
Abstract
We have characterized the induction of the mitogen-inducible form of cyclo-oxygenase, COX-2, during focal cerebral ischemia following permanent middle cerebral artery occlusion (MCAO) in the rat. Marked unilateral induction of COX-2 mRNA was detected in ischemic regions ipsilateral to the occlusion. A significant increase in COX-2 mRNA was detected in “core” and “penumbra” regions of the cerebral cortex between 4 and 24 h after occlusion; this was most marked at 4 h in the penumbra region, in which a 19-fold increase above untreated control levels was detected. A smaller but significant induction was also detected at 4 h in the caudate. A correlation was demonstrated between the extent of COX-2 mRNA induction in cortical regions at 4 h and the severity of tissue damage subsequently detected at 24 h post MCAO. MK-801 significantly attenuated the induction of COX-2 mRNA in the penumbra region at 4 h. The demonstration of COX-2 induction following experimental ischemia highlights the importance of this reaction and its products and by-products, for example, free radicals, in the tissue response to this insult.
Although the mechanisms that give rise to ischemic brain damage have not been fully determined, there is mounting evidence implicating the involvement of glutamate, calcium, and free radicals. It has emerged that excitatory responses mediated through the rapid action of glutamate also have profound effects on gene activation, producing long-term cellular responses. These responses are associated with the induction of immediate early genes (IEGs), which show a distinct time course of activation (Dragunow and Robertson, 1987a,b; Saffen et al.; Bartel et al., 1989; Cole et al., 1989; Janssen-Timmen et al, 1989; Sonnenberg et al., 1989; Simonato et al., 1991; Colląo-Moraes et al., 1994). Many of these IEGs (e.g., c-fos, c-jun, zif-268) in turn regulate the action of a number of other genes, thus initiating a cascade of gene activation (Sheng and Greenberg, 1990; Nathans et al, 1991). An ischemia/reperfusion study has demonstrated, using nuclear run-on assays, that the elevated levels of c-fos, jun-B, and c-jun mRNA are likely to be due to an increased rate of transcription (An et al., 1993). The involvement of these IEGs in the regulation of gene expression in ischemia is also supported by an increase in DNA binding activity of the transcriptional factor AP-1 (formed from fos and jun dimers), determined directly by mobility shift assays.
Identifying the targets of these IEGs is necessary to characterise the cellular response to synaptic activation. Recently, using molecular cloning strategies to identify genes induced by excitatory synaptic activity in brain neurones, Yamagata et al. (1993) identified a mitogen-inducible cyclo-oxygenase (COX) that was markedly activated by synaptic activity. Cyclo-oxygenase, or prostaglandin H-synthetase, is the rate-limiting enzyme in the biosynthetic pathway leading to the production of prostaglandins (PG) and thromboxanes from arachidonic acid. This pathway leads to the formation of PGG2 from arachidonic acid and two molecules of oxygen, which is then reduced to PGH2, and in turn acts as the precursor of various eicosanoids including PGE2, PGF2cα, PGD2, prostacyclin, and thromboxane A2. The importance of this enzyme in ischemic damage is indicated by the observation that pretreatment with a COX inhibitor markedly decreases the ischemic damage affecting the CA1 region of the hippocampus in gerbils (Sasaki et al., 1988).
Two isoforms of COX have been identified: COX-1, a constitutively expressed gene (De Witt and Smith, 1988; Funck et al., 1991), and, more recently, a mitogen-inducible form, COX-2, whose expression in mouse fibroblasts is induced by growth factors, phorbol esters, and serum (Herschman, 1991; Kujubu et al., 1991; Xie et al., 1991; O'Banion et al., 1992). COX-1 and COX-2 exhibit approximately 75% amino acid sequence homology (Funk et al., 1991; Kujubu et al., 1991), with the important catalytic amino acid residues conserved (Shimokawa et al., 1990). Neuronal expression of COX-2 is rapidly and transiently induced by seizures or N-methyl-
In this study, we characterized the time course of COX-2 mRNA induction during focal cerebral ischemia using the intravascular suture model of middle cerebral artery occlusion (MCAO) in the rat. The pharmacological sensitivity of the response was investigated by using the NMDA receptor antagonist MK-801.
