
Abstract
Interleukin-1 (IL-1) receptor antagonist (IL-1ra) markedly reduces infarct volume induced by middle cerebral artery occlusion (MCAO) in the rat, when injected either centrally (intracerebroventricularly) or peripherally. The site or sites of action of IL-1 in stroke pathology, however, are not known. The present study investigated the site(s) of action of IL-1/IL-1ra in ischemic brain damage by studying the effects of local injection of IL-1ra into the cortex or striatum following permanent MCAO in the rat. Cortical injection of IL-1ra (5 µg) did not affect infarct volume in the cortex or striatum measured 24 h after MCAO. In contrast, striatal injection of IL-1ra ipsilateral to the infarction caused a significant and highly reproducible reduction of cortical (37%, p < 0.001) and striatal damage (27%, p < 0.001, corrected for edema) compared with vehicle-injected animals. Injection of IL-1ra (5 µg) into the striatum, contralateral to the infarction, resulted in a small (9%) but significant (p < 0.001) reduction of ipsilateral cortical damage, with no effect on ipsilateral striatal damage. Injection of a higher dose of IL-1ra (7.5 µg) in the contralateral striatum caused a further inhibition of ipsilateral cortical damage (24%, p < 0.001) and a significant reduction of ipsilateral striatal damage (16%, p < 0.001). In separate groups of rats, it was established that core temperature (measured continuously in free-moving animals with remote radiotelemetry) was not affected by striatal or cortical injection of IL-1ra. These data show that injection of IL-1ra into the striatum but not the cortex reduces infarct volume in both the striatum and the cortex, independently of effects on core temperature. These results imply that blocking striatal IL-1 contributes to IL-1ra-protective effects. We hypothesize that IL-1 may influence striatal distal cortical damage through either the release of specific substances or activation of polysynaptic pathways.
The cytokine interleukin-1 (IL-1) has been implicated in neuronal damage caused by cerebral ischemia, brain trauma, and excitotoxic damage (Relton and Rothwell, 1992; Lawrence and Rothwell, 1994; Garcia et al., 1995; Toulmond and Rothwell, 1995). IL-1β is induced rapidly (mainly in glia) shortly after the onset of cerebral ischemia in the rat (Liu et al., 1993; Wang et al., 1994; Yabuuchi et al., 1994; Hopkins and Rothwell, 1995). Application of IL-1β has protective effects on cultured neurons in vitro (Strijbos and Rothwell, 1995), but the expression of IL-1β in vivo is believed to promote neuronal death and formation of edema (Relton and Rothwell, 1992; Yamasaki et al., 1992; Loddick and Rothwell, 1996).
Injection of recombinant IL-1β does not cause damage in the normal rat brain but markedly exacerbates damage caused by permanent or reversible middle cerebral artery occlusion (MCAO) (Yamasaki et al., 1992; Loddick and Rothwell, 1996). In contrast, injection of IL-1 receptor antagonist (IL-1ra), a naturally occurring IL-1 antagonist, markedly inhibits neuronal damage caused by focal ischemia (MCAO), traumatic injury, excitotoxicity, hypoxia, or heat stroke (Lawrence and Rothwell, 1994; Martin et al., 1994; Garcia et al., 1995; Lin et al., 1995; Toulmond and Rothwell, 1995; Loddick and Rothwell, 1996). Whereas most of the neuroprotectants studied in cerebral ischemia (e.g., excitatory amino acid antagonists) inhibit only cortical damage (Muir and Lees, 1995), IL-1ra inhibits damage in both the cortex and the striatum (Loddick and Rothwell, 1996). This protection of the striatum suggests that IL-1ra affects mechanisms that are either nonglutamatergic or are downstream of glutamate receptors.
In previous studies on cerebral ischemia, IL-1ra has been administered either into the cerebral ventricles or systemically. While these routes of administration may allow diffusion of IL-1ra throughout the brain, they do not reveal the primary sites of action within the brain. It has been reported that induction of IL-1β mRNA is seen throughout the brain following cerebral ischemia, but the most remarkable induction following transient ischemia is observed in the striatum (Yabuuchi et al., 1994). Recent studies (Lawrence and Rothwell, 1994; Allan et al., 1995) have shown that localized striatal infusion of IL-1β paired with cortical infusion of excitotoxins (N-methyl-D-aspartate or AMPA agonists) exacerbate cortical excitotoxic damage. This distant effect of IL-1 on cortical damage led us to hypothesize that IL-1/IL-1ra may act specifically in the striatum and that localized striatal infusion of IL-1ra would result in the inhibition of both striatal and cortical damage following focal cerebral ischemia. This hypothesis was tested in the present study by investigating the effects of localized injection of IL-1ra into either the cortex or striatum on cortical and striatal infarct volumes caused by permanent MCAO in the rat.
METHODS
All experiments were performed on male Sprague-Dawley rats (Charles River, U.K.) weighing 230–270 g. Animals were housed at 21°C with ad libitum access to food and water. Guide cannulae were implanted stereotaxically into the cortex (coordinates, 2.7 mm anterior, 3.0 mm lateral, 2.4 mm ventral to bregma) or striatum (0.3 mm posterior, 4.0 mm lateral, 5.5 mm ventral to bregma) using coordinates from Paxinos and Watson (1986) of rats under halothane anesthesia (4% induction, 2–3% maintenance in oxygen) 7 days before the induction of ischemia.
Cerebral ischemia was produced by a proximal permanent occlusion of the MCA by electrocautery as described by Tamura et al. (1981). Anesthesia was induced by 4% halothane in oxygen and maintained at 2–3% halothane during the procedure. The total period from induction to recovery was ∼30 min. Body temperature was maintained after the surgical procedure by placing animals under heat lamps.
