
Abstract
Increasing evidence suggests that cyclin-dependent kinases participate in neuronal death induced by multiple stresses in vitro. However, their role in cell death paradigms in vivo is not well characterized. Accordingly, the authors examined whether cyclin-dependent kinase inhibition resulted in functionally relevant and sustained neuroprotection in a model of global ischemia. Intracerebroventricular administration of the cyclin-dependent kinase inhibitor flavopiridol, immediately or at 4 hours postreperfusion after a global insult, reduced injury in the CA1 of the hippocampus when examined 7 days after reperfusion. No significant protection was observed when flavopiridol was administered 8 hours after reperfusion. The tumor-suppressor retinoblastoma protein, a substrate of cyclin-dependent kinase, was phosphorylated on a cyclin-dependent kinase consensus site after the global insult; this phosphorylation was inhibited by flavopiridol administration. Importantly, flavopiridol had no effect on core body temperature, suggesting that the mechanism of neuroprotection was through cyclin-dependent kinase inhibition but not through hypothermia. Furthermore, inhibition of cyclin-dependent kinases improved spatial learning behavior as assessed by the Morris water maze 7 to 9 days after reperfusion. However, the histologic protection observed at day 7 was absent 28 days after reperfusion. These results indicate that cyclin-dependent kinase inhibition provides an extended period of morphologic and functional neuroprotection that may allow time for other neuroprotective modalities to be introduced.
Keywords
Delayed neuronal death is an important component of neuronal injury that occurs after a limited period of ischemic insult. Although many vital cellular functions such as membrane potential, energy metabolism, and protein synthesis appear to be reestablished after reperfusion, selectively vulnerable neurons can die by a process displaying elements of both necrotic and apoptoticlike characteristics (MacManus and Buchan, 2000; Majno and Joris, 1995). Ischemic and reperfusion injuries induce stresses that include delayed excitotoxicity, free radical damage, and damage to the genome (Choi, 1988; Dirnagl et al., 1999; Graham and Chen, 2001; Iadecola and Alexander, 2001; Szatkowski and Attwell, 1994). These injuries, in turn, initiate a complex series of signaling cascades that lead to death. Although several death-promoting signals that participate in ischemic delayed death have been identified (e.g., p53, poly ADP-ribose polymerase, nitric oxide), the mechanism by which these and other molecules modulate death is not fully appreciated (Eliasson et al., 1997; Endres et al., 1997; Lipton, 1999; McGahan et al., 1998). The effort to understand delayed death after ischemia is important because its protracted nature may allow for potential neuroprotective interventions.
One potential target that may control neuronal death after ischemia is cyclin-dependent kinase (CDK) (Osuga et al., 2000). Cyclin-dependent kinases are best characterized by their regulation of the cell cycle and are activated by binding to the appropriate cyclin partner (Morgan, 1995). For example, cyclin D1/Cdk4/6 and E/Cdk2 complexes participate in the G1/S transition, whereas cyclin B/Cdk1 controls M-phase progression. Cyclin-dependent kinases phosphorylated selected substrates needed for cell-cycle progression. Pertinent to this process, G1-related CDKs phosphorylate the tumor-suppressor retinoblastoma protein (pRb). This action results in the dissociation of pRb from transcription factors of the E2F family, which then proceed to activate genes needed for cell-cycle progression (La Thangue, 1994; Lavia and Jansen-Durr, 1999; Wu et al., 1996). In addition to its role in cell-cycle progression, however, accumulating evidence suggests that CDKs may also regulate neuronal death. In this regard, it is hypothesized that the inappropriate activation of CDKs is inconsistent with survival of a terminally differentiated neuron. In support of this hypothesis, several groups have reported the upregulation of cell-cycle molecules during neuronal death in vitro and in vivo (Freeman et al., 1994; Gao and Zelenka, 1995; Gill and Windebank, 1998; Padmanabhan et al., 1999). Importantly, functional studies have indicated that the inhibition of CDKs by the administration of CDK inhibitors or the expression of dominant negative forms of CDKs blocks neuronal death evoked by trophic factor deprivation (Park et al., 1996, 1997b), DNA damage (Park et al., 1997a, 1998a, b ), extracellular K+ deprivation (Padmanabhan et al., 1999), and β-amyloid toxicity (Copani et al., 1999; Giovanni et al., 2000).
Specific to the role of CDKs in ischemic and excitotoxic injury, several groups have reported that cyclin D1 and Cdk4 are upregulated in kainic acid-induced injury, retinal ischemia, and models of cerebral ischemia (Guegan et al., 1997; Li et al., 1997b; Timsit et al., 1999). Importantly, we and others have reported that pRb is phosphorylated on a CDK consensus site in dying neurons after a focal ischemic insult (Hayashi et al., 2000; Osuga et al., 2000). In addition, we have recently shown that periischemic administration of a CDK inhibitor, flavopiridol, inhibits CDK activation and promotes neuronal survival after a mild focal insult (Osuga et al., 2000). Moreover, it has been reported that E2F1-deficient mice are resistant to injury induced by focal ischemia (MacManus et al., 1999). Taken together, the evidence suggests the positive role the CDK/pRb/E2F pathway plays in the promotion of neuronal death after focal ischemia. However, the role of this potential death pathway in global ischemia, which may enact different stress pathways than with focal ischemia, is unknown. In addition, whereas previous studies implicate the importance of CDKs in ischemia-induced neuronal death, the relevance of CDKs as a target for neuroprotective intervention is unclear for many reasons. Principally, it is unknown whether protected neurons after CDK inhibitor administration lead to improved behavioral performance. In addition, it is unclear whether delayed CDK inhibitor administration still affords protection, and if so, whether this protection is sustained. The latter question is important because many neuroprotectants only delay cell death (Colbourne et al., 1999b; Corbett and Nurse, 1998). To examine these questions, we addressed the neuroprotective potential of CDK inhibition against the delayed neuronal loss at the field of CA1 of Ammon horn of the hippocampus (CA1) in a model of transient forebrain ischemia.
MATERIALS AND METHODS
Global ischemic model
All studies were performed in male Wistar rats weighing 180 to 220 g. Surgeries were performed with anesthesia (1%–2.5% halothane carried in oxygen and delivered by a facemask). Rats were allowed to breathe spontaneously during all surgical procedures. All incisions were infiltrated with 2% bupivacaine gel except when associated with a craniotomy. Body temperature was monitored using a rectal probe and was maintained between 36.5 and 37.5°C with the use of a heating pad and radiant heat. Temperature was maintained during all surgical procedures during which inhalational anesthesia was used and during subsequent emergence, four-vessel occlusion (4VO), and for 30 minutes after 4VO when the animal recovered. Acetaminophen suppositories were administered to all rats for the first 3 days of recovery. Global ischemia was performed as described previously, with modifications (Buchan et al., 1991; Pulsinelli et al., 1982). Briefly, 1 day before global ischemia, the common carotid arteries were exposed by a ventral midline neck incision and looped with silk suture. The vertebral arteries were cauterized at the first vertebra by a dorsal neck incision. All wounds were closed with surgical clips and rats were allowed to recover from anesthesia. The next day, rats were anesthetized and a ligature was passed ventral to the cervical and paravertebral muscles but dorsal to the trachea, esophagus, external jugular veins, and common carotid arteries. Rats were allowed to recover from anesthesia until pain responses to tail-pinching returned. Both the common carotid arteries were quickly occluded by tightening the suture placed the previous day. Signs of an effective acute global ischemia (i.e., loss of responsiveness within 10–15 seconds associated with running behavior and loss of righting reflexes) were observed and recorded. The ligature surrounding the paravertebral musculature was then tightened to prevent the opening of collateral blood flow. Dilation and fixation of the pupils were observed in all animals. After 10 minutes, ligatures in the neck and around the common carotid arteries were removed and the neck wound was closed with surgical clips. Rats that did not remain unresponsive during the ischemic period and immediately after the release of ligatures, or those that developed seizures or pulmonary edema, were excluded from further study. Sham-4VO rats underwent the same surgical procedures, except that the common carotid arteries were not occluded and the ligature surrounding the paravertebral muscular was not tightened.
