glucose and lower lactate concentrations in the ipsilateral cortex than the HI group (Table 3; P = 0.04, 0.004, 0.001, and 0.001, respectively).
Metabolite levels (μmol/g) in pieces of the left/ipsilateral and right/contralateral cortex 3 hours after hypoxia-ischemia (HI), and in non-HI controls
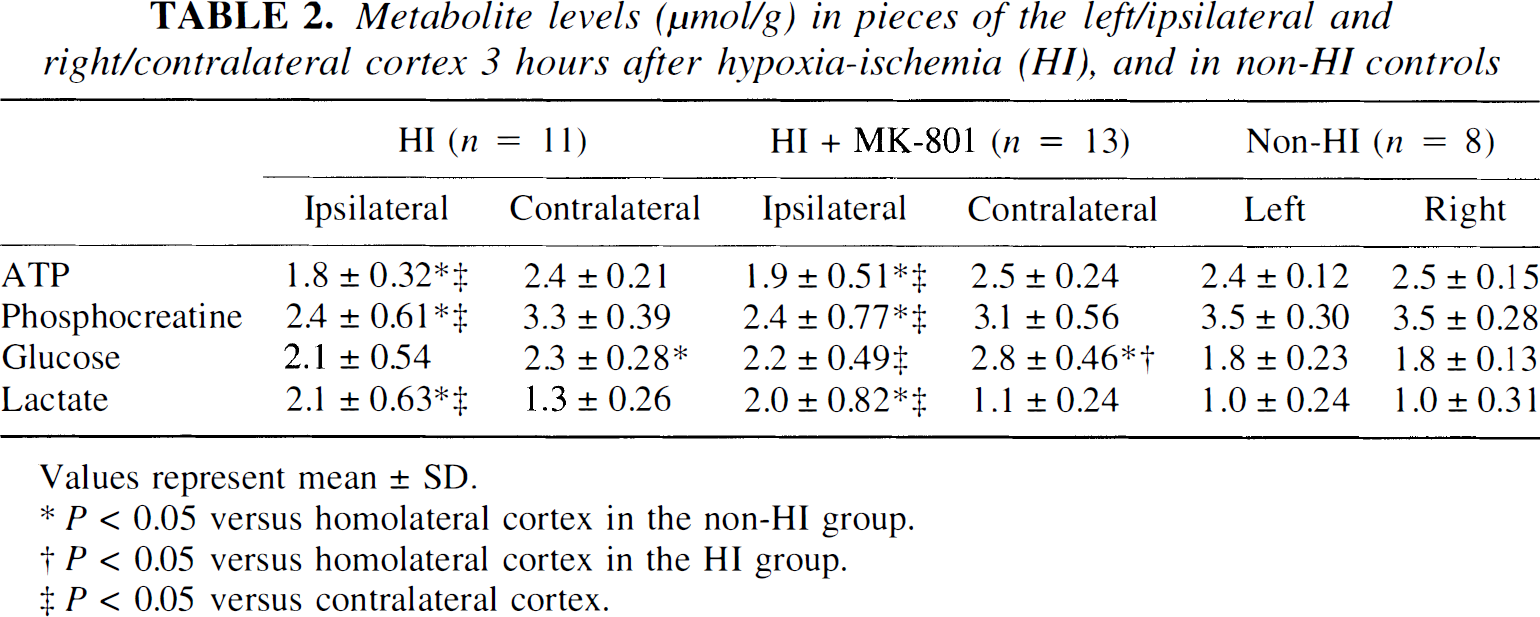
Values represent mean ± SD.
P < 0.05 versus homolateral cortex in the non-HI group.
P < 0.05 versus homolateral cortex in the HI group.
P < 0.05 versus contralateral cortex.