Either vehicle (saline) or human recombinant IL-1ra (Amgen, U.S.A.; 5 µg/µl saline) was infused at a rate of 1 µl/2 min through an injection cannula placed within the guide cannula. The injection cannula was withdrawn 2 min after the end of the infusion. A separate group of animals was infused with a larger dose of 7.5 µg of IL-1ra into the contralateral striatum.
Sham animals had operations with the preimplanted guide cannula. The same surgical protocol as stroked animals was followed until the coagulation of the MCA. At this point, the coagulating forceps were placed around the artery, but no current was passed. The animals were then returned to the surgical protocol and subsequent death and staining.
Infarct volume was determined 24 h after ischemia by staining 500-µm fresh brain sections with 2% triphenyltetrazolium chloride (Sigma). This procedure stains for mitochondrial viability, giving a clear representation of the lesion (Bederson et al., 1986). The lesion area from each section was charted onto standardized stereotaxic maps (Paxinos and Watson, 1986) using anatomical landmarks. These maps were then quantified by automated image analysis (SeeScan). Total lesion volumes were calculated by integration of the areas of infarction.
To determine the effects of L-1ra on core temperature, further groups of rats were implanted with guide cannula in the ipsilateral striatum or cortex, and temperature-sensitive radio-transmitters (Data Sciences International, U.S.A.) were inserted into the peritoneal cavity 5–7 days before MCAO. Animals were allowed to recover from anesthesia, and once conscious they were then placed in a separate telemetry room (22°C) and allowed to remain for at least 18 h before death. Data from the individual animals were compared at the time of onset of stroke. Body temperature data were compared as the area under the curve for each group. Temperature recordings from individual animals were then compared with their respective infarct volumes to determine any correlation between body temperature and infarct volume.
All data are expressed as group means ± SD. Statistical differences between lesion volumes were assessed using either Student's t test or Welch's alternate t test (where standard deviations were significantly different), with probability of < 5% for statistical significance.
RESULTS
Following MCAO, animals sustained damage in the left parietal cortex and striatum. The pattern of damage was consistent with the area of the brain supplied by the left MCA and with previously published data (Loddick and Rothwell, 1996). Animals showed moderate behavioral changes consisting of abnormal postural reflex and enhanced turning (circling) to the side contralateral to the infarction. Some animals exhibited seizure activity of vertical “hopping” and a few animals had episodes of barrel rolling.
Sham operations
Sham-operated animals showed no behavioral changes. Staining of the brain with triphenyltetrazolium chloride showed no obvious damage at any level.
Cortical injections
Injection of IL-1ra (5 µg) into the cortex ipsilateral to infarction had no effect on cortical infarct volume (vehicle 109.9 ± 5.5 mm3, IL-1ra 111.6 ± 4.3 mm3; n = 9) or damage in the striatum (vehicle 33.2 ± 2.1 mm3, IL-1ra 34.4 ± 1.8 mm3; n = 9) (see Fig. 1).
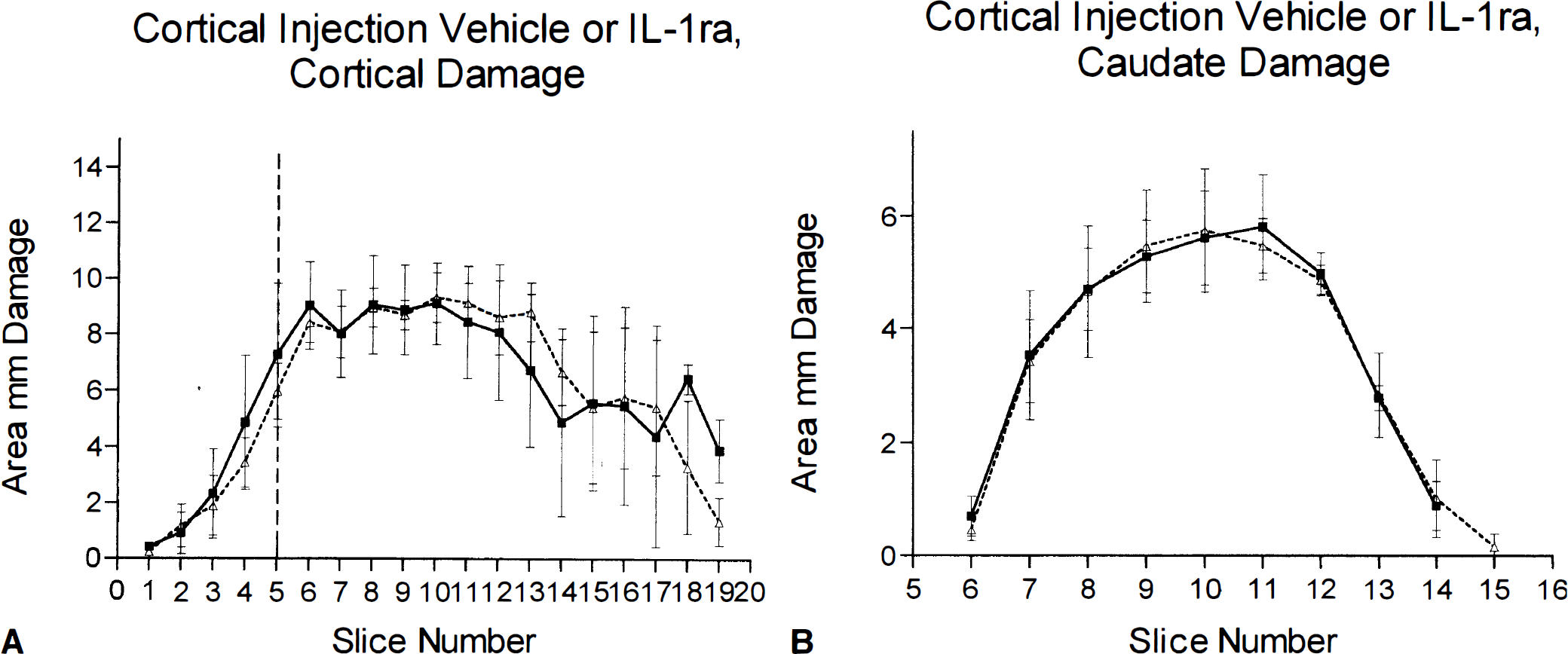
Infarct volume (mm2) measured 24 h after MCAO in rats that received cortical injection of IL-1ra (5 µg) or vehicle. Filled squares represent saline-injected animals, open triangles show IL-1ra-treated animals. Brain sections are shown on the abscissa at 0.5-mm intervals with bregma at slice 10. Vertical dashed line is the location of the injection cannula. There was no difference between vehicle-and IL-1ra-treated animals. Data shown as means ± SD, n = 9. See text for abbreviations.