Lateral intraventricular infusion
In rats receiving intracerebroventricular infusion, a midline sagittal incision was made in the scalp. A cannula was stereotactically planted into the left lateral ventricle through a 1-mm burr hole (from bregma: −0.35-mm anterioposterior, 1.4-mm lateral, 3.5-mm deep). Flavopiridol (a gift from Peter J. Worland) in a stock concentration of 50 mmol in dimethyl sulfoxide was diluted with vehicle (Kreb-Ringer solution; Sigma-Aldrich, Toronto, ON, Canada) and was administered in a single dose at the indicated times after reperfusion. The cannula was then left in place for 10 minutes after the infusion. In rats receiving long-term intracerebroventricular infusion, Alzet osmotic pumps (Model 2001, 200-μL capacity, 1-μL/h pump rate; Alza Corporation, Mountain View, CA, U.S.A.) filled with 500-μmol/L flavopiridol were implanted 24 hours before 4VO. Pump function was confirmed by weighing the pump before placement and after experiment. Concentration of flavopiridol was based on previous reports showing that delivery of a 100- to 500-μmol/L flavopiridol solution provided protection that correlated with inhibition of phosphorylation of pRb after focal stroke (Osuga et al., 2000).
Core temperature telemetry
A separate group of animals was treated similarly to those in the global ischemic model, except sterilized telemetry probes (Model TA10TA; Data Sciences, St. Paul, MN, U.S.A.) were implanted in the peritoneal cavity using anesthesia immediately before 4VO. After global ischemia, rats were individually housed in a cage placed on a telemetry receiver (RLA-1020; Data Sciences). Temperature was sampled every 30 seconds and was continuously monitored for 48 hours. Data collected were binned into 5-minute intervals and the temperature data set was analyzed using multivariate analysis of variance for statistical significance using a computer program (Statistica 4.1; StatSoft, Cupertino, CA, U.S.A.).
Morris water maze
The Morris water maze test was performed as previously described (Xu et al., 1999). Briefly, a hidden clear plastic platform (18-cm diameter) was placed 47 cm away from the wall of the water maze (170-cm diameter, 45-cm deep) and 2 cm below the water surface. The platform remained in the same location for all sessions and trials during pretraining and testing. The water maze was divided into four quadrants and the starting quadrant was randomized daily, with all rats using the same daily order. Rats were released into the maze head-up and facing the wall of the maze. If an animal failed to find the platform in 120 seconds, it was then placed on the platform for 10 seconds. Each session consisted of five trials, and data from these five trials were averaged to form the daily score. Rats were allowed to rest for a minimum of 5 minutes between trials. All animals were pretrained for 4 consecutive days in the week preceding 4VO. Seven days after reperfusion, rats were tested for 3 consecutive days (7, 8, and 9 days) using the same paradigm as in the pretraining. On the last day of testing (day 10), the platform was wrapped in black and raised 1 cm above the water surface. The escape latency, trajectory, and length of trajectory were recorded in all trials. The behavioral data set was analyzed using multivariate analysis of variance followed by post hoc analysis with Fisher least significant difference test as implemented in Statistica (StatSoft).
Western blots
Western blot analysis was carried out on the nuclear extract as described previously (Sonnenberg et al., 1989) from the dorsal hippocampus. The hippocampi were extracted 3, 6, 12, and 24 hours after reperfusion and the tissue from sham studies was extracted 24 hours after the sham 4VO. Rats were anesthetized intraperitoneally with 50 mg/kg pentobarbital and perfused with 200 mL ice-cold heparinized phosphate-buffered saline. After isolating the hippocampi, the dorsal hippocampus was separated from the rest of the hippocampus along the hippocampal fissure and was immediately placed in 750 μL ice-cold sucrose buffer (0.3 mol/L sucrose, 1 mmol/L ethyleneglycoltetracetic acid, 25 mmol/L sodium chloride, 15 mmol/L Tris-hydrochloride [pH 6.8], 1 mmol/L phenylmethylsulfate fluoride, 2 μg/mL leupeptin, 5 μg/mL aprotinin, 0.1 mmol/L sodium vanadate, and 20 mmol/L β-glycerophosphate). Samples were homogenized with a Polytron (Kinematica, Cincinnati, OH, U.S.A.) and centrifuged (Eppendorf Model 5147R, Eppendorf AG, Hamburg, Germany) for 1 minute at 13,000 g. The supernatant was then removed with minimal disturbance to the pellet and 100 μL high-salt buffer (0.75 mol/L sodium chloride, 0.05 mol/L HEPES, 12.5% glycerol, 0.75 mmol/L magnesium chloride, 0.5 mmol/L ethyleneglycoltetracetic acid, 5 mmol/L dithiothreitol, 1 mmol/L phenylmethylsulfate fluoride, 2 μg/mL leupeptin, 5 μg/mL aprotinin, 0.1 mmol/L sodium vanadate, and 20 mmol/L β-glycerophosphate) was added. The mixture was then vortexed five times for 10 seconds at an interval of at least 5 minutes and centrifuged (Eppendorf Model 5147R) for 10 minutes at 13,000 g. The nuclear extracts were mixed with solubilization buffer (62.5 mmol/L Tris, 2.5 mmol/L edetic acid, 2.5 mmol/L ethyleneglycoltetracetic acid, 10% glycerol, 2% sodium dodecyl sulfate, 0.001% bromphenol blue, and 5% β-mercaptoethanol). Samples were loaded on to sodium dodecyl sulfate-polyacrylamide gels and transferred to nitrocellulose or polyvinylidene fluoride membranes (NEN, Boston, MA, U.S.A.). Membranes were probed with anti-Phospho(ser795)pRb (Cell Signaling Technology, Beverly, MA, U.S.A.) and with anti–β-actin (Sigma, St. Louis, MO, U.S.A.) as the indicator for equal loading.
Histology
Sections were stained for hematoxylin and eosin as described previously (Stevens, 1982). At the predetermined reperfusion intervals, rats were anesthetized and perfusion fixed with 200 mL heparinized phosphate-buffered saline followed by 200 mL 4% paraformaldehyde buffered with 0.1 mol/L phosphate (pH 7.4). The brains were removed, postfixed in 10% formalin for 7 days, and paraffin embedded. Brain sections (7 μm) were collected at the level of the middorsal hippocampus (bregma −3.0 – −3.3 mm) and paraffin was removed. Surviving CA1 neurons were identified using standard criteria in 4VO and sham-4VO studies (Buchan et al., 1991). Cell counts from the left and right hippocampi were averaged and expressed as counts per millimeter for CA1. The cell count data were analyzed with an independent t-test (two tailed) or analysis of variance followed by Tukey honestly significant difference test using Statistica (StatSoft).
RESULTS
Cyclin-dependent kinase inhibition is neuroprotective
To characterize the profile of neuronal loss in the CA1 region after a 10-minute 4VO in our experimental settings, an observer who was blinded to the design examined the neuronal survival at various intervals after reperfusion (Fig. 1). Normal CA1 neurons are characterized by a large round soma with a conspicuous nucleus. Single or multiple nucleoli are often visible in the nucleus. CA1 neurons showing a shrunken appearance with eosinophilic cytoplasm and pyknosis, or fragmented nuclei were excluded from the count of surviving neurons. The onset of the neuronal loss was delayed and abrupt, and neuronal death was nearly complete by 48 hours (Fig. 1) (P < 0.001).