Metabolite levels (μmol/g) in pieces of the left/ipsilateral and right/contralateral cortex 8 hours after hypoxia-ischemia (HI)
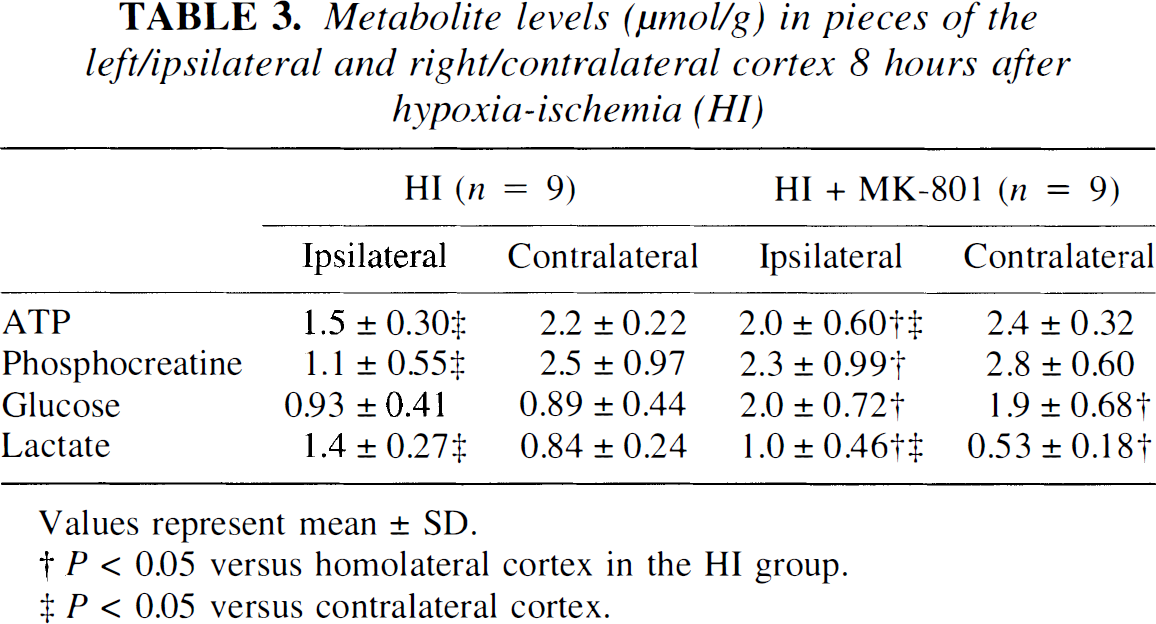
Values represent mean ± SD.
P < 0.05 versus homolateral cortex in the HI group.
P < 0.05 versus contralateral cortex.
Cerebral energy utilization
There was no significant change of cerebral energy utilization in the HI group compared with the non-HI controls, although there was a tendency toward reduced values in the former (Table 4). The HI plus MK-801 group had reduced cerebral energy utilization in the ipsilateral cortex (Δ~P = 2.6 ± 1.8 μmol/min/g) compared with the contralateral cortex (4.2 ± 0.92 μmol/min/g; P = 0.004), with the ipsilateral cortex in the HI group (4.6 ± 2.3 μmol/min/g; P = 0.04), and with the left parietal cortex in the non-HI controls (4.9 ± 0.61 μmol/min/g; P = 0.03; Table 4). The cerebral energy utilization in the right (contralateral) cortex did not differ between the non-HI, HI, and HI plus MK-801 groups.
Metabolite fluxes (μmol/min per g) after decapitation in pieces of the left/ipsilateral and right/contralateral cortex 3 hours after hypoxia-ischemia (HI), and in non-HI controls
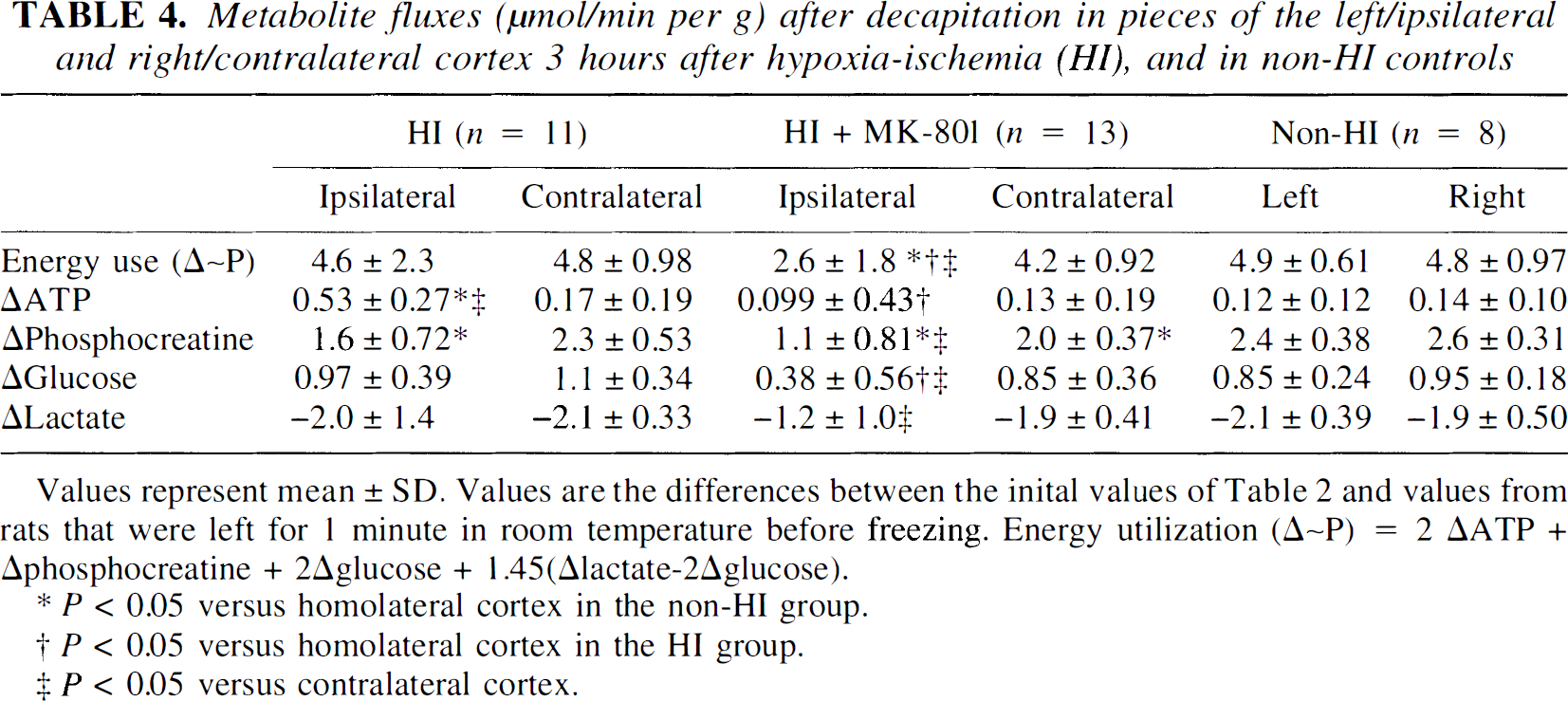
Values represent mean ± SD. Values are the differences between the inital values of Table 2 and values from rats that were left for 1 minute in room temperature before freezing. Energy utilization (Δ~P) = 2 ΔATP + Δphosphocreatine + 2Δglucose + 1.45(Δlactate-2Δglucose).