Ipsilateral striatal injections
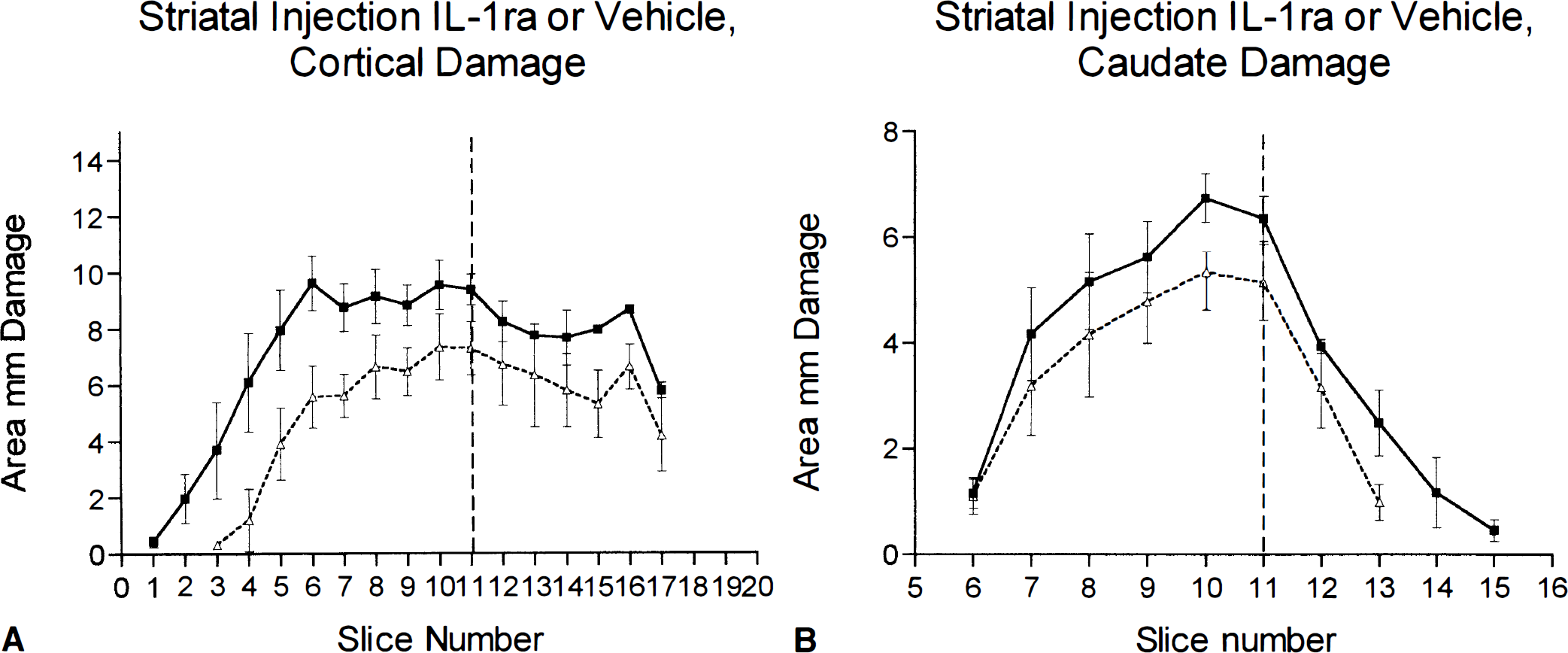
In contrast, injection of IL-1ra into the striatum ipsilateral to the occlusion produced a significant reduction (38%) in the cortical infarction volume (vehicle 120.8 ± 3.6 mm3, IL-1ra 75.2 ± 8.2 mm3 n = 9, p < 0.001, Welch's t test) (see Fig. 2). Protection was seen primarily in the anterior cortex in the motor regions (Fig. 3). There was also a reduction (21%) in the striatal infarction volume in IL-1ra-treated animals (vehicle 34.7 ± 3.2 mm3, IL-1ra 27.5 ± 2.7 mm3; n = 9, p < 0.001).
Infarct volume (mm2) measured 24 h after MCAO in rats that received striatal injection of IL-1ra (5 µg) or vehicle. Filled squares represent saline-injected animals, open triangles show IL-1ra-treated animals. Brain sections are shown on the abscissa at 0.5-mm intervals with bregma at slice 10. Vertical dashed line is the location of the injection cannula. Animals with injection of IL-1ra in the striatum ipsilateral to MCAO had significantly less damage in the cortex (p < 0.001, Welch's t test) and the striatum (p < 0.001, Student's t test) when compared with vehicle-treated animals. Data shown as means ± SD, n = 9. See text for abbreviations.