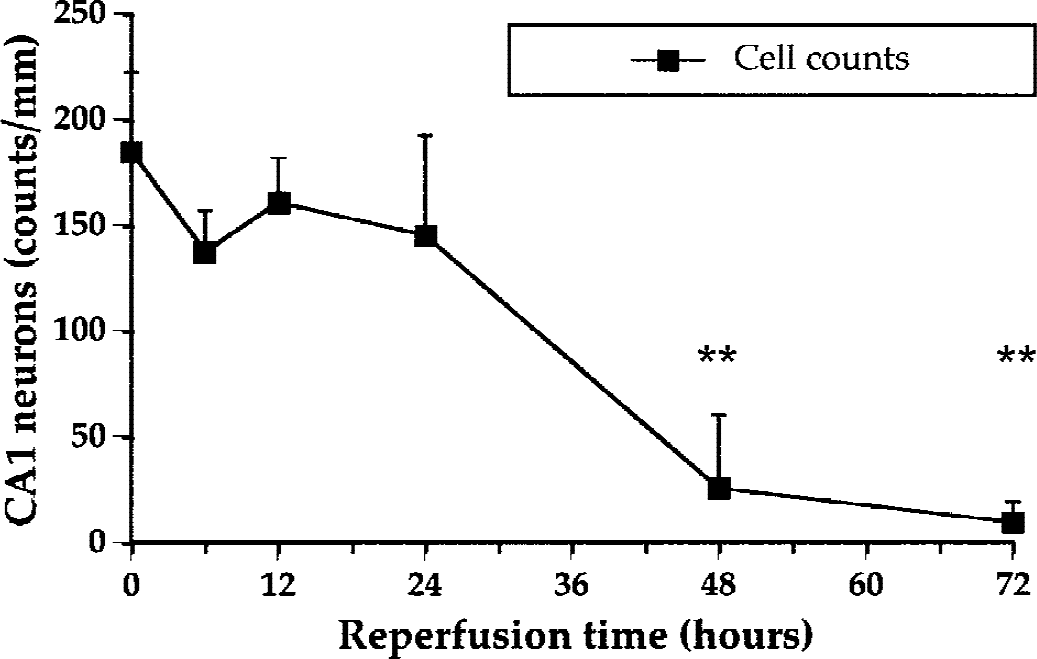
Surviving neuronal counts in the CA1 region of the hippocampus after 10-minute four-vessel occlusion at various intervals after reperfusion. Neuronal density in the CA1 was determined as described in Methods. Note the significant loss of the CA1 neurons at 48 hours. Data are mean + SD (n = 4 at 0 [sham], 6, 12, and 48 hours; n = 5 at 24 and 72 hours). For clarity, downward SD bars are omitted. **P < 0.01, compared with sham four-vessel occlusion studies (at time zero, analysis of variance followed by Tukey honestly significant difference test).
To determine whether CDK inhibition would ameliorate the delayed neuronal loss observed after 10-minute 4VO, we used a CDK inhibitor, flavopiridol. This agent has been previously shown to inhibit the CDK family of kinases (Cdk1–7) but not tyrosine kinases (EGFR, SRC) or signaling kinases such as PKC and ERK-1 (Sedlacek, 2001). CA1 neuron survival was assessed on 7 days after reperfusion. Intracerebroventricular flavopiridol administration (300 μmol/L, 10 μL for 10 minutes) alone did not result in changes in neuronal morphology in any region of the hippocampus when compared with vehicle-treated animals (Fig. 2A). In addition, no changes in neuronal numbers in the CA1 region were detected with flavopiridol treatment when examined 7 days after infusion (Fig. 2B). The neuronal counts per millimeter were 167.6 ± 19.3 (n = 4) in the animals treated with vehicle and 171.6 ± 5.1 (n = 4) in those treated with flavopiridol (Fig. 2B). However, animals treated with a single infusion of vehicle or flavopiridol immediately before the onset of 4VO displayed a significant increase in CA1 neuronal survival (78.8 ± 52.0, n = 7) compared with those treated with vehicle (28.7 ± 26.8, n = 7, P < 0.05) (Figs. 2A and 2B). Postischemic administration of flavopiridol also resulted in significant neuroprotection. As shown in Fig. 2, animals receiving a CDK inhibitor 4 hours after reperfusion still showed significant neuroprotection when compared with vehicle-treated animals (71.4 ± 38.3, n = 7 vs. 26.3 ± 26.3, n = 7; P < 0.05) (Figs. 2A and 2B) and was comparable with that observed in animals treated with flavopiridol immediately before the onset of 4VO (Figs. 2A and 2B). However, when the delay in flavopiridol administration was extended to 8 hours, no significant protection was observed (flavopiridol, 24.8 ± 18.1, n = 7 vs. vehicle, 20.6 ± 14.9, n = 7). These data indicate that intracerebroventricular administration of flavopiridol improved neuronal survival 7 days after a 10-minute global ischemic insult. In addition, the neuroprotective window of opportunity, at least in this dosage paradigm, extends to a time between 4 and 8 hours after reperfusion.
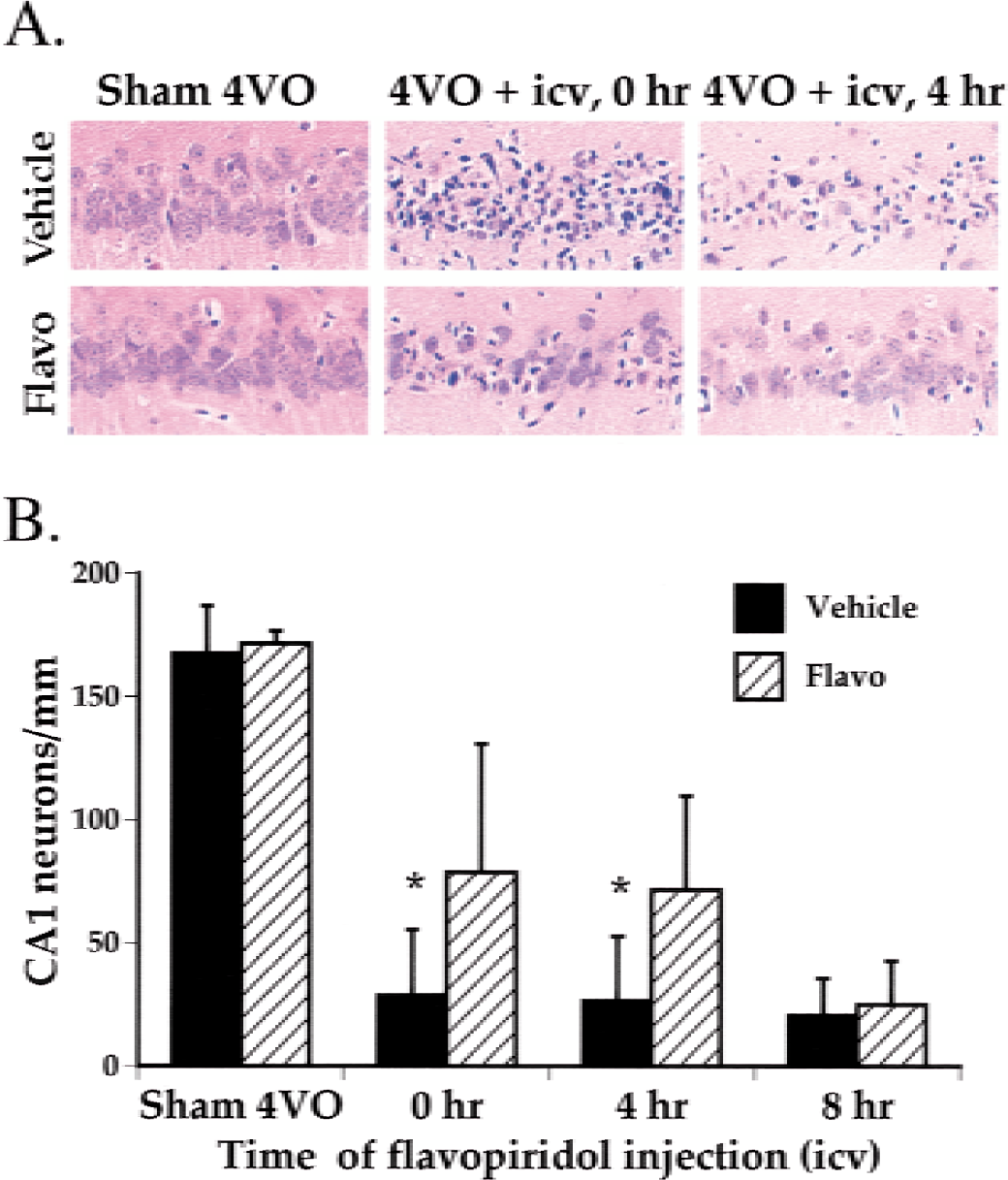
Increased CA1 neuronal survival at 7 days after 10-minute four-vessel occlusion and intracerebroventricular flavopiridol infusion.