P < 0.05 versus homolateral cortex in the non-HI group.
P < 0.05 versus homolateral cortex in the HI group.
P < 0.05 versus contralateral cortex.
Mitochondrial respiration
After HI, ADP-stimulated and uncoupled respiration was significantly reduced, regardless of substrate (Fig. 1; Table 5 and Table 6). Also, after HI, the +ADP respiration was approximately 54% (P < 0.0001, n = 14), 68% (P = 0.01, n = 5), and 75% (P = 0.007, n = 5) of the respiration in non-HI controls with pyruvate plus malate, glutamate plus malate, or glutamate plus succinate in the presence of rotenone as substrates, respectively. There was no linear change in mitochondrial +ATP pyruvate—supported respiration over time between 1 and 4 hours after HI (R2 = 0.083). There were no significant differences in –ADP respiration for any of the substrates used between the HI and the non-HI groups. Treatment with MK-801 after HI caused a normalization of pyruvate-supported +ADP respiration to 77% of non-HI controls (P = 0.006 compared with HI group, n = 14; Table 5). There was a significant difference between the HI group (4.8 ± 1.1, n = 5) and the HI plus MK-801 group (8.1 ± 1.9, n = 6; P = 0.01), even if including only the non-HI controls and the rats of the HI and HI plus MK-801 groups that were decapitated between 1 to 2 hours after HI. Treatment with MK-801 also tended to increase pyruvate-supported uncoupled respiration (not significant; Table 5).