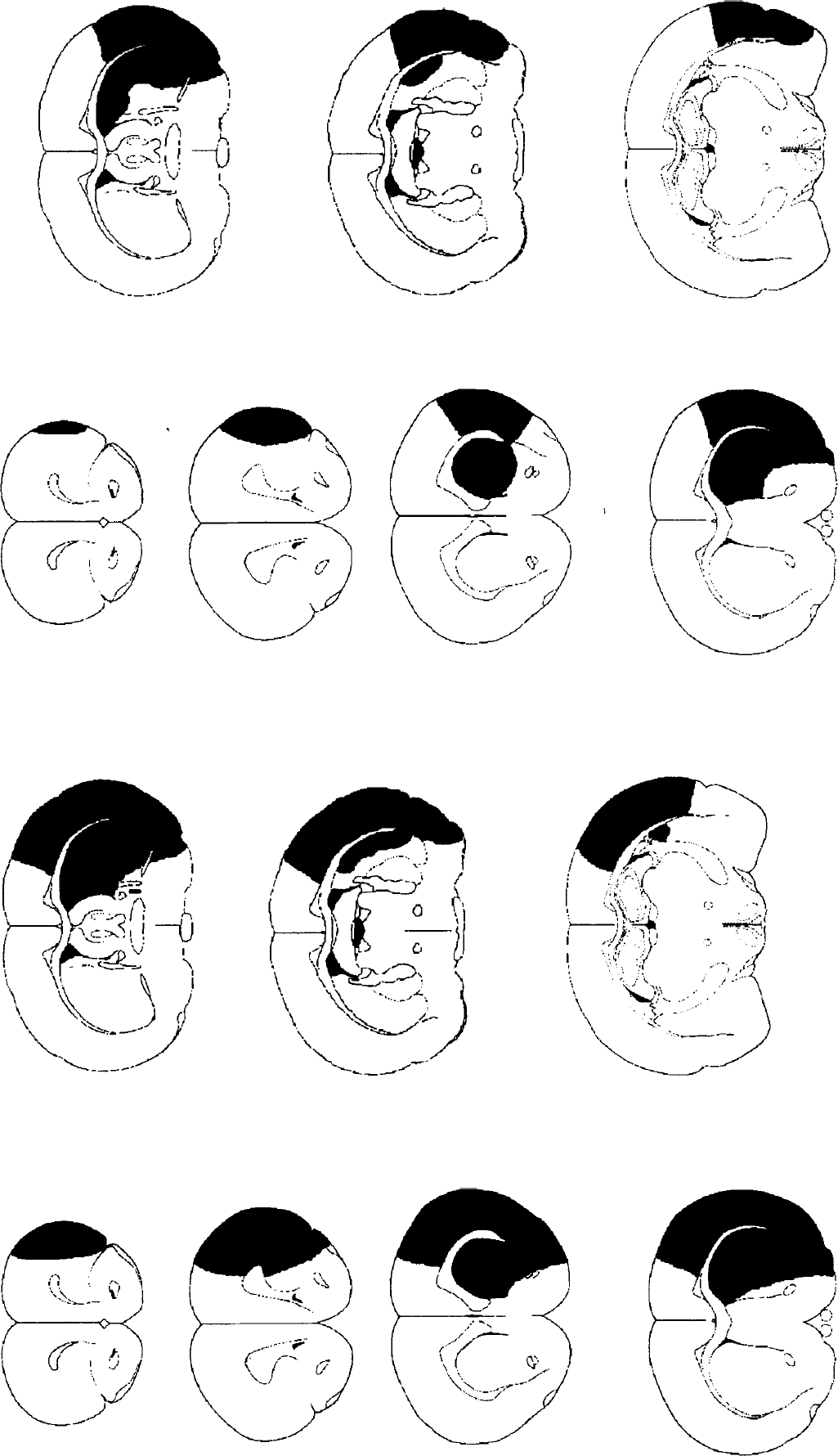
Brain slices depicting regions of damage (shaded areas) in representative animals measured 24 h after MCAO injected into the striatum with either vehicle (
Contralateral striatal injections
Injection of IL-1ra (5 µg) into the contralateral striatum reduced the volume (9%) of cortical infarction (vehicle 112.9 ± 3.6 mm3, IL-1ra 102.5 ± 4.9 mm3; n = 9, p < 0.001) (see Fig. 4). The difference in cortical damage was again observed primarily in the anterior cortex. In contrast, injection of IL-1ra at this dose into the contralateral striatum did not affect the volume of striatal damage ipsilateral to the infarction (vehicle 33.1 ± 1.9 mm3, IL-1ra 30.9 ± 1.9 mm3; n = 9, NS) (see Fig. 4).
Infarct volume (mm2) measured 24 h after MCAO in rats injected with IL-1 ra (5 µg) or vehicle into the contralateral striatum. Filled squares represent saline-injected animals, open triangles show IL-1ra-treated animals. Brain sections are shown on the abscissa at 0.5-mm intervals with bregma at slice 10. Vertical dashed line is the location of the injection cannula. Animals with contralateral injection of IL-1ra had significantly less damage in the cortex (p < 0.001, Student's t test) but no difference in infarct volume in the striatum. Data shown as means ± SD, n = 9. See text for abbreviations.
