
Abstract
The authors previously reported that mRNA for macrophage inflammatory protein-1α (MIP-1 α), a member of the CC chemokines, was expressed in glial cells after focal cerebral ischemia in rats. However, the function of chemokines in the ischemic brain remains unclear. Recently, viral macrophage inflammatory protein-II (vMIP-II), a chemokine analogue encoded by human herpesvirus-8 DNA, has been demonstrated to have antagonistic activity at several chemokine receptors. In the present study, the effects of vMIP-II and MIP-1α on ischemic brain injury were examined in mice to elucidate the roles of chemokines endogenously produced in the ischemic brain. Intracerebroventricular injection of vMIP-II (0.01–1 μg) reduced infarct volume in a dose-dependent manner when examined 48 hours after 1-hour middle cerebral artery occlusion followed by reperfusion. However, 1 μg MIP-1α increased infarct volume in the cortical region. These results supported the possibility that chemokines endogenously produced in the brain are involved in ischemic injury, and that chemokine receptors are potential targets for therapeutic intervention of stroke.
Chemokines constitute a large family of structurally related cytokines that have chemotactic activity for leukocytes (Gale and McColl, 1999; Zlotnik and Yoshie, 2000). This family includes more than 40 members, and has been divided into four subfamilies: CXC, CC, C and CX3 C (Rollins, 1997). The expression and function of chemokines have been intensively investigated in inflammatory and allergic responses in peripheral organs such as the kidney, lung, liver, and skin (Feng et al., 1995; Frevert et al., 1995; Maltby et al., 1996; Barker et al., 1990). Recently, it has been reported that chemokines are also expressed in the brain under various pathological conditions, including mechanical injury (Hausmann et al., 1998), Alzheimers disease (Horuk et al., 1997), and multiple sclerosis (McManus et al., 1998). We and other groups have demonstrated the expression of mRNAs of chemokines such as macrophage inflammatory protein-1α (MIP-1α), monocyte chemoattractant protein-1 (MCP-1), and cytokine-induced neutrophil chemoattractant in the ischemic brain (Takami et al., 1997; Wang et al., 1995; Kim et al., 1995; Liu et al., 1993). Chemokines activate astrocytes and microglia to increase their migration (Tanabe et al, 1997). Chemokines are also implicated in the extravasation of leukocytes into the brain parenchyma after ischemia (Weiss et al., 1998). However, little is known about whether the chemokines produced in the ischemic brain are neuroprotective or neurodegenerative.
Viral macrophage inflammatory protein-II (vMIP-II) is a chemokine analogue peptide encoded by Kaposi sarcoma-associated herpes virus (Moore et al., 1996), and has 45.1% and 40.0% identity to human and mouse MIP-1α, respectively, at the amino-acid sequence level (Nicholas et al., 1997). vMIP-II shows antagonism against the effects of various chemokines, including MIP-1α and monocyte chemoattractant protein-1, at a broad range of chemokine receptors (Table 1), and some in vivo effects of this peptide have been reported (Table 2). In the present study, the effects of vMIP-II on the ischemic brain injury resulting from 1-hour occlusion of the middle cerebral artery (MCA) were examined to elucidate whether the chemokines produced in the ischemic brain are neuroprotective or neurodegenerative.
In vitro activity (antagonism at chemokine receptors) of vMIP-II

In vivo activity of vMIP-II

MATERIALS AND METHODS
Focal cerebral ischemia model
Male ddY mice (4-weeks old; Japan SLC, Hamamatsu, Japan) were housed under a diurnal lighting conditions with access to food and water ad libitum. Animals were anesthetized with 1.5% halothane for induction and maintained with 1.0% halothane in 70% nitrous oxide/30% oxygen using a vaporizer (Halowick, Murako Medical Co., Tokyo, Japan). Rectal temperature was kept at 37°C during the surgical procedure with a thermostatically controlled heating pad (NS-TC, Neuroscience, Tokyo, Japan; BAT-12, Physitemp Instruments, Clifton, NJ, U.S.A.). Middle cerebral artery occlusion was induced with an 8–0 nylon monofilament (11 mm) coated with a mixture of silicone resin and hardener (Xantopren and Optosil Activator; Heraeus Kulzer, Dormagen, Germany). After ligation of the common and external carotid arteries, the filament was introduced into the left internal carotid artery through the incision at the external carotid artery, and pushed up to the anterior cerebral artery to occlude the MCA. After 1 hour of occlusion, the filament was withdrawn and the external carotid artery was coagulated with bipolar forceps, and the common carotid artery was released from ligation. Regional cerebral blood flow of each animal was monitored by laser-Doppler flowmetry (FLO-C1, Omegawave, Tokyo, Japan) using a flexible probe affixed to the skull (2 mm posterior and 6 mm lateral to the bregma) with cyanoacrylate glue (Aron Alpha, Toagousei Co., Tokyo, Japan). The animals in which regional cerebral blood flow in the MCA region was reduced to less than 25% by occlusion and recovered to more than 65% by reperfusion were used for further evaluations. Mice were kept in a warming chamber (Thermocare, Incline Village, NV, U.S.A.) for 3 hours after surgery. Animal experiments were performed in accordance with the Guideline for Animal Experimentation in National Cardiovascular Center (November 1, 1989).