Cyclin-dependent kinase inhibitor administration does not alter body temperature
Pervious studies have indicated that hypothermia is neuroprotective in animals subjected to global insult (Colbourne et al., 1999a, 1997). In addition, numerous studies have suggested that certain neuroprotectants such as dizocilpine (MK-801) may work by lowering body temperature rather than by selectively blocking an intended target (Buchan and Pulsinelli, 1990; Corbett et al., 1990). Accordingly, to examine whether the observed neuroprotection by CDK inhibitor administration was the result of a lowered body temperature, we evaluated the effects of flavopiridol on core body temperature over an extended period. Temperature was monitored using a wireless probe implanted in the peritoneal cavity. Published reports have indicated that temperature monitored in this manner accurately reflects brain temperature (Colbourne et al., 2000; Corbett et al., 2000). Two paradigms were used to study the effect of intracerebroventricular flavopiridol administration (500 μmol/L, 15 μL for 10 minutes) on core body temperature. The first paradigm used animals that underwent sham 4VO, whereas the second paradigm used animals undergoing 4VO. Flavopiridol was administered 4 hours after 4VO or sham 4VO. In both groups, the core body temperature was initially controlled to 37°C during and for 30 minutes after the sham 4VO or 4VO. The temperature was allowed to fluctuate without any intervention for the rest of the 48-hour observation period. In both paradigms, no significant difference was observed with flavopiridol treatment (Fig. 3). It is noteworthy that a spontaneous mild hyperthermic episode followed the global insult as reported by others, despite the lower temperature set-point during the intracerebroventricular infusion (Fig. 3) (Colbourne et al., 1993; Kim et al., 1996). In addition, the natural set-point of all animals was between 37.5 and 38°C, higher than the average set point of 37°C used during the surgical procedures. These temperature data indicate that intracerebroventricular infusion of flavopiridol, in the dosing regime tested, did not alter core temperature regulation, thus supporting the notion that hypothermia is not the likely explanation for neuroprotection by flavopiridol.
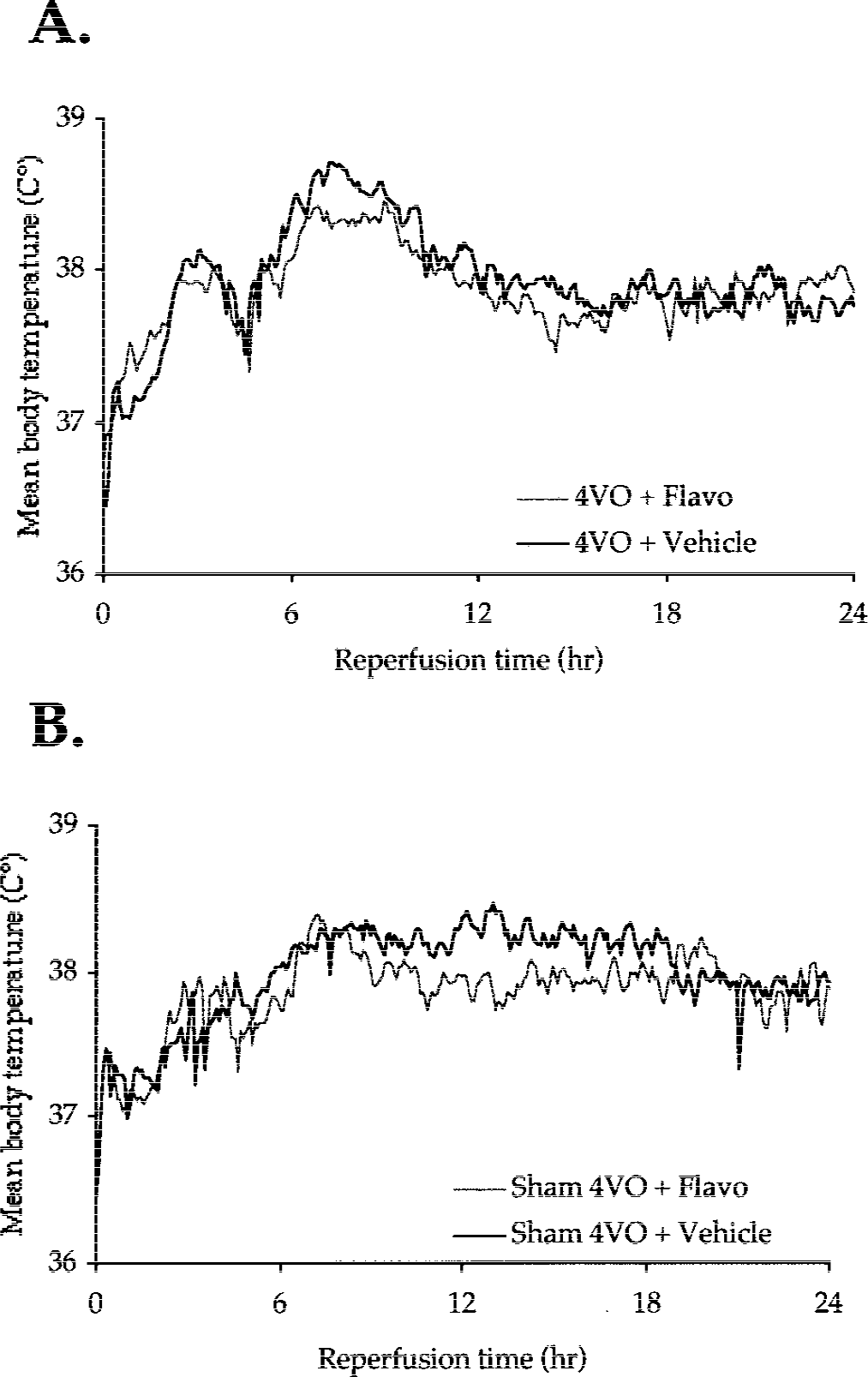
Core body temperature during a 24-hour period after 10-minute four-vessel occlusion and intracerebroventricular flavopiridol infusion. Body temperature was measured with a wireless sensor as described in Methods. Data were sampled every 30 seconds and binned into 5-minute intervals. For clarity, SD bars are omitted and only means are shown.