METHODS
Animal model of MCAO and reperfusion
Male Sprague-Dawley rats weighing 300 ± 25 g were anaesthetized with 4% halothane in a 1:1 mixture of nitrous oxide and oxygen after an overnight fast. With the halothane then maintained at 1.5% in an open anaesthetic system without tracheotomy, the origin of the left middle cerebral artery (MCA) was occluded with an intravascular suture following the method of Koizumi et al. (1986), described in an English version by Nagasawa and Kogure (1989). Briefly, this involved introducing a 4/0 nylon suture (Ethylon, Ethicon Ltd., U.K.) with 7 mm of its shaft from the tip thickened with silicone rubber (Silasatic sealant 732 RTV, Merck Ltd., U.K.) to an outside diameter of 0.28 mm, into the left external carotid artery in a retrograde fashion toward the carotid bifurcation, and then directed distally up the left internal carotid artery to a distance of 17.5 ± 0.5 mm from the carotid bifurcation to the tip of the suture. Postmortem examination of the brains for the exact location of the intravascular suture in pilot studies (Calląo-Moraes et al., unpublished data) showed that the suture fully occluded the left anterior portion of the circle of Willis, from the anastomosis of the left internal carotid artery to the origin of the left anterior cerebral artery (ACA). In the process, the origins of the left MCA and penetrating branches supplying the caudate nucleus/putamen were blocked off. Perfusion of the left ACA territory is maintained by an azygous continuation of the union of both left and right ACA in the rat brain (Green, 1968). Access to the left posterior cerebral artery (PCA) from the basilar artery via the posterior communicating artery was uninterrupted. The suture was secured at two points: first, to the stump of the ligated left external carotid artery; second, within the ligated internal carotid artery, proximal to the pterygopalatine branch. Rectal temperature was maintained throughout at 37.0 ± 0.5°C with a heat lamp, and fluid loss during surgery was compensated by a postoperative i.p. injection of saline (10 ml/kg). The cutaneous wound was sutured and cleaned, and the rats were left to recover with free access to food and water.
To investigate the time course of COX-2 mRNA induction during permanent cerebral ischemia, the rats were killed by cervical dislocation at various times between 2 and 24 h postocclusion (3–6 rats at each time point). A separate group of four animals was pretreated with MK-801 (3 mg/kg) 20 min before MCAO, killed 4 h after occlusion, and similarly compared with animals receiving a vehicle injection before MCAO and killed at the same time.
Immediately after cervical dislocation, the brain was rapidly removed, anatomically dissected, and frozen in liquid nitrogen. Up to five regions, both ipsilateral and contralateral to MCAO, were selected. These were cortical tissue from the centre of the MCA territory and the caudate nucleus/putamen (both regions shown to be part of the ischemic “core” from previous ink perfusion studies) (Colląo-Moraes et al., 1994; unpublished data); and three cortical regions surrounding the “core”, including frontal cortex and dorsomedial cortex, both containing tissue supplied by the ACA and MCA, and occipital cortex containing tissue supplied by both the MCA and PCA. The last three regions contain tissue at the border zone between MCA and adjacent arterial territories, where the depth of ischemia may be only moderate, representing an ischemia penumbra where neurones are alive but nonfunctional but whose survival is limited to only a few hours (Hossmann, 1994). In most of the RNA analyses, the dorsomedial cortex sample was used as a source of such moderately ischemic tissue, and for this purpose it has been referred to as the penumbra throughout this study. Similarly, the sample of cortex from the centre of the MCA territory has been referred to as the core. These samples were stored at −70°C until required. Nonoperated rats were similarly investigated and served as baseline controls.