Intracerebroventricular administration of vMIP-II and MIP-1α
vMIP-II and MIP-1α were purchased from R&D Systems (Minneapolis, MN, U.S.A.) and dissolved in 0.1 mol/L phosphate-buffered saline (PBS) (pH 7.4) containing 0.1% bovine serum albumin. These peptides (0.01–1 μg in 5 μL) were administered by intracerebroventricular injection (1.0 mm lateral, 0.5 mm posterior, 3.0 mm ventral to bregma) at 1 hour before MCA occlusion and after 1 hour of reperfusion. Previously, Chen et al. (1998) reported that repeated intravenous injection of vMIP-II at a dose of 25 μg/d for four days (total 100 μg/rat) significantly attenuated glomerulonephritis in rats. In the present study, as mice were used instead of rats and local instead of systemic administration was employed, 1/50-1/5000 doses of vMIP-II were selected. Vehicle-treated mice were injected with 5μL of PBS / 0.1% bovine serum albumin.
Measurement of infarct volume
Forty-eight hours after reperfusion, the animals were killed by transcardial perfusion with 10% formalin in 0.1 mol/L PBS (pH 7.4) with anesthesia by intraperitoneal injection of sodium pentobarbital (100 mg/kg). The brains were removed quickly and kept in 10% formalin/0.1 mol/L PBS for 24 hours. After incubation in PBS containing 20% sucrose for 12 hours, the brains were cut into coronal sections 50-μm thick at an interval of 500 μm throughout the cerebrum (from bregma 3.2 mm to bregma −3.8 mm) (Franklin and Paxinos, 1997) on a freezing microtome (HM400R, Microm, Walldorf, Germany). The sections were mounted onto gelatin-coated glass slides and stained with hematoxylin and eosin. The infarct areas in the cortical and subcortical regions of each section were measured using the Olympus Image Analysis System (Olympus Optical Co., Tokyo, Japan), and integrated to give the total infarct volume. The total infarct volume and contralateral hemispheric volume of the vehicle-injected group were 55.2 ± 17.2 and 129.2 ± 12.6 mm3, respectively.
Measurement of brain-shrinkage volume
The animals were killed by transcardial perfusion 14 days after reperfusion and the brains were cut into coronal sections as described previously. After staining with 0.1% cresyl violet, the areas of ipsilateral (ischemic side) and contralateral (control side) hemispheres on each section were measured and integrated to give the hemispheric volume of each side. Brain-shrinkage volume was calculated by the following formula: brain shrinkage volume (%) = {(contralateral hemispheric volume − ipsilateral hemispheric volume) / contralateral hemispheric volume}× 100 (%)
Physiologic parameters
In four mice selected at random from each group, rectal temperature was measured from the time of the first intracerebroventricular injection to that of death. In the same mice, temporal muscle temperature was monitored during the surgical procedure for MCA occlusion. In another four mice from each group, the left femoral artery was cannulated with PE-10 polyethylene tubing to monitor mean arterial blood pressure, and 100 μL arterial blood was collected through the cannula during ischemia (30 min after the MCA occlusion) and analyzed for pH and partial pressures of carbon dioxide and oxygen.
Statistical analysis
Data are presented as means ± SD. Statistical analyses were carried out using StatView (version 5.0; SAS Institute Inc., Cary, NC, U.S.A.). Infarct volumes were analyzed by one-way analysis of variance followed by the Bonferroni post hoc test. Infarct areas, brain shrinkage, and physiologic parameters were analyzed with the t-test.