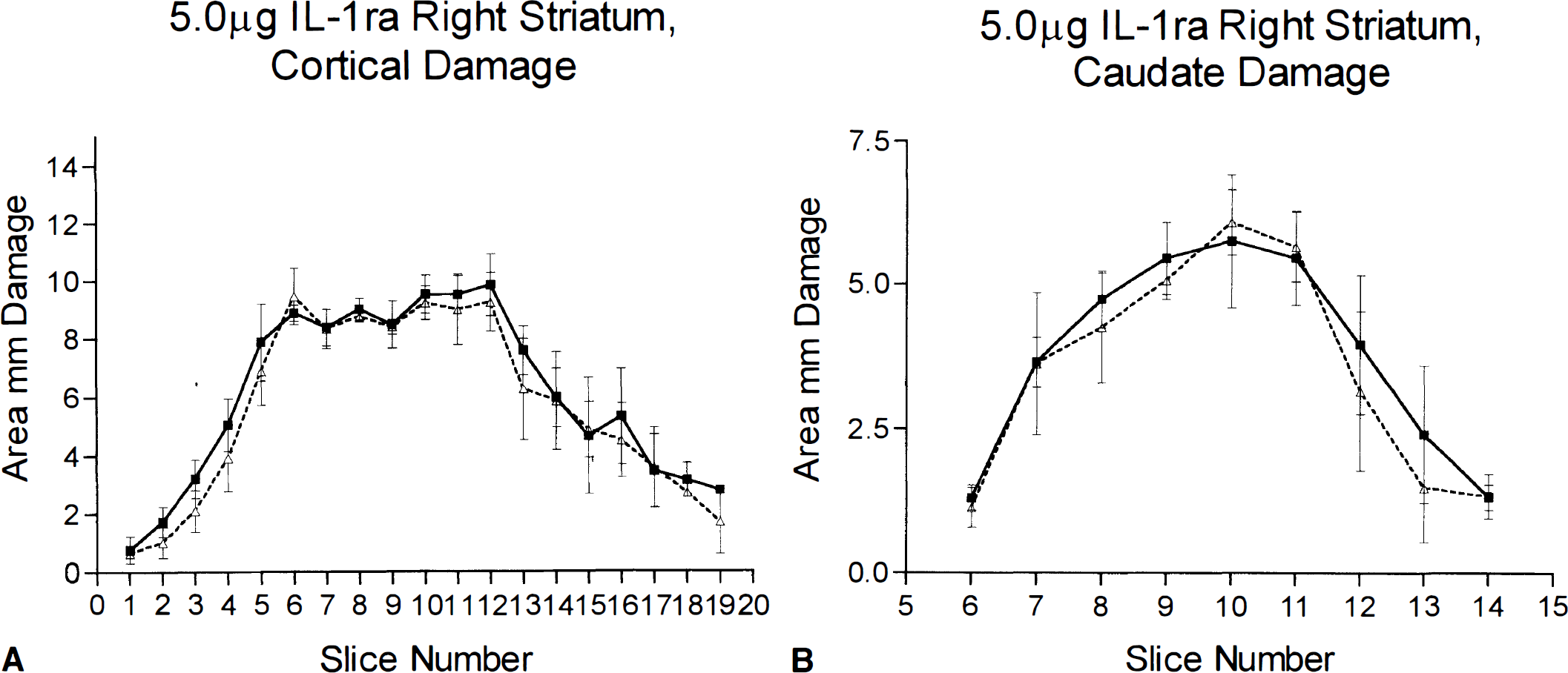
Injection of a higher dose of IL-1ra (7.5 µg) into the contralateral striatum produced a significant reduction (24%) in cortical infarction damage (vehicle 131.3 ± 5.0 mm3, IL-1ra 99.2 ± 4.1 mm3; n = 9, p < 0.001) (Fig. 5), which was seen at all levels of the cortex. There was also a significant reduction in striatal infarction damage (vehicle 32.1 ± 2.0 mm3, IL-1ra 26.9 ± 1.1 mm3; n = 9, p < 0.001) (Fig. 5), which was apparent in the more anterior levels of the striatum.
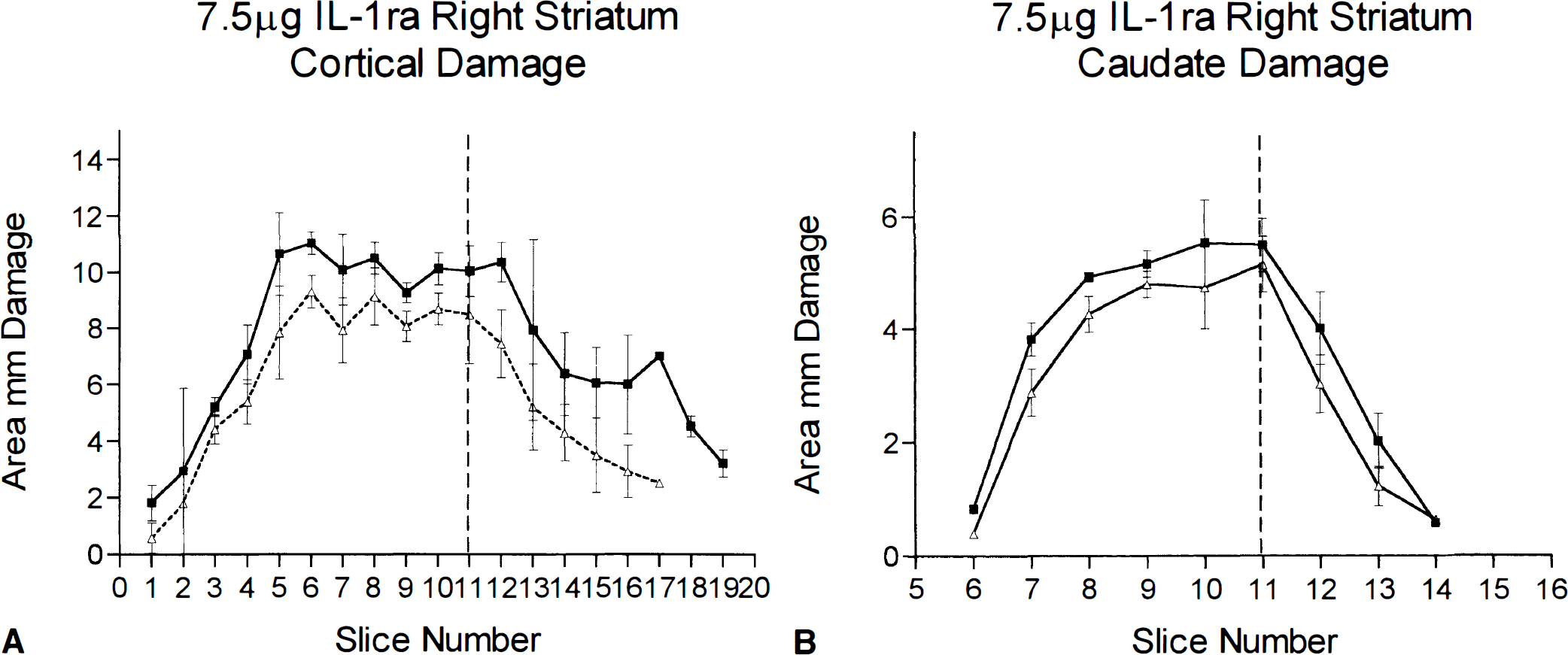
Infarct volume (mm2) measured 24 h after MCAO in rats that received contralateral striatal injection of IL-1ra (7.5 µg) or vehicle. Filled squares are saline-injected animals, open triangles are IL-1ra-treated animals. Brain sections are shown on the abscissa at 0.5-mm intervals with bregma at slice 10. Vertical dashed line is the location of