Global ischemia leads to activation of CDK-pRb pathway that is suppressed with flavopiridol
Previous reports have indicated that key cell-cycle regulators such as cyclin D1 and Cdk4 are upregulated in CA1 neurons after focal and global ischemia (Guegan et al., 1997; Li et al., 1997a; Osuga et al., 2000; Timsit et al., 1999). Accordingly, we examined whether CDK was activated after 4VO by determining whether pRb, a substrate of Cdk4/6, was phosphorylated on a CDK consensus site after reperfusion as a marker for CDK activity. We also examined whether flavopiridol treatment would attenuate this activity, as one would predict if flavopiridol-induced neuroprotection acted by inhibiting CDK activity. After sham surgery or 4VO, the dorsal hippocampus was isolated (Fig. 5A) and subjected to Western blot analyses using an antibody specific to pRb phosphorylated on ser 795, a consensus site for CDK phosphorylation (Fig. 4A). A low basal level of phosphorylated pRb was observed in sham-treated animals (Fig. 4B). No significant increase in pRb phosphorylation was observed 3 hours after reperfusion. However, a dramatic increase in pRb phosphorylation was detected 12 and 24 hours after reperfusion (Fig. 4B). These data suggest that CDKs are activated after the global insult. Importantly, this upregulation was attenuated in animals that received flavopiridol treatment (Fig. 4C). Equal loading of the samples was confirmed with β-actin detected on the same blots. Taken together, these data indicate that pRb is phosphorylated after 4VO and provide evidence that flavopiridol is neuroprotective by inhibiting CDK activity.
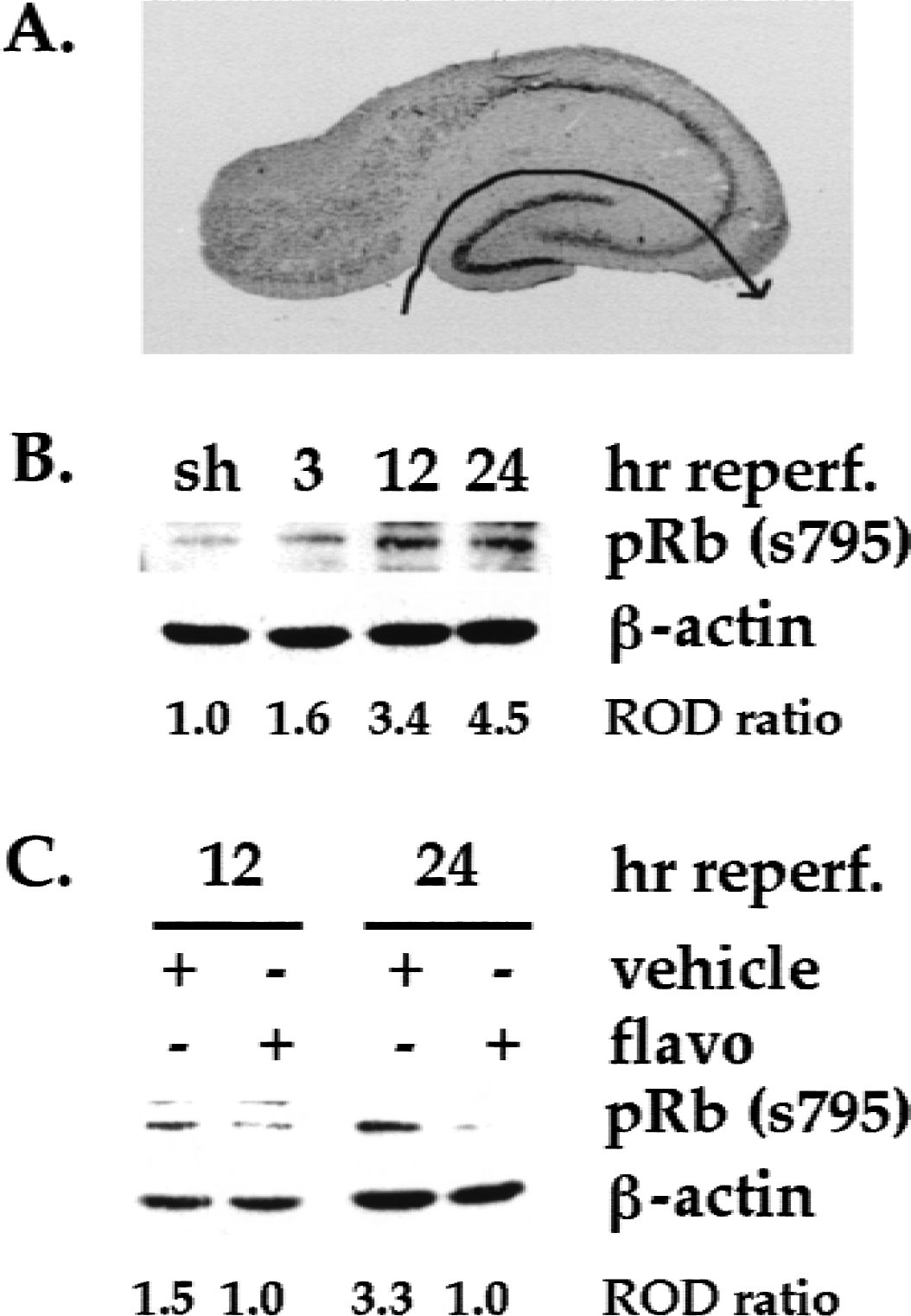
Upregulation of pRb (s795) in the hippocampus after four-vessel occlusion and its attenuation in animals treated with intracerebroventricular flavopiridol.