RESULTS
Physiologic parameters
Regional cerebral blood flow was monitored in all mice. Injection of 1 μg vMIP-II or MIP-1α did not affect regional cerebral blood flow during or after ischemia (Table 3). As shown in Table 4, there were no significant differences in rectal or temporal muscle temperature among the groups. Neither vMIP-II nor MIP-1α affected cardiovascular parameters such as mean arterial blood pressure, pH, and partial pressures of carbon dioxide and oxygen.
Regional cerebral blood flow (rCBF)
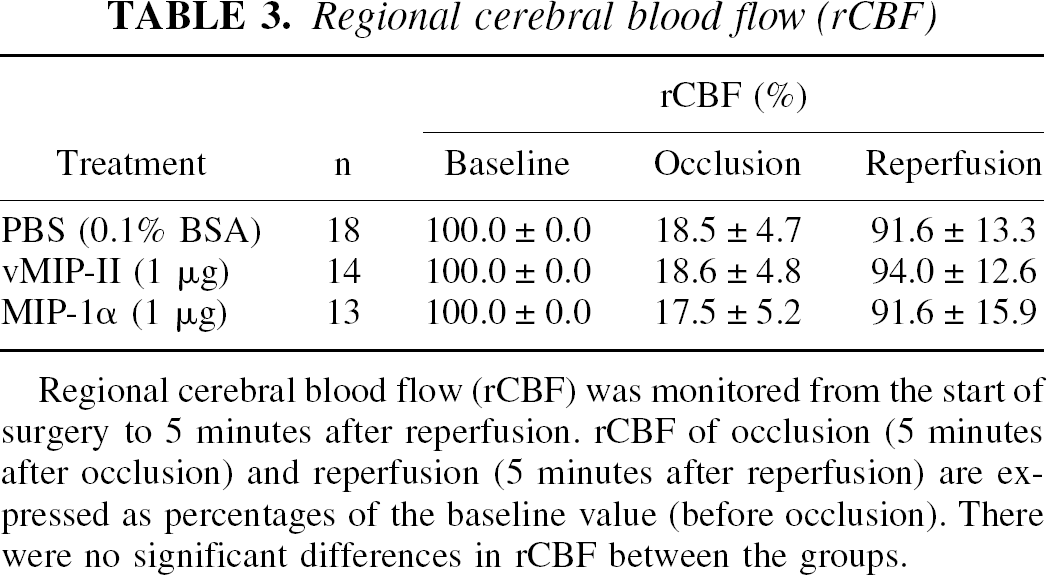
Regional cerebral blood flow (rCBF) was monitored from the start of surgery to 5 minutes after reperfusion. rCBF of occlusion (5 minutes after occlusion) and reperfusion (5 minutes after reperfusion) are expressed as percentages of the baseline value (before occlusion). There were no significant differences in rCBF between the groups.
Physiologic parameters
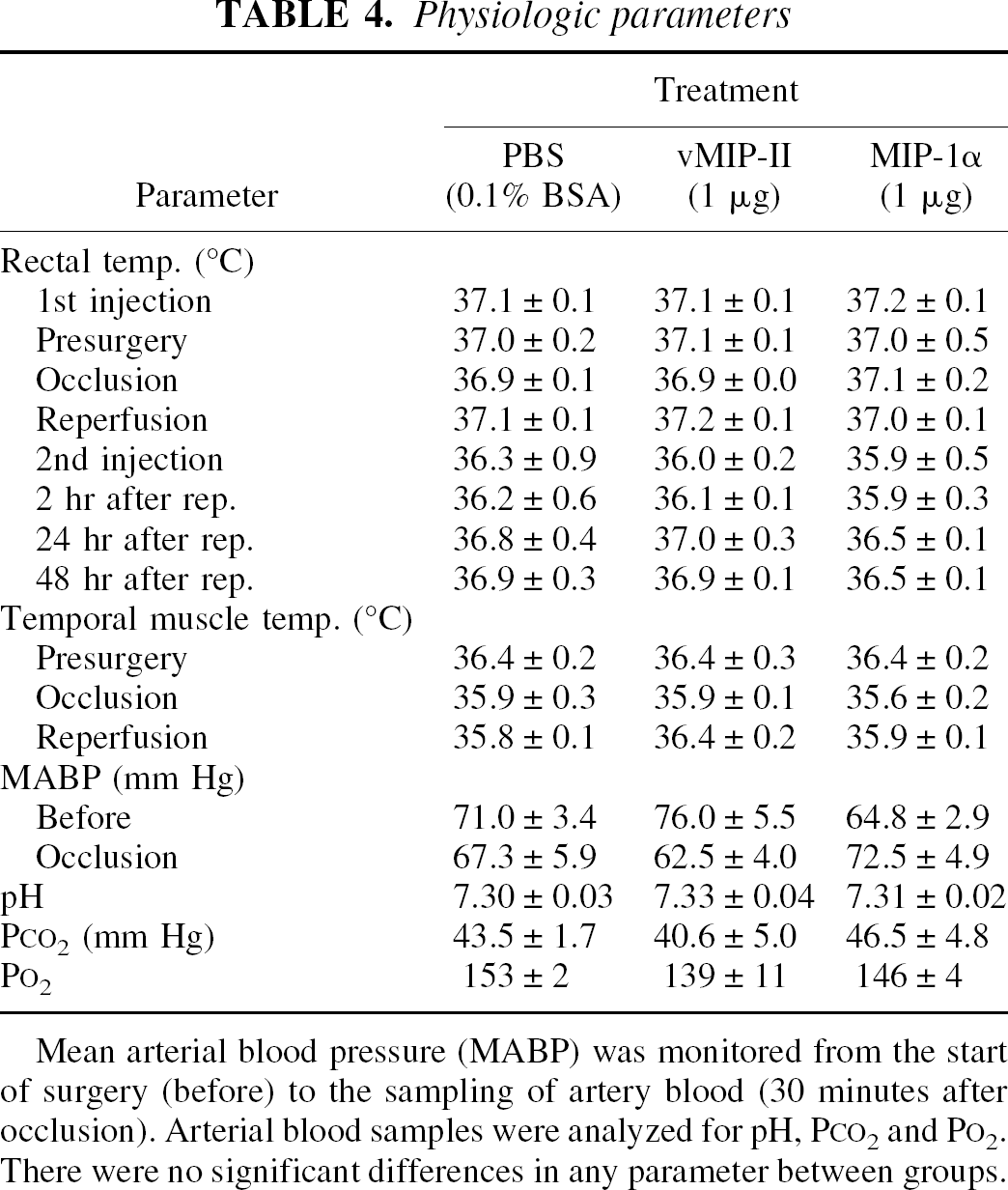
Mean arterial blood pressure (MABP) was monitored from the start of surgery (before) to the sampling of artery blood (30 minutes after occlusion). Arterial blood samples were analyzed for pH, PCO2 and PO2. There were no significant differences in any parameter between groups.
Effects of vMIP-II on brain infarction 48 hours after MCA occlusion
Representative brain sections from the groups injected with vehicle or vMIP-II are shown in Fig. 1. In vehicle-injected mice, brain infarction was primarily observed in the cerebral cortex and striatum. Injection of 1 μg vMIP-II reduced the infarct areas. The effects of vMIP-II on brain infarction were evaluated by measuring the infarct areas (Fig. 2A) and calculating the total infarct volumes (Fig. 2B). vMIP-II decreased the total infarct volume in a dose-dependent manner. Total infarct volume of the vehicle-injected group was 43.1 ± 14.1%, but was decreased to 31.6 ± 12.9%, 29.3 ± 12.3%, and 26.0 ± 6.1% in the groups injected with 0.01, 0.1, and 1μg vMIP-II, respectively. The reduction in infarct volume by vMIP-II was more prominent in the subcortical region than in the cortical region (Fig. 2C). vMIP-II at doses of 0.01 to 1 μg significantly decreased the infarct volume in the subcortical region, but significant reduction of infarct volume in the cortical region was observed in the group injected with 1 μg vMIP-II but not 0.01 or 0.1 μg vMIP-II.