Histochemical measurement of cerebral infarction
To compare the induction of COX-2 mRNA with brain tissue damage in this animal model, the extent of cerebral infarction was measured using the histochemical technique of triphenyltetrazolium chloride (TTC) perfusion staining as previously described (Park et al., 1988). Following 24 h of permanent ischemia in a separate group of 12 rats, TTC (2% in saline) was perfused by a transcardiac route to the brain with the rats under terminal anaesthesia (i.p. 0.8 g/kg of urethane), followed by perfusion of formal saline. The brains were removed and sectioned into 1-mm-thick coronal slices. Unstained areas of cerebral gray matter in photographs of the slices were then measured by computerized planimetry and integrated volumetrically.
RNA extraction and detection
Total RNA was isolated from tissue samples by acid guanidinium thiocyanate-phenol-chloroform extraction (Chomczynski and Sacchi, 1987). The resulting RNA was analysed by Northern blotting and slot blotting as previously described (de Belleroche et al., 1990). Tissue levels of COX-2 mRNA were quantitated by reference to ß-tubulin mRNA using cDNA probes for detection. We used a 449-bp EcoRI fragment of pCRII to detect COX-1 mRNA, a 608-bp EcoRI fragment of pCRII to detect COX-2 mRNA (Feng et al., 1993; donated by Simon Tate, Glaxo Group Research), and an 800-bp fragment of ß-tubulin (Hall et al., 1983) to detect ß-tubulin mRNA.
Northern and slot blot filters were preincubated in hybridization buffer [50% deionized formamide, 5 × SSPE (0.9 M NaCl, 0.05 M sodium pyrophosfate, pH 7.4, 5 mM EDTA), 5 × Denhardt's solution (0.1% bovine serum albumin, 0.1% Ficoll, 0.1% polyvinylpyrrolidone), 0.5% (wt/vol) sodium dodecyl sulfate, 100 μg/ml salmon sperm DNA] for 2 h at 42°C. Filters were then hybridized overnight at 42°C in hybridization buffer containing [32P]-labeled cDNA (3–5 × 106 dpm/ml) to a specific activity of 2.5–3.5 × 109 dpm/μg with [α-32P]-dCTP using the oligolabeling method of Feinberg and Vogelstein (1983, 1984). Blots were washed with 3 × SSC, 0.1% SDS for 30 min at 65°C and 1 × SSC, 0.1% SDS for a further 30 min at 65°C and exposed to Hyperfilm-MP (Amersham International PLC) with intensifying screens at −70°C.
The COX-2 cDNA probe hybridized to a single mRNA species of 4.2 kb as previously reported (Yamagata et al., 1993). The COX-1 cDNA probe hybridized predominantly to a single mRNA species of 2.8 kb and a minor species of 5.1 kb (O'Banion et al., 1992). The intensity of hybridization on slot blots was measured with a scanning densitometer (Joyce Loebl Chromoscan) at 530 nm. The results are expressed as a ratio of the COX-2 mRNA signal: ß-tubulin mRNA signal. Statistical analysis was carried out by analysis of variance (ANOVA) and multiple t tests (Student's t test).
RESULTS
Time course of the induction of COX-2 mRNA during permanent cerebral ischemia and its sensitivity to MK-801
Marked unilateral induction of COX-2 mRNA was detected in the core ischemic region ipsilateral to the occlusion as shown by Northern analysis (Fig. 1). A single species of mRNA of 4.2 kb was detected at 2, 8, and 24 h. Levels of COX-2 mRNA in nonoperated control animals (t0 time = 0) were not clearly detectable despite greater RNA loading for these samples (Fig. 1). Similarly, there was no induction of COX-2 mRNA detected in contralateral cerebral cortex by this method. Quantitation of COX-2 mRNA was carried out by slot-blot analysis with reference to levels of ß-tubulin mRNA in the same samples. A significant increase in COX-2 mRNA was detected in the core and penumbra regions between 4 and 24 h after occlusion, most marked at 4 h in the penumbra region, where a 19-fold increase above control t0 levels was detected (Fig. 2). A smaller but significant induction was also detected at 4 h in the caudate (which also undergoes ischemic changes in this model). Although MCAO caused a small induction in COX-2 mRNA levels in the contralateral cortex, they were not significantly elevated compared with t0 control levels (Fig. 2). Northern analysis of COX-1 mRNA was carried out in parallel, but no significant increase was detected in the brain following MCAO (data not shown), indicating the specificity of the induction of COX-2 mRNA.