the injection cannula. Animals with contralateral injection of IL-1ra (7.5 µg) had significantly less damage in both the cortex and the striatum (p < 0.001, Student's t test). Data shown as means ± SD, n = 9. See text for abbreviations.
Temperature recordings
After MCAO there was a modest (0.5–0.8°C) reduction in body temperature in all animals (striatal or cortical injections) over the first 3 h. The minimum body temperatures were observed 1 h after occlusion. By 3–4 h after surgery, body temperatures had returned to normal (i.e., within the range of unoperated controls, data not shown) and remained so for the of the experiment. Sham-operated animals exhibited hypothermia for ∼1 h with a minimum temperature of 36.6 ± 0.3°C (Fig. 6) but thereafter exhibited normal body temperatures.
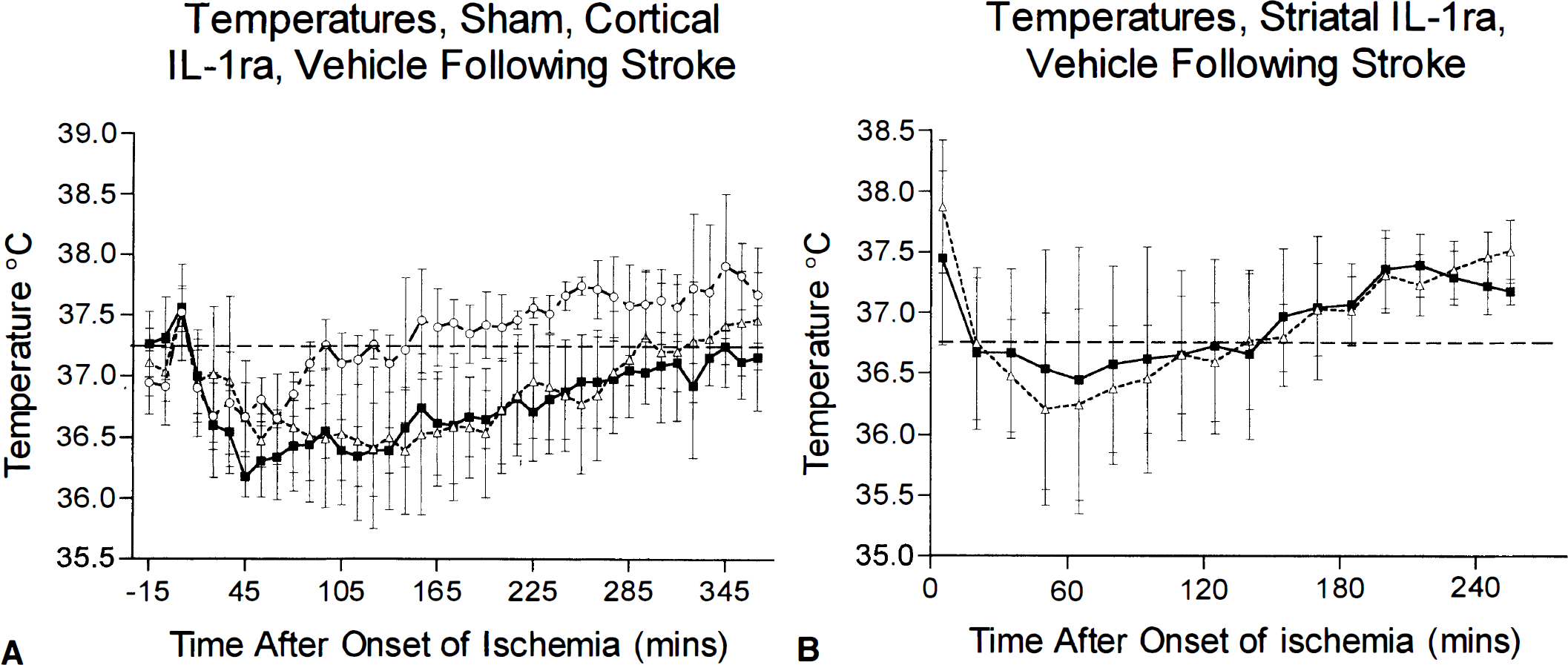
Core temperatures of saline- and IL-1ra-treated animals after MCAO.