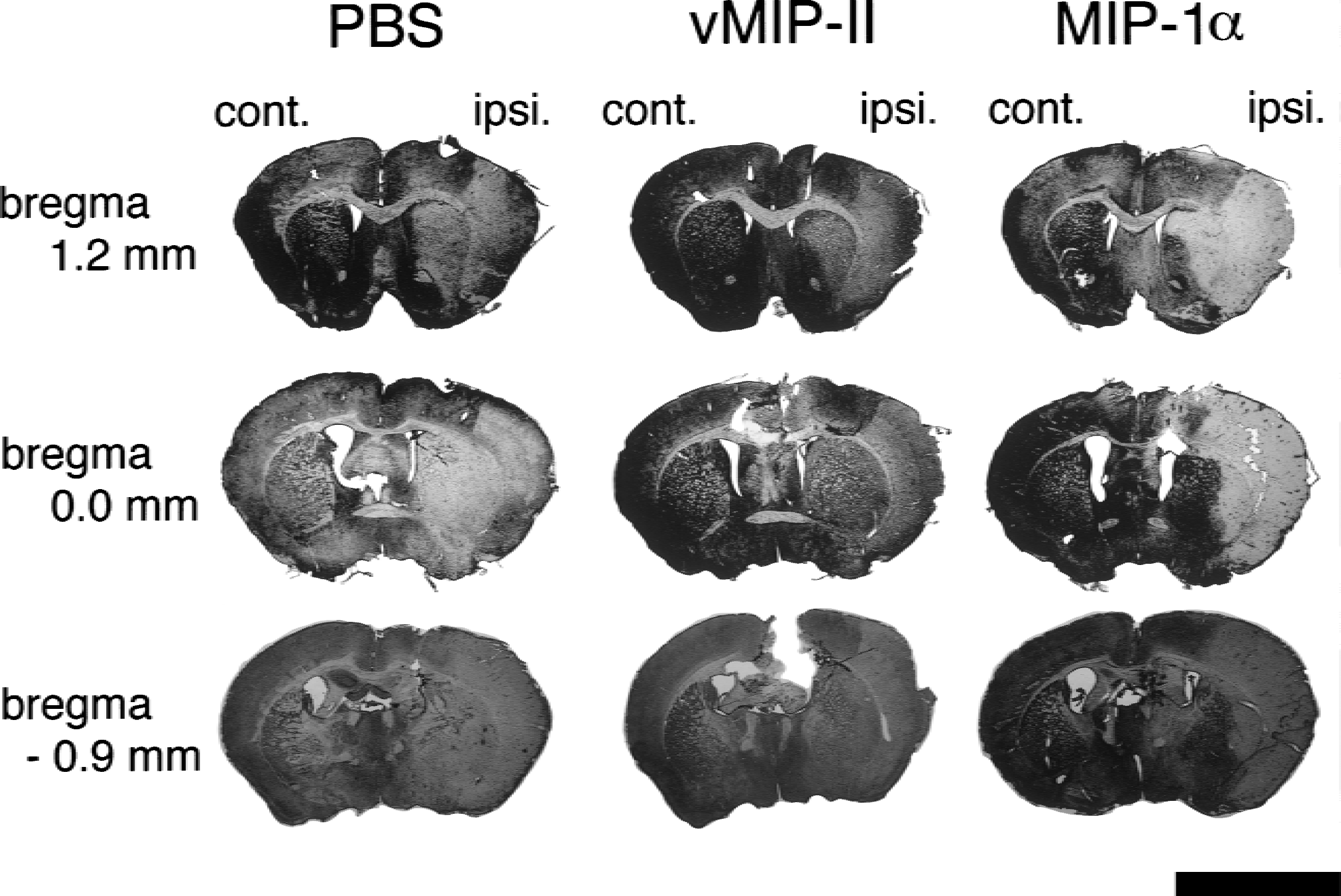
Photographs of representative brain sections from mice injected with vehicle (left), 1 μg vMIP-II (middle), or 1 μg MIP-1α (right). The brains were removed 48 hours after 1-hour MCA occlusion. Fourteen sections from bregma 3.2 mm to bregma −3.8 mm were prepared for each animal and stained with hematoxylin and eosin. Three sections at bregma 1.2, 0.0, and −0.9 mm are presented. Scale bar = 5mm.