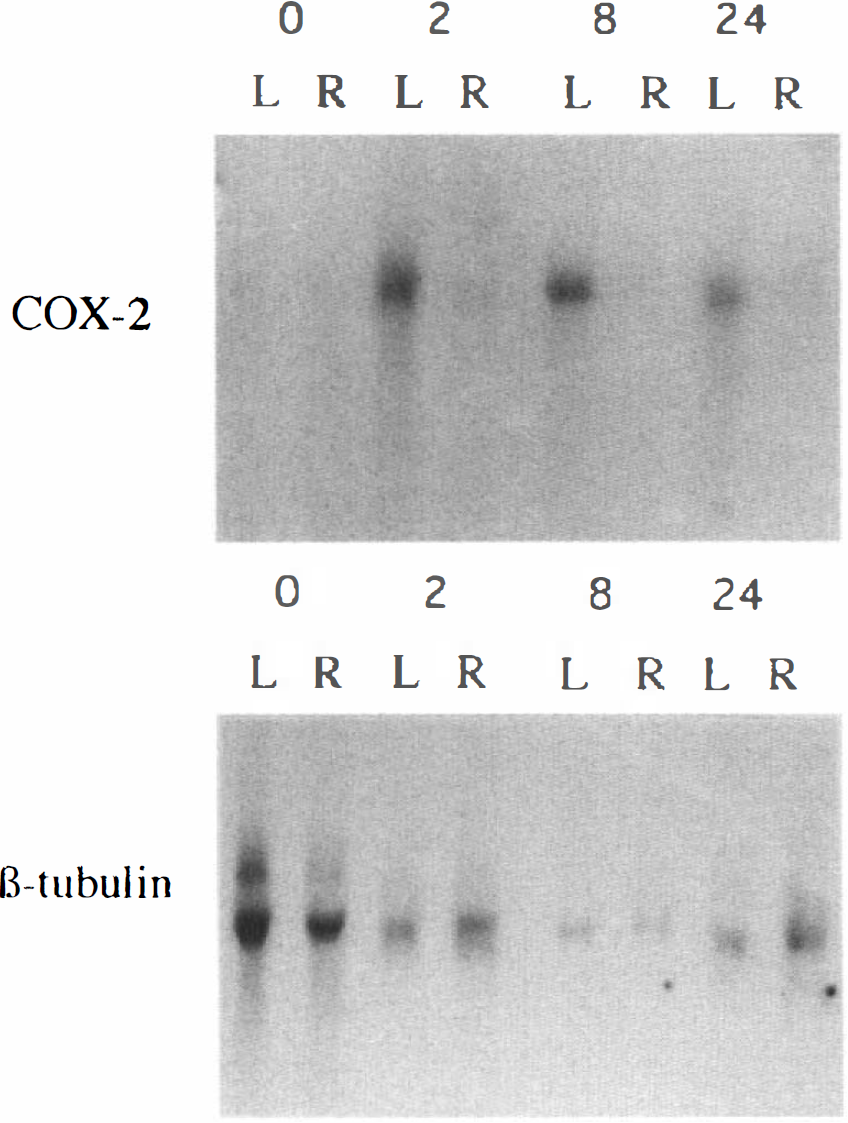
Effect of permanent left middle cerebral artery occlusion MCAO on levels of cyclo-oxygenase (COX)-2 mRNA related to levels of ß-tubulin mRNA. MCAO territory (core) cerebral cortex ipsilateral (L) and contralateral (R) to the occlusion was dissected at various times (2, 8, and 24 h) after occlusion and frozen in liquid nitrogen. Then mRNA was prepared and separated by electrophoresis, blotted, and hybridized with cDNA probes for COX-2 and ß-tubulin. Loading in nonoperated control rat samples t0 controls, shown by 0, was greatest to allow detection of low basal levels of COX-2 mRNA. A marked induction of COX-2 mRNA was detected in ipsilateral cortex at 2, 8, and 24 h after ischemia. The contralateral and ipsilateral cortex at each time point was derived from the same rat. Quantitation was carried out at each time point (n = 3–6) by slot-blot analysis (see Fig. 2).