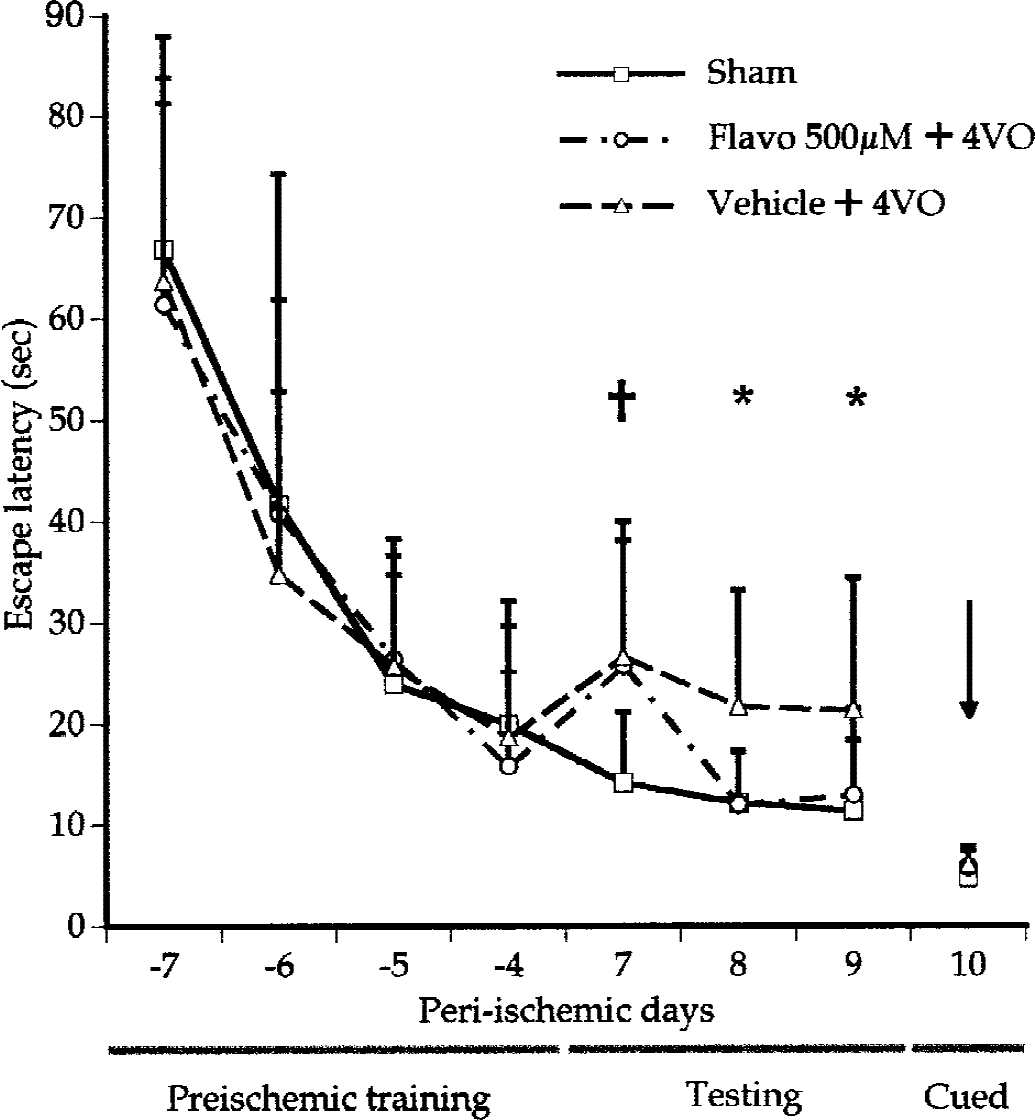
Improved escape latency in the Morris water maze at 7 to 10 days in rats subjected to four-vessel occlusion and intracerebroventricular flavopiridol infusion. Animals are pretrained in the water maze for 4 consecutive days 1 week before the ischemic insult (preischemic training, day −7 – −4) as described in Materials and Methods. All animals were then tested 1 week after the ischemic insult for 4 consecutive days (testing, 7–9 days; cued, 10 days). Intracerebroventricular flavopiridol (500 μmol/L, 15 μL) or vehicle was administered to animals at 4 hours after reperfusion (0 day). Data are mean + SD (n = 10 for each group). For clarity, SD bars are omitted. Data from the sham four-vessel occlusion groups (treated with flavopiridol or vehicle) are combined. For clarity, SD bars are plotted upwards for all groups. Flavo, flavopiridol; 4VO, global ischemia using the four-vessel occlusion method. †P < 0.05, compared with the four-vessel occlusion sham group; *P < 0.05, compared with vehicle-treated animals (multivariate analysis of variance followed by Fisher least significant difference test).
Cyclin-dependent kinase inhibitor administration results in improved performance in the Morris water maze
This evidence suggests an obligatory role for CDKs in neuronal death after global ischemic insult. However, it is critical to determine whether animals subjected to neuroprotective treatments have improved behavioral performance, the ultimate goal of a neuroprotective treatment. Previous evidence has shown that damage in brain regions that include the hippocampus results in impaired spatial learning and memory as measured with the Morris water maze (Block, 1999; Briones and Therrien, 2000; Xu et al., 1999). Accordingly, we tested whether neuroprotection imparted by CDK inhibition translated to improved Morris water maze performance. All rats were pretrained to find a hidden platform invisible to the swimming rats, as indicated in Materials and Methods (Fig. 5). These pretrained animals were then randomly assigned to one of four groups. Two groups of animals were then subjected to 4VO and then treated with flavopiridol (n = 10) or vehicle (n = 10) (500 μmol/L, 15 μL for 10 minutes) 4 hours after reperfusion. The remaining two groups were subjected to sham 4VO and then treated with flavopiridol or vehicle in an identical manner. Starting 7 days after reperfusion, the animals were subjected to the water-maze test on 3 consecutive days and the cued test on the fourth day. There was no statistical difference in any of the parameters observed (escape latency, length and pattern of trajectory, and swimming velocity) between the two sham-4VO groups and their data were combined. As shown in Fig. 5, animals subjected to 4VO and treated with vehicle spent nearly twice as much time finding the hidden platform (escape latency) when compared with sham-treated animals. This difference was observed in all three testing days (14.1 ± 7.1,12.1 ± 5.1, and 11.3 ± 7.1 seconds, n = 10 vs. 26.6 ± 13.4, 21.7 ± 11.5, and 21.4 ± 13.0 seconds, n = 10; P < 0.05) after reperfusion. The escape latency of animals subjected to 4VO and treated with flavopiridol did not differ on the first day of testing (day 7) when compared with animals subjected to 4VO and treated with vehicle (25.7 ± 12.4, n = 10 vs. 26.6 ± 13.4, n = 10; P > 0.05). Interestingly, however, the pattern changed on the second and third days of testing (days 8 and 9) when the animals treated with flavopiridol improved significantly compared with those treated with vehicle (e.g., 12.0 ± 5.3 vs. 21.7 ± 11.5 on day 8). To control for possible motor or visual deficits in all groups, a cued test was performed on day 10 by wrapping the platform in black and raising it above the water level so that it was visible to the swimming rats. No difference was observed in escape latency times in any groups tested, indicating that observed differences 7 to 9 days after reperfusion could not be accounted for by any possible differences in motor or visual function (Fig. 5). No difference was observed in any other parameter (e.g., length and pattern of trajectory, swimming velocity). These observations indicate that CDK inhibitors administered even 4 hours after the onset of reperfusion improve behavioral performance associated with ischemic injury, and that protected neurons may have at least partially retained their function.
Neuroprotection afforded by cyclin-dependent kinase inhibition is not permanent
This evidence indicates that CDK inhibition provides neuroprotection against a global insult. The protection is associated with a decrease in pRb phosphorylation but is not related to hypothermic events. In addition, neuroprotection by CDK inhibitor administration is associated with improved behavioral outcome and has at least a 4-hour window of protection. A critical consideration, however, for any potential neuroprotective agent is whether the neuroprotection is sustainable. Accordingly, we examined whether the neuroprotection observed with flavopiridol treatment 7 days after reperfusion was present on day 28. At 28 days after reperfusion, the neuronal density in the CA1 region of sham-treated animals was 146 ± 27.4 (n = 10), similar to densities observed in other sham groups. Vehicle-treated animals displayed almost no CA1 neurons (5.3 ± 6.3, n = 10) (Fig. 6) whereas animals subjected to 4VO and administered flavopiridol 4 hours after reperfusion had a slightly higher average number of CA1 neurons (12.0 ± 26.5, n = 10) 28 days after reperfusion (Fig. 6). However, this difference was not statistically significant when compared with that of vehicle-treated animals. Importantly, the average number of neurons in flavopiridol-treated animals declined between days 7 and 28 (Figs. 2B and 6B). These data indicate that the protection afforded by a single dose of flavopiridol given 4 hours after reperfusion was transient.