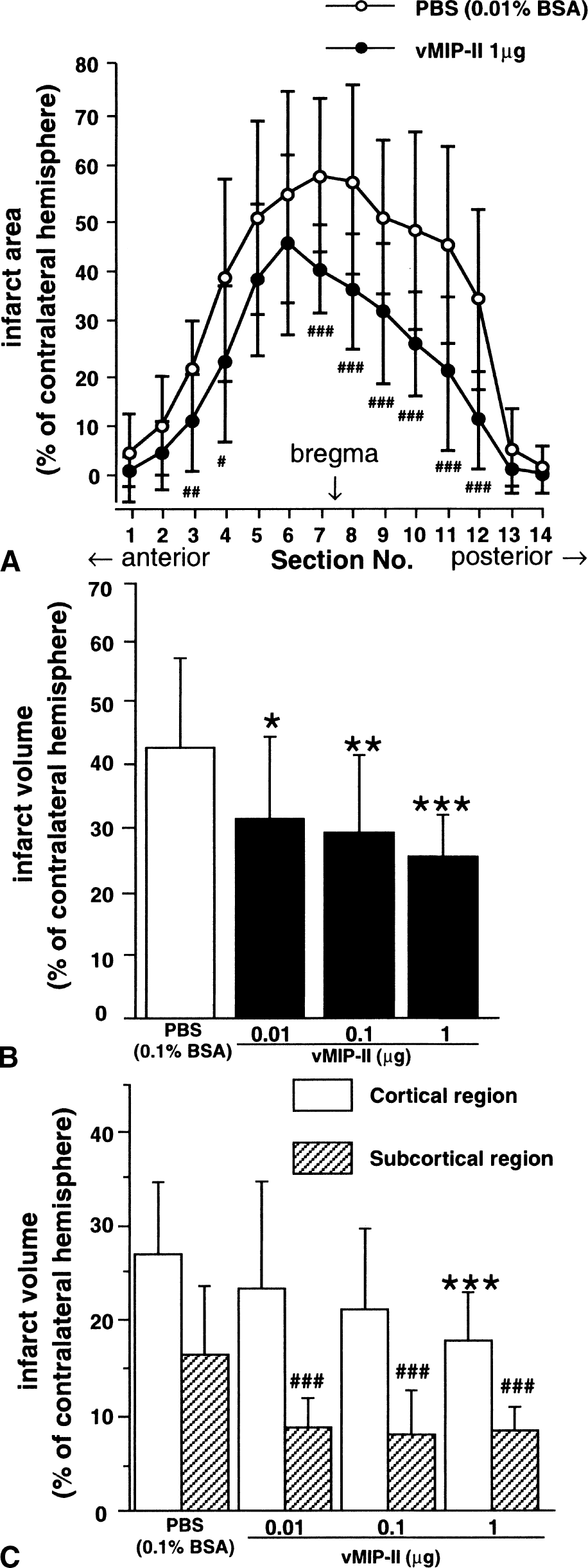
Effects of vMIP-II on brain infarction 48 hours after 1-hour MCA occlusion. Infarct areas (
Effects of MIP-1α on brain infarction 48 hours after MCA occlusion
Representative brain sections from the groups injected with vehicle or MIP-1α are compared in Fig. 1. MIP-1α at a dose of 1 μg significantly increased the infarct volume in the cortical region, but no significant effect was observed in the subcortical region (Fig. 3).
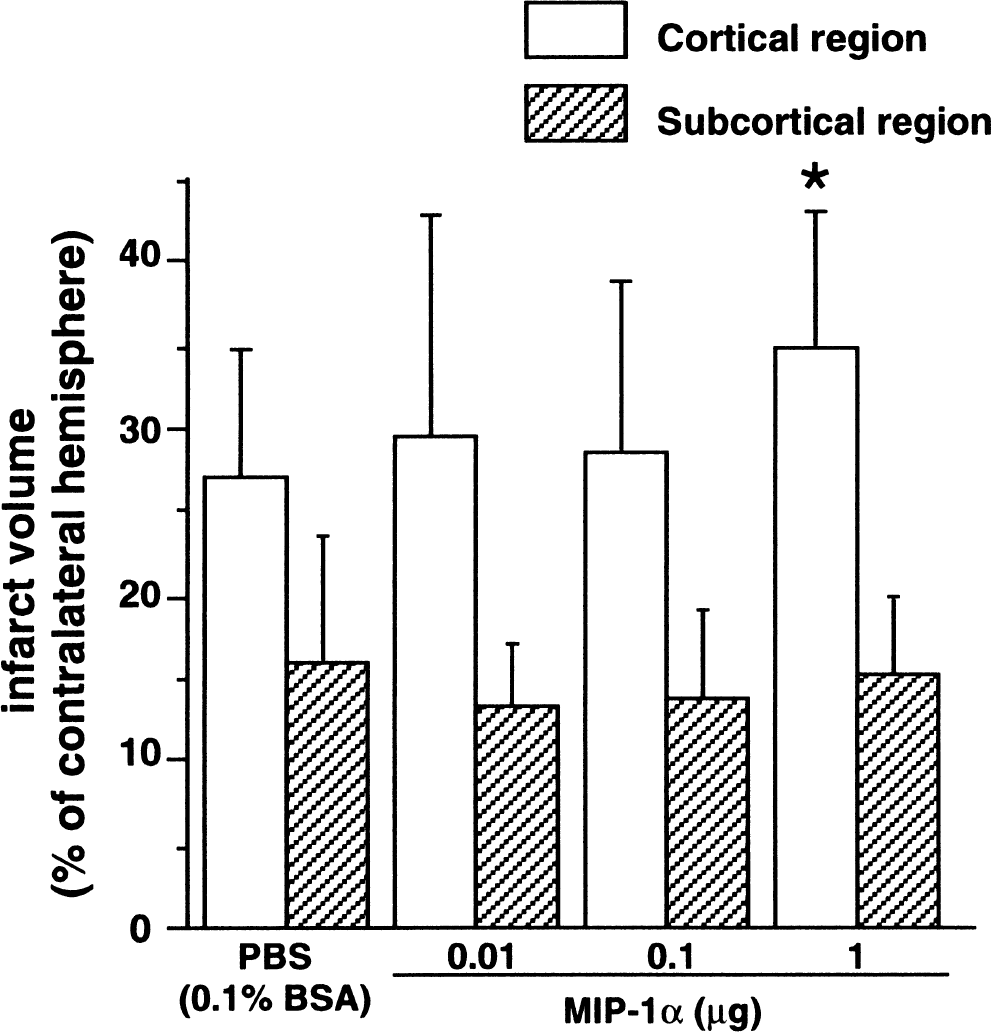
Effects of MIP-1α on brain infarction 48 hours after 1-hour MCA occlusion. The infarct volumes were evaluated separately in the cortical and subcortical regions and compared between the vehicle-and MIP-1α-injected groups. Infarct volumes are presented as percent of contralateral hemispheric values. Data are expressed as means ± SD. *, P < 0.05 compared with the vehicle-injected group (analysis of variance followed by the Bonferroni post hoc test). N = 13 to 18 in each group.
Effects of vMIP-II on brain damage 14 days after MCA occlusion
Brain-shrinkage volume was assessed at 14 days after reperfusion to elucidate whether vMIP-II attenuated or merely delayed brain injury (Fig. 4). Brain shrinkage volume was 23.6 ± 3.9% of the contralateral hemispheric volume in vehicle-injected mice. Injection of 1 μg vMIP-II significantly decreased the brain shrinkage to 17.4 ± 3.7%.
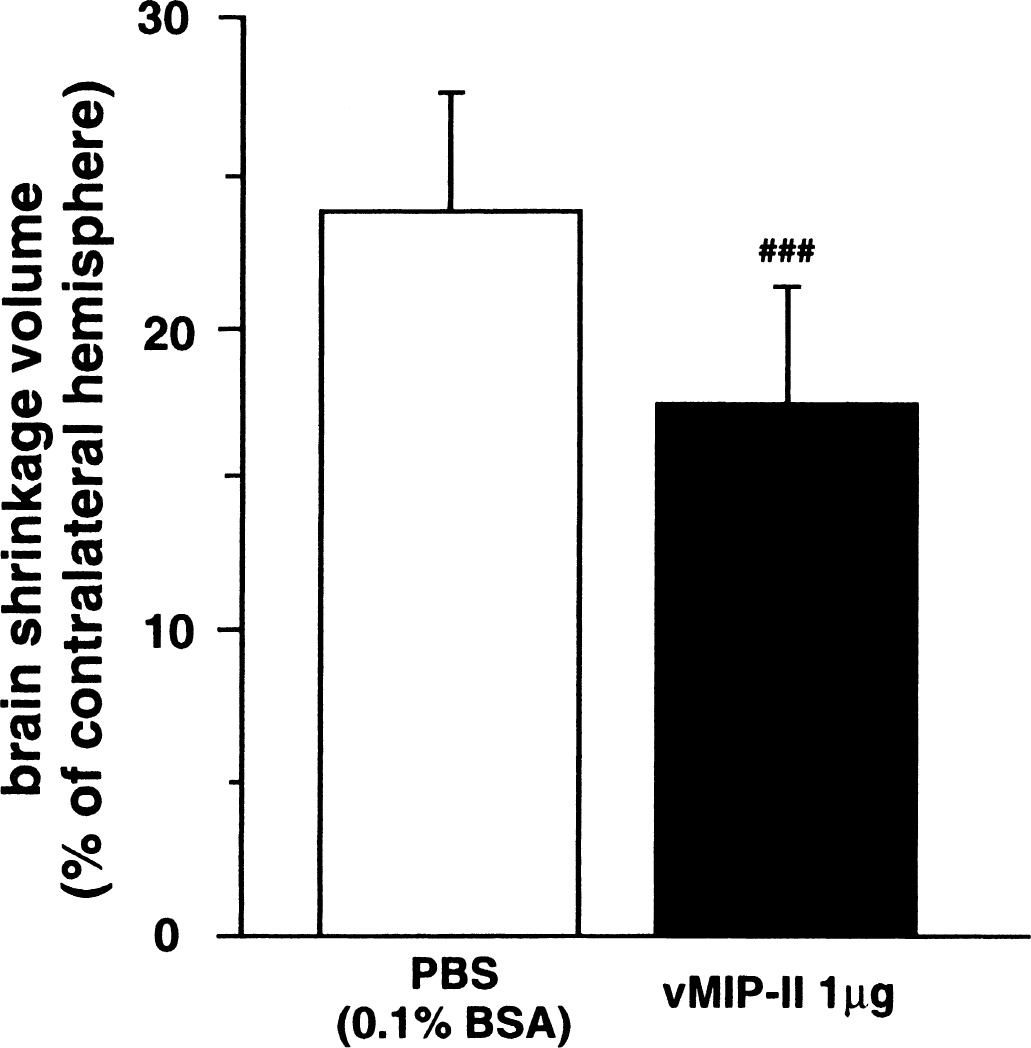