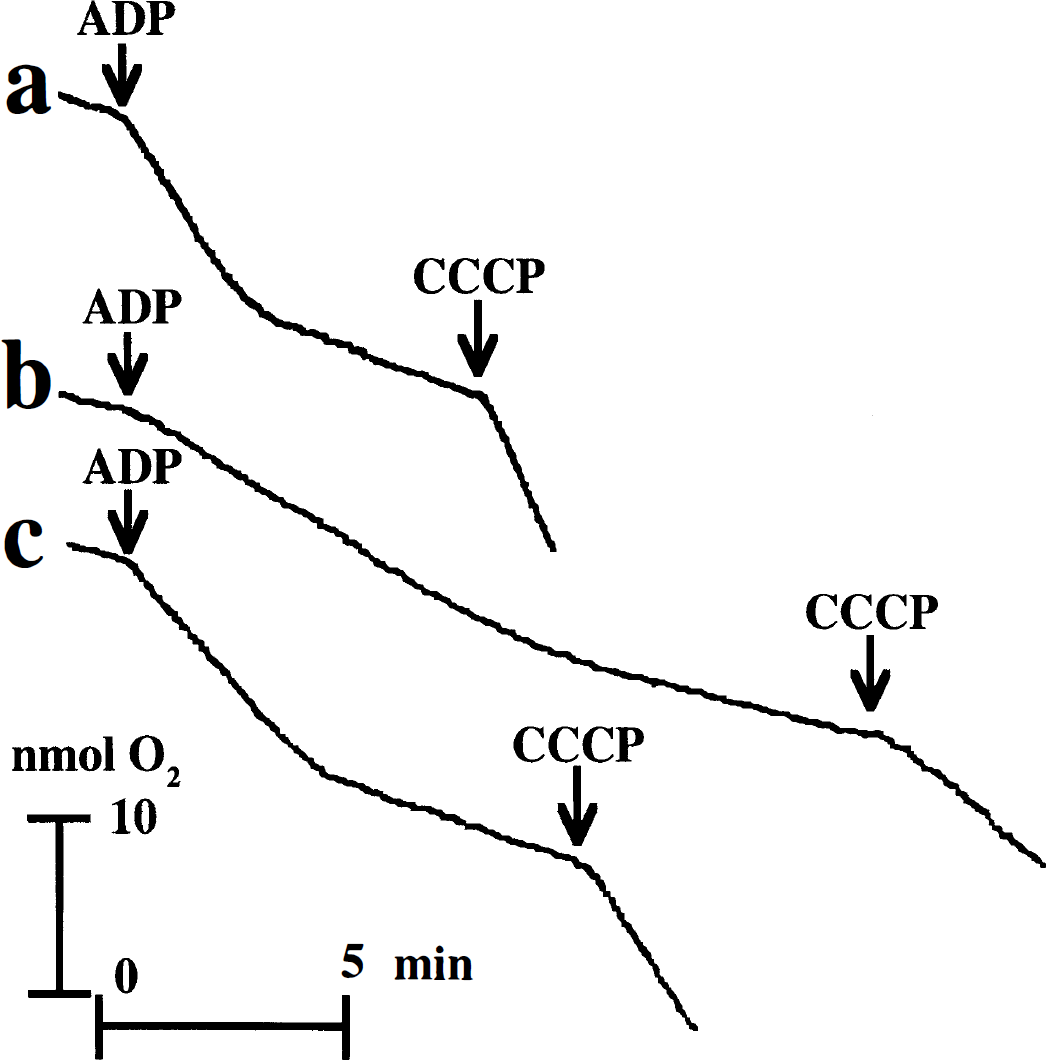
Pyruvate (5 mmol/L) plus malate (2.5 mmol/L)–supported mitochondrial oxygen consumption tracings from the left cortex of nonhypoxia—ischemia (non-HI) control (
Pyruvate (5 mmol/L) + malate (2.5 mmol/L) supported mitochondrial oxygen consumption (nmol O2/min/mg protein) in pieces of the left/ipsilateral cortex 1 to 4 hours after hypoxia-ischemia (HI), and in non-HI controls

CCCP, carbonyl cyanide m-chlorophenyl-hydrazone; RCR, respiratory control ratio. Values represent mean ± SD.
P < 0.05 versus the non-HI group.
P < 0.05 versus the HI group.
Mitochondrial oxygen consumption (nmol O2/min/mg protein) in pieces of the left/ipsilateral cortex 1 to 4 hours after hypoxia-ischemia (HI), and in non-HI controls

Values represent mean ± SD. All concentrations are final concentrations. CCCP, carbonyl cyanide m-chlorophenylhydrazone; RCR, respiratory control ratio.