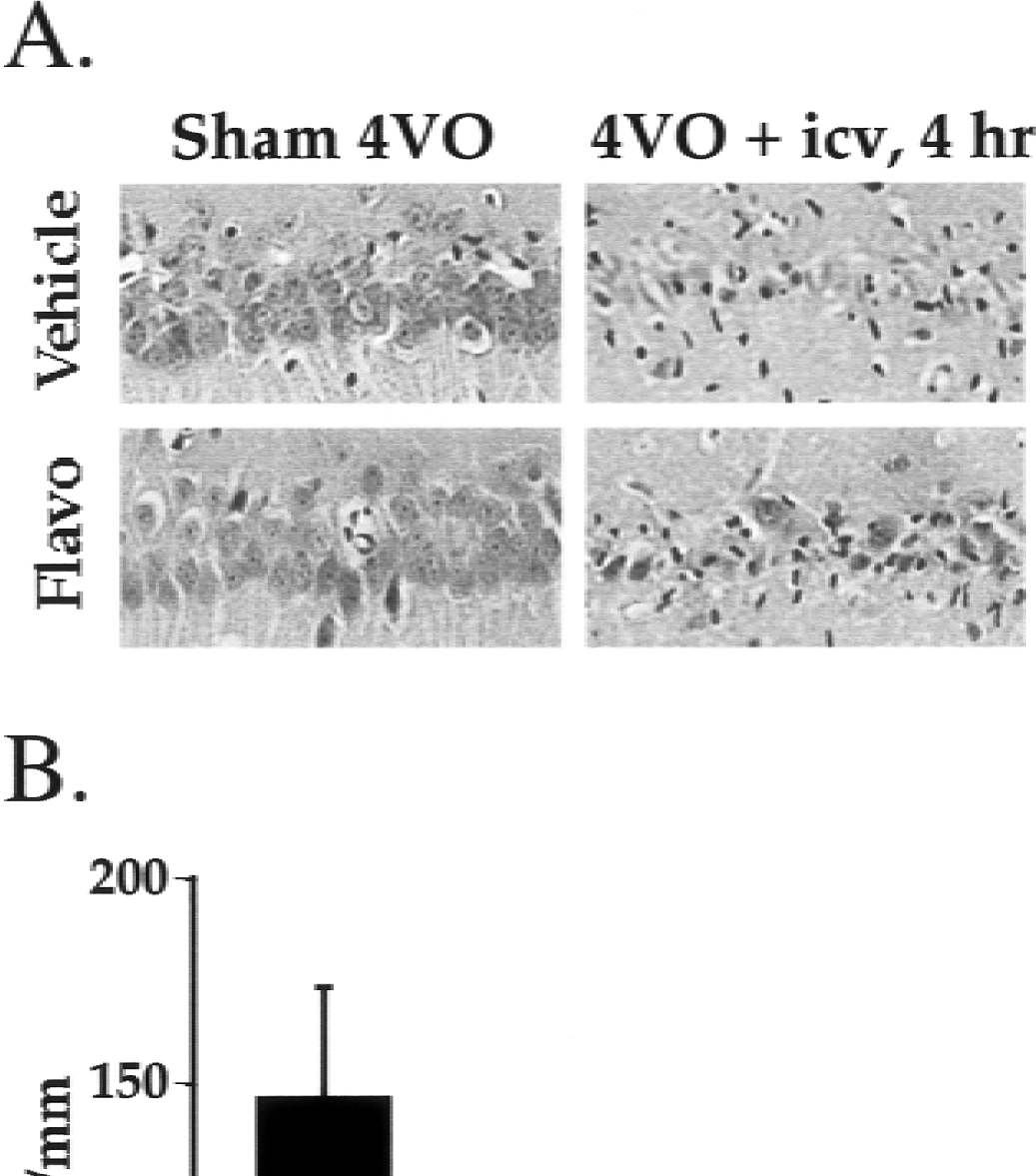
Surviving neuronal counts at 28 days in the CA1 region of the hippocampus after 10-minute four-vessel occlusion and intracerebroventricular flavopiridol infusion.
To examine whether the failure of long-term neuroprotection may be the result of an insufficient dose and duration of the flavopiridol treatment, we used an Alzet osmotic pump to provide extended administration of flavopiridol (Osuga et al., 2000). The osmotic pump had a capacity of 200 μL and delivered flavopiridol at a rate of 1 μL/h for 7 days. The pumps were implanted 1 day before 4VO at the time of the cauterization of the vertebral arteries. Histologic studies were conducted 28 days after reperfusion. However, even with this extended dosing paradigm, no significant neuroprotection was observed at day 28 (data not shown). These data suggest that the CDK inhibitor-based neuroprotection postpones neuronal death and does not provide permanent histologic protection.
DISCUSSION
Delayed neuronal death is prevalent in selectively vulnerable regions of the central nervous system after global insult and in penumbra regions after focal ischemia (Dirnagl et al., 1999; Pulsinelli et al., 1982). The protracted timeline of neuronal loss has provided hopes for neuroprotective intervention to reduce ischemic injury. However, whereas numerous death signals have been identified to control this process, no convincing neuroprotective strategy, with the possible exception of hypothermia, has withstood more stringent preclinical animal testing (Colbourne et al., 2000). The reasons for this are complex and likely result from the participation of multiple death-related events that effect ischemic damage (Dirnagl et al., 1999; Graham and Chen, 2001; Lewen et al., 2000; MacManus and Buchan, 2000; Stys, 1998; Szatkowski and Attwell, 1994). Accordingly, it has become increasingly important to characterize novel signals induced by the ischemic insult and to evaluate their role in ischemic injury.
We propose that one of such events is the activation of CDKs. Mounting evidence indicates that CDKs participate in death of injured adult neurons in vivo. For example, we and others have observed increases in cell-cycle regulators (e.g., cyclin D1, Cdk4) on focal and global ischemic insult (Guegan et al., 1997; Li et al., 1998; Osuga et al., 2000; Timsit et al., 1999). Consistent with this upregulation, we and others have also observed increases in pRb phosphorylation in a focal model of stroke injury (Hayashi et al., 2000; Osuga et al., 2000) and other models of neuronal death (Gill and Windebank, 1998; Giovanni et al., 1999; Padmanabhan et al., 1999; Park et al., 2000a). Increased cyclin and CDK levels have also been reported in hippocampal neurons after global ischemia and kainic acid exposure, and after retinal ischemia (Liu et al., 1996; Park et al., 2000b; Timsit et al., 1999). Activation of this pathway does not appear to be limited to ischemic-excitotoxic neuronal death. For example, increases in cyclin D1 or Cdk4 have also been reported in vulnerable brain regions of patients with Alzheimer disease (Busser et al., 1998; McShea et al., 1997; Nagy, 2000; Vincent et al., 1997).
We presently demonstrate that delayed administration of the CDK inhibitor flavopiridol, up to 4 hours after reperfusion, significantly protects CA1 neurons from delayed neuronal death when examined 7 days after reperfusion. This observation is significant because it suggests a potential window of therapeutic delivery. Additionally, this observation indicates that the involvement of CDKs in global and focal ischemia (Osuga et al., 2000) and in different neuronal types. This finding is noteworthy because regions affected in human stroke vary widely. Our present results also reflect reports indicating increased cell-cycle molecules in several different ischemic paradigms (Frade, 2000; Guegan et al., 1997; Li et al., 1997a; Osuga et al., 2000; Timsit et al., 1999). Finally, the induction of phosphorylated pRb in both focal (Osuga et al., 2000) and global (present data) models of stroke is consistent with the more general involvement of CDKs in ischemic damage.
It is important to emphasize that protection is not complete, indicating that other pathways must participate in the delayed ischemic neuronal death. In support of this idea, numerous groups have reported upregulation of a variety of death-related proteins, such as the tumor suppressor p53 (Copani et al., 2001; Crumrine et al., 1994; Martin, 2001; McGahan et al., 1998; Morrison and Kinoshita, 2000; Xiang et al., 1996) and the stress-activated JNK/c-Jun pathway (Takagi et al., 2000; Wu et al., 2000). Upregulation of p53 has been shown in both focal and global models of cerebral ischemia (Crumrine et al., 1994; Li et al., 1997a; McGahan et al., 1998). It will be important to determine whether and how various death-promoting signals such as p53 act in relation to CDK activity, and whether combination strategies for inhibiting multiple death signals will provide synergistic protection after an ischemic insult. We have begun to make these linkages in in vitro models of cortical neuronal death after DNA damage. Relevant to this, we have shown that CDKs and p53 act independently to control caspase activity (Morris et al., 2001), whereas c-Jun activity is regulated by CDKs (Ghahremani et al., 2000). Whether this process occurs in vivo in adult neurons after stroke remains to be determined.