Time course of induction of cyclo-oxygenase (COX)-2 mRNA following permanent left middle cerebral artery occlusion (MCAO). At various time points (4, 8, and 24 h) after occlusion, MCA territory [core ) to the occlusion, were dissected and used for preparation of mRNA. Quantitation of COX-2 mRNA was carried out by slot-blot analysis by reference to levels of ß-tubulin mRNA. Values are means with the SDs shown by error bars for three to six rats at each time point. Analysis of variance showed a significant effect of time in ipsilateral cortex in core, penumbra, and caudate regions (p = 0.003, p = 0.003, and p = 0.039, respectively) and compared with contralateral cortex in the core and penumbra regions (p = 0.0001 and p = 0.001, respectively), †, ††, and ††† indicate that MCAO significantly increased levels of COX-2 mRNA compared with t0 controls (p < 0.003, p < 0.001 and p < 0.0001). * and ** indicate that the values in ipsilateral cortex were significantly greater than contralateral cortex of the same rats (p < 0.025 and p < 0.005).
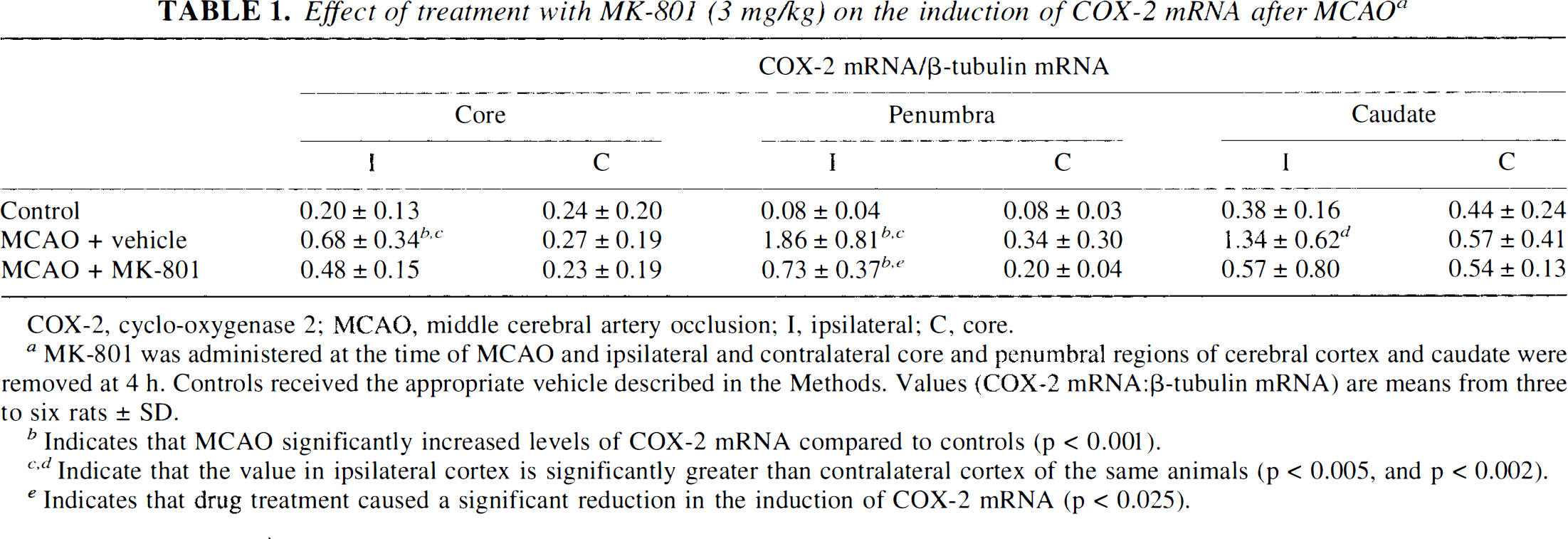
MK-801 significantly attenuated the induction of COX-2 mRNA in the penumbra region at 4 h (p < 0.025, Table 1). The induction of COX-2 mRNA in the core region was smaller than that in the penumbra and showed only a slight attenuation by MK-801. No effect was detected in caudate.