P < 0.05 versus the non-HI group.
DISCUSSION
Energy metabolites and utilization
The parietal cortex is the area that is most likely to be damaged after HI in the 7-day-old rat (Hagberg et al., 1994). In this study, vehicle-treated rats that were allowed to survive until 2 weeks after HI had an average tissue reduction of 38% of the ipsilateral hemisphere, which agrees with a 38% weight deficit of the ipsilateral hemisphere in vehicle-treated rats in a recent neuroprotection study from our laboratory (Gilland and Hagberg, 1997). With this severity of insult, it is most likely that the tissue sampled from the parietal cortex in the HI group would eventually have undergone infarction, which also is supported by the secondary deterioration of the phosphocreatine levels. In the HI group, the levels of high-energy phosphate compounds in the brain were partially recovered at 3 hours after HI, and the ipsilateral phosphocreatine levels were 75% of the contralateral; however, at 8 hours after the insult, a secondary energy failure was observed with the ipsilateral phosphocreatine levels being only 47% of the contralateral. This secondary energy failure has been described in human newborns after asphyxia and in several animal models and probably reflects a progression of tissue infarction or necrosis (Azzopardi et al., 1989; Palmer et al., 1990; Yager et al., 1992; Lorek et al., 1994; Buchli et al., 1995).
In accordance with Vannucci and others (1994), HI and reperfusion did not increase the rate of energy utilization in the brain. On the contrary, there was a small tendency toward reduced energy use after HI (Table 4). Treatment with MK-801 after HI reduced the energy utilization in the ipsilateral parietal cortex to 57% of the rate in vehicle-treated rats after HI (Table 4) but did not affect the energy utilization in the contralateral cortex. This indicates that the metabolic rate is more dependent on NMDAR activation during reperfusion after HI than under normal conditions. The mechanism for this NMDAR dependency may relate to increased extracellular levels of excitatory amino acids during early reperfusion (Puka-Sundvall et al., 1996), and to enhanced sensitivity of the NMDAR. The latter effect can be caused by removal of the Mg2+ block from the receptor channel when intracellular energy levels are low (Novelli et al., 1988; Beal et al., 1993; Greene and Greenamyre, 1996) or by other changes in the function of the NMDAR after ischemia (Andiné et al., 1988; Hammond et al., 1994; Heurteaux et al., 1994; Miyazaki et al., 1994). These effects, which could act synergistically, cause increased influx of Ca2+ and Na+ through the NMDAR channels, resulting in elevated energy use for active ion transport. The lack of effect of MK-801 treatment on energy utilization in the contralateral cortex is in agreement with an unaltered CBF in the contralateral cortex after MK-801 treatment (Gilland and Hagberg, 1996). In contrast, MK-801 treatment reduces the CMRglc also in the contralateral cortex (Gilland and Hagberg, 1996). This might reflect an increased use of lactate and ketone bodies as substrates for energy production in the contralateral hemisphere (Nehlig and Pereira de Vasconcelos, 1993; Vannucci et al., 1994).
Mitochondrial oxygen consumption
Hypoxia—ischemia induced a reduction of both NAD-linked, pyruvate plus malate—supported, and glutamate plus malate—supported respiratory rates and of FAD-linked, succinate-supported respiration (Table 5 and Table 6). Thus, the mitochondrial dysfunction cannot be explained by a selective inhibition of the pyruvate dehydrogenase complex or the NADH-CoQ reductase complex (complex I of the electron transport chain), as has been suggested in the adult rat (Zaidan and Sims, 1993). For all three substrates, the uncoupled respiration was reduced as much as +ADP respiration, precluding a selective reduction in ATP-synthetase (F0-F1-ATPase; complex V) activity.