Importantly, our observations support the idea that flavopiridol-mediated CA1 neuronal survival is not due to induced hypothermia. Hypothermic effects of some neuroprotectants, such as dizocilpine and diazepam, have been reported to complicate the interpretation of their neuroprotective properties (Buchan, 1990; Corbett et al., 1990; Dowden et al., 1999; Nurse and Corbett, 1996). However, our evidence suggests that the mechanism of protection is likely through CDK-inhibitory properties rather than temperature effects. This view is further supported by our observations that pRb is phosphorylated after global insult and that flavopiridol administration attenuates this signal. Moreover, the timing of delayed flavopiridol treatment correlates with pRb phosphorylation. For example, minimal pRb phosphorylation is observed 3 hours after 4VO, a time when flavopiridol can still be administered and is effective. Finally, in support of this interpretation, we have shown that expression of dominant negative Cdk4/6 and the mutant form of pRb with the CDK site removed is neuroprotective against DNA damage-induced neuronal death in vitro (Park et al., 1997a, 1998a, 2000a). Significantly, flavopiridol also inhibits death induced by DNA damage (Park et al., 1997a, 1998a). The importance of pRb in neuronal death is further supported by observations that pRb-deficient mice show severe neuronal death and inactivation of pRb by large T-antigen expression, which result in death of Purkinje neurons in vitro (Feddersen et al., 1995; Lee et al., 1994). Our data favor the interpretation that the CDK inhibitor works by blocking the Cdk4/6/pRb pathway. We cannot, however, rule out the possibility that flavopiridol may be acting on other CDKs or by another unspecified mechanism unrelated to hypothermia. Regarding the former possibility, Cdk5—a member of the CDK family not involved in cell-cycle control—has been shown to play an important role in neuronal death (Lee et al., 2000; Patrick et al., 1999). Importantly, it has been shown that cleavage of the Cdk5 activator p35 to a more stable p25 form occurs after focal ischemia (Lee et al., 2000).
What are the potential downstream mechanisms that mediate the death signal generated from deregulated pRb? Retinoblastoma protein is best characterized for its role in regulation of E2F family members. Interestingly, E2Fs have been implicated in ischemic damage. For example, we have previously shown that E2F1 levels are increased after focal insult (Osuga et al., 2000), and it has been reported that E2F1-deficient mice are more resistant to focal ischemia (MacManus et al., 1999). Additional support for the importance of E2F1 in neuronal death in nonischemic models has also been reported. For example, ectopic neuronal death in pRb-deficient mice is dependent on E2F1 (Jacks et al., 1992; Lee et al., 1992). In addition, E2F1 expression is sufficient to promote death of cerebellar granule neurons (O'Hare et al., 2000) and cortical neurons (Hou et al., 2000). Finally, E2F1-deficient mice are resistant to death of cultured cortical and cerebellar granule neurons evoked by β-amyloid toxicity and low extracellular K+, respectively (Giovanni et al., 2000; O'Hare et al., 2000). In support of this idea, flavopiridol has been shown to inhibit cytochrome c release in neurons treated with DNA damage in vitro (Stefanis et al., 1999). In addition, Sugawara et al. (1999) have shown that cytochrome c is similarly released after global insult in CA1 neurons. However, it is critical to note that neuronal death and stress pathways are context dependent. Accordingly, it is a possibility that the mechanism by which CDKs or pRb regulate death is different depending on the neuronal death paradigm. This is especially relevant with death induced in the global model of ischemia that may have both necrotic and apoptoticlike characteristics.
An important basic consideration of any neuroprotective strategy is whether protection afforded by inhibition of a particular death signal results in improved behavioral outcome (Corbett and Nurse, 1998). We demonstrate that CDK inhibitor administration significantly improves spatial learning and memory as measured by the Morris water maze. Performance in the Morris water maze is also affected by the functional and morphologic integrity of other cerebral structures (e.g., neocortex, amygdala). The water maze data are nonetheless consistent with the interpretation that improved hippocampal morphology may contribute to improved water maze performance. Although significant improvements were observed, the behavioral test results had a different pattern from that of the sham-4VO studies. Of significance, no improvement in flavopiridol-treated animals was detected on the first day of testing, indicating that the retention or acquisition of learned memory was not completely normal. The reason for this finding could include incomplete neuroprotection observed with flavopiridol treatment or alterations in functional connectivity of hippocampal neurons. However, we observed almost complete normalization of behavioral outcomes on subsequent testing days. Because behavioral outcome is the ultimate measure of neuroprotection for an effective treatment strategy, our findings of improved water-maze behavior are of clear significance.
Recent evidence has suggested that the majority of neuroprotective strategies, although effective at earlier survival times, fail to provide sustained neuroprotection (Colbourne et al., 1999b; Corbett and Nurse, 1998). Our present data reflect these earlier reports. There are several possible explanations for the lack of sustainable neuroprotection afforded by CDK inhibition. One possible explanation is that the lack of protection is associated with inappropriate quantity or timing of drug delivery. Although we cannot definitely rule out this possibility, our efforts to increase delivery by continuous infusion did not result in increased prolonged survival of protected neurons.
An alternative explanation for the observed lack of long-term protection is that different pathways of death may become unmasked over time as selected pathways of death are inhibited. We have made similar observations in several different in vitro models of neuronal death. For example, caspase inhibition provides only transient protection from DNA damage (Stefanis et al., 1999). In this case, we have suggested that mitochondrial defects that persist even in the presence of caspase inhibitors may promote the more protracted delayed death. Similar types of delayed death have been observed in Bax-deficient neurons treated with β-amyloid (Giovanni et al., 2000).
The possibility of unmasking alternative pathways of delayed death becomes important if one entertains the suggestion that stress cues resulting from ischemia may be chronic, even long after the acute insult has been eliminated after reperfusion (Corbett and Nurse, 1998; Nurse and Corbett, 1996). In this view, neurons that survive the initial ischemic event may be exposed to an altered environment that includes extrinsic chronic stress cues (e.g., inflammatory cytokines) and intrinsic stress signals (e.g., loss of synaptic integrity). Accordingly, it is likely that any single neuroprotective strategy will have to be combined with other therapeutic modalities to eliminate or ameliorate these stresses before sustained neuroprotection can be achieved.
Although the potential to exploit CDK inhibition as a neuroprotective strategy against delayed neuronal death is present, the limitations of this approach, and others like it, should be considered carefully (Corbett and Nurse, 1998). At minimum, preclinical data for any single target neuroprotective therapy must establish (1) efficacy in multiple animal models of cerebral ischemia, (2) a delayed window of protection, (3) behavioral improvements after neuroprotection, and (4) long-term protection. We have demonstrated that CDK inhibitors fulfill the first three criteria, but fail on the last. However, neuroprotectant strategies such as CDK inhibition are critical because they will provide an opportunity for delayed application of other potential neuroprotective modalities. We propose that these additional modalities include strategies to alleviate more chronic stress cues that a neuron may experience after reperfusion, such as inflammation and axonal degeneration. It is likely that future neuroprotective strategies may take a more holistic and multifaceted approach to include other neuroprotective modalities.
Footnotes
Acknowledgments:
The authors thank Dr. Kenneth Marshall for his suggestions and comments regarding this project, and Susan Evans and Kathy McKay for their excellent technical assistance with temperature telemetry.