Effect of treatment with MK-801 (3 mg/kg) on the induction of COX-2 mRNA after MCAO a
COX-2, cyclo-oxygenase 2; MCAO, middle cerebral artery occlusion; I, ipsilateral; C, core.
MK-801 was administered at the time of MCAO and ipsilateral and contralateral core and penumbral regions of cerebral cortex and caudate were removed at 4 h. Controls received the appropriate vehicle described in the Methods. Values (COX-2 mRNA: ß-tubulin mRNA) are means from three to six rats ± SD.
Indicates that MCAO significantly increased levels of COX-2 mRNA compared to controls (p < 0.001).
Indicate that the value in ipsilateral cortex is significantly greater than contralateral cortex of the same animals (p < 0.005, and p < 0.002).
Indicates that drug treatment caused a significant reduction in the induction of COX-2 mRNA (p < 0.025).
Correlation between changes in gene expression and infarct volume
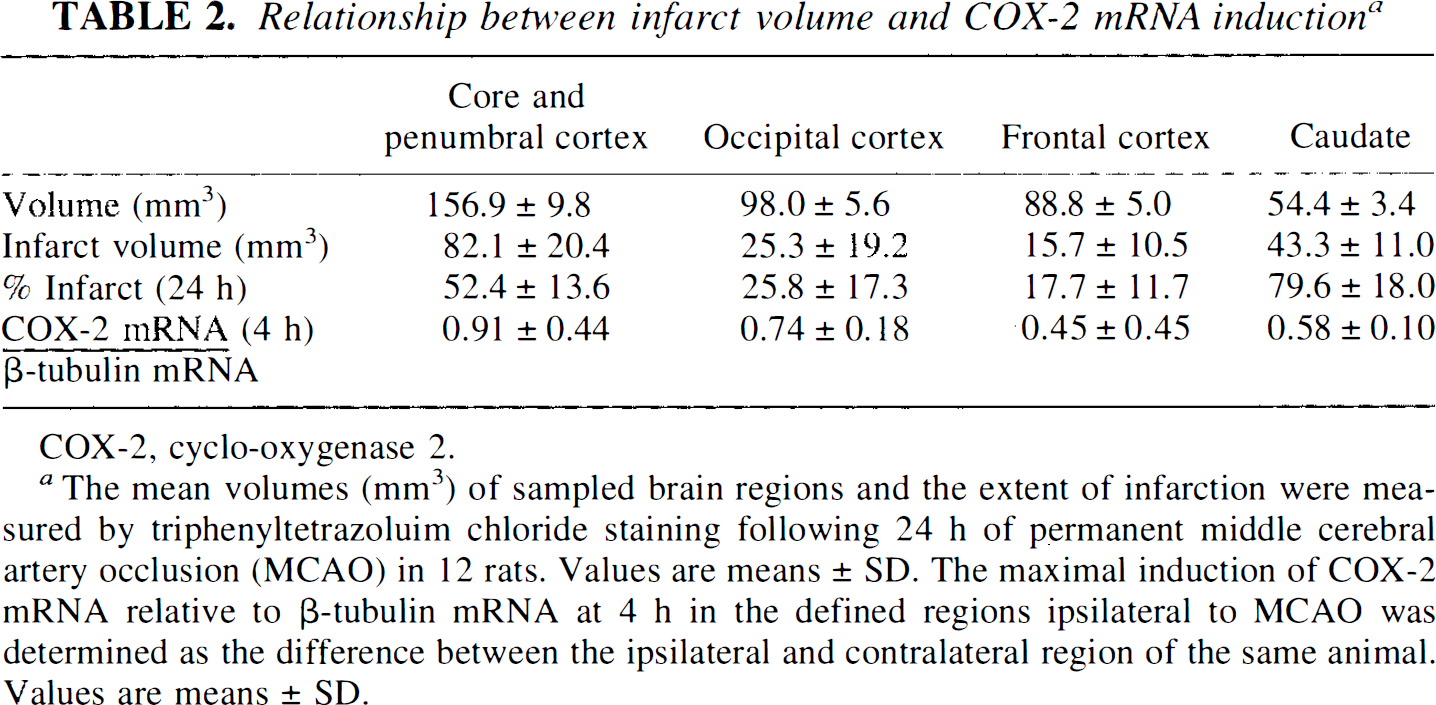
All five tissue regions contained infarcted tissue at 24 h. The volume of infarction in the frontal cortex region, measured in the anterior four 1-mm coronal slices, represented 17.7% of the total volume of cortex for that region. The percentage volume of cortical infarction in the next five 1-mm coronal slices, containing the central MCA territory and dorsomedial cortex region (core and penumbra combined) was 52.4%. In the caudate nucleus/putamen, 79.6% of the tissue was infarcted, and the posterior five 1-mm coronal slices, the occipital cortex region, contained 25.8% infarcted cortex (Table 2). A correlation was seen between the extent of COX-2 mRNA induction in cortical regions at 4 h with the severity of tissue damage subsequently detected [coefficient of correlation (r) = 0.901], both being highest in the core/penumbra regions compared with the frontal and occipital regions; however, this relationship was not borne out in the caudate, where the greatest proportion of tissue volume was affected, but only intermediate levels of COX-2 mRNA were induced.
Relationship between infarct volume and COX-2 mRNA induction a
COX-2, cyclo-oxygenase 2.
The mean volumes (mm3) of sampled brain regions and the extent of infarction were measured by triphenyltetrazoluim chloride staining following 24 h of permanent middle cerebral artery occlusion (MCAO) in 12 rats. Values are means ± SD. The maximal induction of COX-2 mRNA relative to ß-tubulin mRNA at 4 h in the defined regions ipsilateral to MCAO was determined as the difference between the ipsilateral and contralateral region of the same animal. Values are means ± SD.