Injection of IL-1ra into the striatum of rats after MCAO had no significant effect on body temperature when compared with the saline-injected ischemic animals (Fig. 6) (minimum core temperature IL-1ra 36.2 ± 0.8°C, saline 36.5 ± 0.9°C). Striatal injection of IL-1ra again significantly reduced lesion volumes in both the cortex (vehicle 99.1 ± 5.1 mm3, ± 7.1 mm33; n = 6, p < 0.0001) and the striatum (vehicle 29.4 ± 1.3 mm3, IL-1ra 16.6 ± 2.1 mm3; n = 6, p < 0.0001). When the lesion volumes and temperatures of the individual animals were linearly regressed, there was no correlation between core temperature and infarct volume in either the vehicle-treated group (r = 0.08) or the IL-1ra-treated group (r = 0.014).
Injection of IL-1ra into the cortex also had no significant effect on core temperature (Fig. 6) (minimum core temperature vehicle 36.3 ± 0.3°C, IL-1ra 36.6 ± 0.2°C). Cortical injections of IL-1ra did not affect lesion volume in the cortex (vehicle 77.7 ± 15.9 mm3, IL-1ra, 83.8 ± 17.4 mm3; n = 4, NS) or striatum (vehicle 24.0 ± 4.2 mm3 IL-1ra, 25.0 ± 1.0 mm3; n = 4, NS). Again there was no correlation between core temperature and infarct volume in either the vehicle-treated group (r = 0.43) or the IL-1ra-treated group (r = 0.41).
DISCUSSION
We and others have previously demonstrated potent neuroprotective effects of IL-1ra injected either into the cerebral ventricles (Relton and Rothwell 1992; Loddick and Rothwell, 1996) or systemically (Relton and Rothwell, 1992; Garcia et al., 1995). The objectives of the present study were to investigate the site(s) of action of IL-1ra in protecting against ischemic brain damage.
Injection of IL-1ra (5 µg) into the cortex (the primary site of infarction) did not affect damage caused by permanent MCAO. The cortical coordinates were chosen to target a region of the cortex that is protected by intracerebroventricular administration of IL-1ra (10 µg) (Loddick and Rothwell, 1996), suggesting that this area is within the ischemic penumbra and thus is salvageable. Although a higher dose of IL-1ra (10 µg) has been injected intracerebroventricularly in previous studies (Relton and Rothwell, 1992; Loddick and Rothwell, 1996), a dose of 5 µg IL-1ra injected intracerebroventricularly also markedly inhibits (>50%) damage caused by MCAO (J. K. Relton and N. J. Rothwell, unpublished data).
The site of cortical IL-1ra injection used here (in the frontal and parietal cortex) is the origin of corticostriatal projections that terminate at the site of the coordinates for the striatal injections (McGeorge and Faull, 1989). We have also ested the efficacy of a higher dose (7.5 µg) of IL-1ra and at another cortical site (0.2 mm anterior, 3.0 mm lateral, 2.0 mm ventral to bregma; 5 µg IL-1ra) (Paxinos and Watson, 1986) and did not observe any cortical or striatal protection (data not shown). The failure of IL-1ra administered into the cortex to reduce ischemic damage suggests that cortical production of IL-1β does not mediate ischemic damage in the cortex or the striatum.
In marked contrast, IL-1ra injected (at the same dose, 5 µg) into the ipsilateral striatum markedly inhibited damage in both the striatum and the cortex. IL-1ra reduced damage throughout the striatum, which may reflect diffusion within the structure, whereas inhibition of cortical damage was greatest in the more anterior region of the cortex, where the corticostriatal projections originate. In later experiments, we observed cortical protection (20–40%) in animals with different striatal injection sites (0.2 mm anterior, 3.6 mm lateral, 4.0 mm ventral or −0.3 mm anterior, 3.0 mm lateral, 7.0 mm ventral to bregma) (Paxinos and Watson, 1986), and the extent of protection corresponded approximately to the areas of origin of the corticostriatal projections in the ischemic penumbra (i.e., greater protection in the caudal parietal cortex) (data not shown). Since IL-1ra injected directly into the cortex failed to inhibit damage, cortical neuronal protection after striatal injection is probably not due merely to diffusion of IL-1ra from the striatum to the cortex.
Injection of IL-1ra (5 µ) into the contralateral striatum due (where no damage was apparent) resulted in a small but significant inhibition of cortical infarct volume. The area protected in this experiment was primarily in the anterior cortex, again at the origin of the cortical projecting fibers terminating at the injection site (McGeorge and Faull, 1989). It has been reported that 13% of cortical neurons projecting to the striatum project bilaterally to both hemispheres (McGeorge and Faull, 1989), suggesting the existence of a direct pathway that could influence contralateral neuronal death. Infusion of IL-1β in the striatum ipsilateral or contralateral to cortical infusions of either N-methyl-D-aspartate or AMPA receptor agonists exacerbates cortical damage without damaging the striatum (C. Lawrence and N. J. Rothwell, unpublished data), again supporting a direct action of IL-1β on cortical nerve terminals.
Injection of a higher dose of IL-1ra (7.5 µg) in the striatum contralateral to the infarction led to a greater inhibition (30%) of damage in the ischemic cortex that was not as site specific as seen with the lower dose. There may have been greater diffusion of IL-1ra throughout the striatum, allowing more projections to be affected. The inhibition of damage in the striatum ipsilateral to the infarction suggests that the IL-1ra either diffused across hemispheres to limit the striatal damage (a possibility with this higher dosage) or that IL-1β expressed contralaterally from the infarction influences striatal damage. We have found expression of IL-1β in the contralateral striatum following stroke (C. Davies et al., unpublished data), suggesting that the “unaffected” hemisphere may be influenced by ischemia in the contralateral hemisphere.