Isolated brain mitochondria represent only 3% to 16% of the total pool of mitochondria in the tissue, and it cannot be excluded that isolation of mitochondria involves a selection of subgroups after HI (Sims and Blass, 1986). Measuring oxygen consumption by tissue homogenates does not involve any selection of mitochondria. However, after homogenization of the brain, both nonsynaptosomal, and synaptosomal mitochondria are present. The +ADP respiratory rate primarily measures the activity of nonsynaptosomal mitochondria, since ADP cannot enter synaptosomes, whereas the –ADP and uncoupler-stimulated respiratory rates measure the activity of the whole mitochondrial population. In the 7-day-old rat, we found a ratio of uncoupled to +ADP respiratory rates of 1.4, which is 25% lower than in the adult (Sims and Blass, 1986; Sims and Pulsinelli, 1987; Sims, 1991), in accordance with fewer dendrites and axons in the immature neuron (Kristt, 1978; Romijn et al., 1991). In adult rat homogenates, nonsynaptosomal mitochondria are of both neuronal and glial origin (Sims and Blass, 1986). In the immature rat, glia are less developed (Privat, 1975) and, presumably, a substantial part of the free mitochondria are of neuronal origin.
The respiratory control ratio (+ADP/–ADP), which reflects the coupling of respiration to ATP synthesis, was lowered after HI for all three substrates, but this did not reflect an uncoupling of the mitochondrial metabolism, since it was caused by a reduction of +ADP respiration and not by an increase in –ADP respiration. In addition, the energy metabolites in the ipsilateral cortex 1 to 4 after HI indicates an impaired flux of metabolites through the citric acid cycle, electron transport chain, or both, combined with lactate production. The levels of the glycolytic intermediates glucose, pyruvate, and lactate are increased, the cytoplasmic NAD+/NADH ratio is reduced, and the mitochondrial NAD+/NADH ratio is normal or reduced (Palmer et al., 1990; Yager et al., 1991; Yager et al., 1992; Gilland and Hagberg, 1996). The concentration of lactate in the ipsilateral hemisphere was higher than in the contralateral hemisphere and higher than the blood lactate levels. The high lactate permeability across the blood—brain barrier in the immature brain (Cremer et al., 1979) and the normal CBF (Gilland and Hagberg, 1996; Gilland et al., 1997) suggest that the increased lactate levels in the brain reflect lactate production and not decreased clearance of lactate accumulated during HI.
Relevance of the metabolic impairment
The decreased mitochondrial respiratory capacity and sustained energy utilization probably is an early step in the NMDAR-dependent injurious processes after HI and not secondary to tissue infarction as is outlined here. At 2 to 3 hours after HI, we did not find a difference in ATP and phosphocreatine concentrations between rats that had received MK-801 or vehicle after HI, implying that there was not a difference in the extent of tissue damage at this time. However, treatment with MK-801 induced a marked improvement in the mitochondrial respiratory capacity already at 2 hours and reduced the energy utilization and the CMRglc at 3 hours after HI (current results; Gilland and Hagberg, 1996).
Mitochondrial impairment as an early step in the neurodamaging cascade after HI agrees well with data from adult rats after forebrain ischemia, where a reduction in mitochondrial function precedes, by several hours, changes in energy metabolite levels (Pulsinelli and Duffy, 1983; Sims, 1991).
There were, however, differences both in ATP and phosphocreatine concentrations between MK-801 and vehicle-treated rats at 8 hours after HI, suggesting that the effects on mitochondrial function and energy utilization are of functional importance for the energy state of the tissue, and that the secondary energy failure depends on NMDAR activation.
Stimulation of the NMDAR might cause mitochondrial damage through elevation of intracellular Ca2+, release of nitric oxide (NO·), and production of free radicals (Greene and Greenamyre, 1996). In vitro exposure of mitochondria to free radicals induces a similar reduction of +ADP and uncoupler stimulated respiration, as does HI (Hillered and Ernster, 1983), and treatment with a free radical scavenger after reversible focal ischemia attenuates a secondary mitochondrial dysfunction in adult rats (Kuroda et al., 1996). Hypothetically, the mitochondrial damage and impaired energy metabolism makes the cells more susceptible to NMDAR stimulation, which further impairs the mitochondrial function through increases in intracellular Ca2+, NO· formation and free radical production (Beal et al., 1993).
CONCLUSION
In conclusion, treatment with MK-801 after HI reduces energy utilization and attenuates the mitochondrial dysfunction and secondary energy failure. These results explain why NMDAR activation induces increased CMRglc during reperfusion, and they may be relevant in understanding NMDAR involvement in the pathogenesis of HI injury in the immature brain.
Footnotes
Abbreviations used
Acknowledgment
The authors thank Neil R. Sims for providing the impetus for measuring mitochondrial respiration.