DISCUSSION
Experimental cerebral ischemia causes a profound effect on the expression of a number of immediate early genes/early response genes and heat-shock proteins. The candidates recruited and the temporal pattern of these responses are similar to those seen during the intense synaptic activation that occurs in seizures, kindling, and excitotoxin injection and appears to be initiated by glutamate, as NMDA receptor antagonists block these IEG responses to excitation and ischemia (Dragunow et al., 1990; Wood and de Belleroche, 1991; Colląo-Moraes et al, 1994; Colląo-Moraes and de Belleroche, 1995; Adams et al., 1996). Further, inhibition of NMDA receptor synthesis, by treatment with antisense oligodeoxynucleotides to NR-1 mRNA (the universal NMDA receptor subunit), protects cortical neurones form neurotoxicity elicited by NMDA in vitro and reduces infarct volume produced by focal ischemia in spontaneously hypertensive rats (Wahlestedt et al., 1993). Understanding the functional consequence of these changes requires the identification of their gene targets in the case of transcription factors because a number of neuronal genes with the appropriate responsive elements in their promoter sequences could potentially be affected. The identification of COX-2 mRNA in this response to experimental ischemia highlights the potential importance of this reaction and its products in the tissue response to this insult subject to the demonstration of a parallel increase in COX-2 protein levels. The enzyme catalyses the key regulatory step in the biosynthesis of prostaglandins and thromboxanes, which are abundant in the CNS and are believed to play an important role in neuronal signaling (Narumiya et al, 1982; Shimizu and Wolfe, 1990). A further consequence of increased COX-2 activity is the generation of highly reactive oxygen free-radical species, with their potent damaging effects on lipids, proteins, and DNA (Siesjö, 1989). Thus, through this mechanism, COX-2 induction could be responsible for a major component of tissue damage resulting from ischemia and would represent an important target for treatment of the pathological consequences of this condition. The induction of COX-2 mRNA could also be part of a protective response to generate local inflammatory reactions, such as vasodilatation and neutrophil recruitment.
Although levels of COX-2 mRNA were raised in most ischemic regions, that is, the cortical “core” and the caudate, higher levels were found in the penumbra and frontal cortex at 4 h. These two regions would have contained zones of moderate ischemia with varying degrees of collateral perfusion from the anterior cerebral artery, and such raised levels of COX-2 enzyme would use the residual supply of oxygen during the acute period of ischemia and, in turn, generate cytotoxic free radicals to augment tissue damage. By 24 h, moderately ischemic neurones are dead (Hossmann, 1994). Reperfusion of ischemic tissue would provide conditions for enhanced oxygen-derived free-radical damage. Reducing glutamate action by the NMDA antagonist, MK-801, produced a powerful inhibitory effect on COX-2 induction during ischemia, particularly in the penumbra region, where the most marked induction of COX-2 was seen.
Both the rat and murine COX-2 promoter share a number of elements that confer phorbol ester and serum inducibility (Sirois and Richards, 1993; Fletcher et al., 1992). An important mediator of a number of neuronal responses is the cAMP-responsive element binding protein (CREB), which is phosphorylated by both a cAMP-dependent protein kinase (Sheng and Greenberg, 1990) and a Ca2+-dependent protein kinase, probably Ca2+/calmodulin-dependent protein kinase II (Sheng et al., 1990). The consensus sequence for CRE to which the phosphorylated protein binds (Xie et al., 1994) is present in the mouse and rat COX-2 promoter (Fletcher et al., 1992; Sirois et al, 1993). The sensitivity of COX-2 induction to a glutamate receptor antagonist could reflect the role of CREB, whose activity would be stimulated by increased intracellular cAMP and Ca2+, especially in the penumbra region. Levels of Ca2+ would be elevated by NMDA receptor gating Ca2+ channels, depolarisation, and neurotransmitter activation of G-protein receptors coupled to phospholipase C and IP3 production, whereas levels of cAMP would be elevated by neurotransmitter activation of adenylyl cyclase-linked G-protein receptors. A cis-acting region of murine COX-2 has also been identified (−300 to −371 from the transcription start site) that is responsive to PAF and retinoic acid (RE), acting both independently and synergistically (Bazan et al., 1994); inhibition of intracellular PAF binding inhibits the induction of zif-268 mRNA and COX-2 mRNA in the rat brain (Marcheselli and Bazan, 1994; Bazan et al., 1994), indicating that PAF generated during ischemia may also stimulate COX-2 mRNA production.
In conclusion, the identification of COX-2 mRNA induction during experimental cerebral ischemia in this study highlights the importance of COX-2 and its products and by-products (e.g., free radicals) in the tissue response to this insult.
Footnotes
Abbreviations used
Acknowledgment:
We thank the BBSRC (SERC) for the award of a studentship to Y.C.M. and Bayer AG for financial support.