Core (peritoneal) temperature (measured continuously by radiotelemetry) in conscious vehicle- or IL-1ra-treated animals declined over the first 3 h after the onset of ischemia. This reduction of body temperature has several possible causes, including anesthesia, surgery, effects of individual housing, and the reduced mobility presenting from stroke damage. However, since sham-operated rats shared only modest (0.5°C) and transient (∼1 h) hypothermia, it is likely that the prolonged hypothermia resulted from the effects of cerebral responses to cerebral ischemia.
In spite of the modest (hypothermic) effects of ischemia on core temperature, no significant differences in temperature were observed between rats administered with vehicle or IL-1ra into either the striatum or the cortex. Although cooling can reduce infarct volume after transient MCAO, the temperatures required to achieve significant protection (30–33°C) (Green et al., 1992; Karibe et al., 1994) are much lower than those seen in the present study. Moyer et al. (1992) demonstrated a reduction (77%) in infarct volume in permanent distal occlusion of the MCA after spontaneous hypothermia at brain temperatures of 33°C. We cannot rule out the possibility that IL-1ra injected into the striatum caused significant changes in brain temperature that were not reflected by changes in core temperature. However, Karibe et al. (1994), using simultaneous monitoring of brain, temporal muscle, and rectal temperatures, showed little difference between brain and rectal temperatures in whole-body hypothermia. The data presented here, together with the fact that marked reductions in core temperature (to 33°C) have only modest effects on infarct volume in a proximal model of permanent MCAO (Ridenour et al., 1992), indicate that the very potent effects of striatal IL-1ra demonstrated here cannot be explained by selective effects on body temperature.
Several hypotheses can be advanced to explain the site-specific protective effects of IL-1ra on ischemic brain damage. First, the main pathophysiological effects of IL-1β may occur in the striatum. IL-1 could influence the terminals of either cortical or thalamic neurons, which may express IL-1 receptors. In separate studies we have shown that biotinylated IL-1ra injected either into the striatum or intracerebroventricularly diffuses rapidly throughout the striatum and to a lesser extent the cortex and binds predominantly to neurons (S. Toulmond et al., unpublished data). The binding of biotinylated IL-1ra is fully displaced by coinfusion of IL-1α or IL-1β. These data indicate the presence of IL-1 receptors in the striatum and cortex but with the greatest density in the striatum. They also show that IL-1ra injected into the striatum does reach the cortex, though it is not clear if this is due to passive diffusion or active (e.g., axonal) transport. The effect(s) of IL-1β on these striatal receptors could then lead indirectly to damage of cell bodies of affected neurons in the cortex. We have conducted preliminary studies of site-specific injections with IL-1β at the onset of ischemia. Striatal injection of IL-1β potentiates ischemic damage in both the striatum and the cortex, while cortical injection of IL-1β does not affect infarct volume (R. P. Stroemer and N. J. Rothwell, unpublished data). It should be noted that IL-1β itself does not kill normal neurons but exacerbates neuronal death caused either by ischemia or by excitotoxicity (Allan et al., 1995; Loddick and Rothwell, 1996).
The second hypothesis of selective IL-1ra protection is that the expression or presence of IL-1ra protection is that the expression or presence of IL-1β in the striatum results in the release or expression of other pathological agents from this region that then diffuse to, or indirectly affect other, distal brain regions, and it is these putative agents, rather than IL-1β itself, that are responsible for producing damage in the cortex. IL-1β is known to generate other cytokines, prostaglandins, nitric oxide, complement, and kinins and promote neutrophil invasion (Kilbourn and Belloni, 1990; Farooqui and Horrocks, 1991; Kluger, 1991; Hewett et al., 1993; Szabo et al., 1993; Rothwell and Hopkins, 1995; Dinarello, 1996). It is possible that the production of striatal IL-1β initiates a cascade of toxic and/or inflammatory agents that then potentiate ischemic damage in the cortex.
There is also evidence favoring an argument for a retrograde rather than an orthograde pathway via the thalamus. Projections from the basal ganglia have some convergence within the thalamus and the subthalamic nucleus in the rat. Projections from the individual nuclei in the thalamus then diverge to different regions of the cortex. This convergence and divergence of neuronal pathways suggest that an orthograde protection through the thalamus would not be as specific as the results seen in this study. Some studies have demonstrated projections from the basal ganglia to the cortex (Dimova and Usunoff, 1989), providing a possible orthograde striatocortical pathway that could explain the results seen in this study. It must be noted, however, that these projections are scarce and diffusely organized.
There are several arguments to support the first hypothesis. The regions with the greatest protection are some distance from the injections, corresponding to the origins of the corticostriatal projections that terminate at the injection site, presumably the areas of the striatum that will have the highest concentration of IL-1ra. This pattern of protection is repeated in the animals with the contralateral striatal injections of IL-1ra. If neuronal protection was dependent upon the reduction of secondary toxins, the protection should have a different distribution, echoing a concentration gradient. The protection seen in the low-dose contralateral injections is almost all in the first few slices, 3–4 mm away from the injection site.
In summary, the results of this study demonstrate that ischemic damage in the cortex is influenced by IL-1β/IL-1ra in the striatum. Blocking striatal IL-1β contributes to IL-1ra's protective effects in the areas of origin of the corticostriatal projections. Although there is a modest hypothermia shortly after stroke, this protection is not caused by the potentiation of hypothermia by striatal injection of IL-1ra. These data provide evidence that striatal circuits, either retrograde or orthograde, influence pathological processes in the cortex in ischemia.
Footnotes
Abbreviations used
Acknowledgements
We are grateful to Amgen, U.S.A., for the supply of IL-1ra and to Anthea Hughes and Helen Anforth for technical support.