Effects of vMIP-II on brain shrinkage 14 days after 1-hour MCA occlusion. Brain shrinkage volumes are presented as percent of contralateral hemisphere values. Data are expressed as means ± SD. ###, P < 0.005 compared with the vehicle-injected group (t-test). N = 7 and 10 for vehicle-and vMIP-II-injected groups, respectively.
DISCUSSION
In this study, we demonstrated that intracerebroventricular administration of vMIP-II attenuated brain infarction after transient focal cerebral ischemia, whereas MIP-1α exacerbated brain infarction in the cortical region. Previously, we and other groups reported the mRNA expression of chemokines such as MIP-1α and monocyte chemoattractant protein-1 (Takami et al., 1997; Wang et al., 1995; Kim et al., 1995) in the ischemic brain. Furthermore, the effects of both of these chemokines are blocked by vMIP-II in vitro (Kledal et al., 1997; Chen et al., 1998). These findings suggest that chemokines produced in the brain during or after ischemia have neurodegenerative roles.
Previous studies showed that systemic administration of antichemokine antibodies against interleukin-8 or cytokine-induced neutrophil chemoattractant attenuated brain infarction resulting from focal cerebral ischemia (Matsumoto et al., 1997; Yamasaki et al., 1997). However, it is not clear whether the antibodies exerted the protective effects by acting in the brain or at the systemic level. In the present study, we injected vMIP-II directly into the cerebroventricle; therefore, it is more likely that vMIP-II exerted its protective effects by acting in the ischemic brain.
Because the intracerebroventricular administration of vMIP-II did not alter physiologic parameters such as cerebral blood flow, body temperature, and blood pressure, the protective effect of vMIP-II against ischemic brain injury was not mediated through actions on body temperature or the cardiovascular system. Furthermore, vMIP-II reduced the brain shrinkage 2 weeks after MCA occlusion, suggesting that this peptide actually attenuated and did not merely delay the brain injury.
The results of the present study show for the first time that central administration of a substance with an antagonistic effect on chemokine receptors protected the brain against ischemic injury resulting from transient MCA occlusion. These results, together with the previous findings that the mRNAs for several chemokines were induced in the ischemic brain, suggest that the chemokines produced in the brain play a crucial role in ischemic injury. Chemokines are probably involved in inflammatory reactions in the brain, which are considered to contribute to brain damage after ischemia (Barone and Feuerstein, 1999), and their receptors could be attractive targets for therapeutic intervention in stroke. In this context, it is interesting to examine whether systemically administered chemokine antagonists exert neuroprotective effects. This issue remains to be clarified in future studies.