
Abstract
A reproducible model of global cerebral ischemia in mice is essential for elucidating the molecular mechanism of ischemic neuronal injury. Such a model is particularly important in the mouse because many genetically engineered mutant animals are available. In C57BL/6 and SV129/EMS mice, we evaluated a three-vessel occlusion model. Occlusion of the basilar artery with a miniature clip was followed by bilateral carotid occlusion. The mean cortical cerebral blood flow was reduced to less than 10% of the preischemic value, and the mean anoxic depolarization was attained within 1 minute. In C57BL/6 mice, there was CA1 hippocampal neuronal degeneration 4 days after ischemia. Neuronal damage depended upon ischemic duration: the surviving neuronal count was 78.5 ± 8.5% after 8-minute ischemia and 8.4 ± 12.7% after 14-minute ischemia. In SV129/EMS mice, similar neuronal degeneration was not observed after 14-minute ischemia. The global ischemia model in C57BL/6 mice showed high reproducibility and consistent neuronal injury in the CA1 sector, indicating that comparison of ischemic outcome between wild-type and mutant mice could provide meaningful data using the C57BL/6 genetic background. Strain differences in this study highlight the need for consideration of genetic background when evaluating ischemia experiments in mice.
Keywords
Transient global cerebral ischemia causes selective neuronal injury in vulnerable regions in the brain, such as the CA1 sector of the hippocampus (Kirino, 1982; Pulsinelli et al., 1982; Smith et al., 1984). Numerous studies have attempted to elucidate the pathophysiology of ischemic injury in CA1 neurons and to use these neurons as a representative model of ischemic neuronal death. Nevertheless, the precise mechanism of CA1 ischemic injury remains largely unknown.
Recent advances in gene manipulation in mice have offered a new opportunity to study the effect of overexpression or targeted disruption of specific genes in vivo. This approach has provided a powerful tool for investigating the molecular mechanism of ischemic brain damage in vivo and demonstrated the involvement of some crucial genes in focal ischemic injury (Huang et al., 1994). However, information obtained by advances in genetic technology is still restricted to focal ischemic brain damage that produces regional pannecrosis of the brain. This is because of a lack of reproducible global ischemia models in the mouse. The inconsistency of the results of ischemic injury mainly derives from individual differences in collateral flow through the circle of Willis, even in the same strain (Fujii et al., 1997; Kitagawa et al., 1998; Sheng et al., 1999; Wellons et al., 2000). The current study was designed to reduce the effect of this flow-related variation by using three-vessel occlusion, specifically occlusion of the basilar artery with a newly developed miniature clip, followed by bilateral carotid occlusion. Furthermore, we provide data for anoxic depolarization and cerebral blood flow changes, the two critical factors for ischemic injury in this small rodent model.
MATERIALS AND METHODS
Animal preparation
Male C57BL/6NCrj (BL/6; Charles River Japan, Yokohama, Japan) and 129/SvEms (SV129; Jackson Laboratory, Bar Harbor, ME, U.S.A.) mice aged 8 to 12 weeks were used in this study. All animal-related procedures were conducted in accordance with the guidelines of the National Institutes of Health (Guide for the Care and Use of Laboratory Animals).
Preparation for cerebral blood flow measurement
The day before ischemia, mice were prepared for cerebral blood flow (CBF) measurement. After induction, anesthesia was maintained with 1.5% halothane in a 30% O2/70% N2O mixture by means of an open face mask. In a lateral position, a preauditory skin incision was made, and the temporal muscle was reflected rostrally. A vinyl tube (inner diameter of 1.0 mm, outer diameter of 3.0 mm, 3.0 mm in length) was attached to the skull bilaterally with cyanoacrylate, 1 mm superior and posterior to the root of the zygomatic arch for measurement of cortical microperfusion by laser Doppler flowmetry (LDF; Advance Laser Flowmetry, model ALF-21, Advance Co., Ltd., Tokyo, Japan). The animals were returned to their cages and allowed free access to food and water until ischemia.
Global cerebral ischemia
After induction, anesthesia was maintained with 1.5% halothane in a 30% O2/70% N2O mixture with laser Doppler microperfusion probes inserted to the guide cannula bilaterally. Atropine sulphate (0.25 mg/kg, i.p.) and amikacin (20mg/kg, i.p.) were administered. Rectal temperature was maintained at 37.0°C by a heating blanket (Animal Blanket Controller, Model ATB-1100, Nihon Kohden, Tokyo, Japan) up to 30 minutes after ischemia. Temporal muscle temperature was monitored with a thermometer (Model BAT-12, Physitemp Instruments, Clifton, NJ, U.S.A.) via a needle microprobe (MT-26/2, Physitemp Instruments) and maintained at 37.0°C by a heating lamp.
After midline cervical incision, the bilateral common carotid arteries were first isolated. Subsequently, the atlanto-occipital membrane was exposed from the left side by retraction of the trachea and esophagus to the right. The dura mater and the underlying arachnoid membrane were incised by a 30-gauge needle and removed to expose the basilar artery, which was then separated from the brain stem by cutting the arachnoid trabeculae (Figs. 1D and 1E). After these preparations, the basilar artery was occluded by a vascular clip (0.2 mm diameter) stainless steel, with a tapered blade tip (Figs. 1C and 2) (FUJITA Medical Instruments Co., Ltd., Tokyo, Japan). After the cessation of blood flow in the basilar artery was visually confirmed, both common carotid arteries were occluded using two Yasargil miniclips (Figs. 1A, 1B, 1F, and 1G). The duration of the surgical procedure to the application of the final clip was usually 20 minutes. Ischemic duration was measured from the application of the last clip to the left common carotid artery. Spontaneous respiration was assisted through a face mask by a rodent respirator (Model 7025, Ugo Basile, Comerio, Italy) during and after ischemia. After a predetermined interval ranging from 8 to 18 minutes, the three clips were removed, and the restoration of blood flow was confirmed in every case by direct inspection of each artery under a microscope. Halothane exposure was discontinued immediately after reperfusion. After wound closure, the animals were returned to their cages and placed in a humidified incubator set at 33.0°C. Rectal temperature was monitored at 2, 6, 24, 48, and 96 hours after ischemia. Acetate Ringer's solution (0.5 mL) was administered subcutaneously in all animals 30 minutes and 24 hours after ischemia.
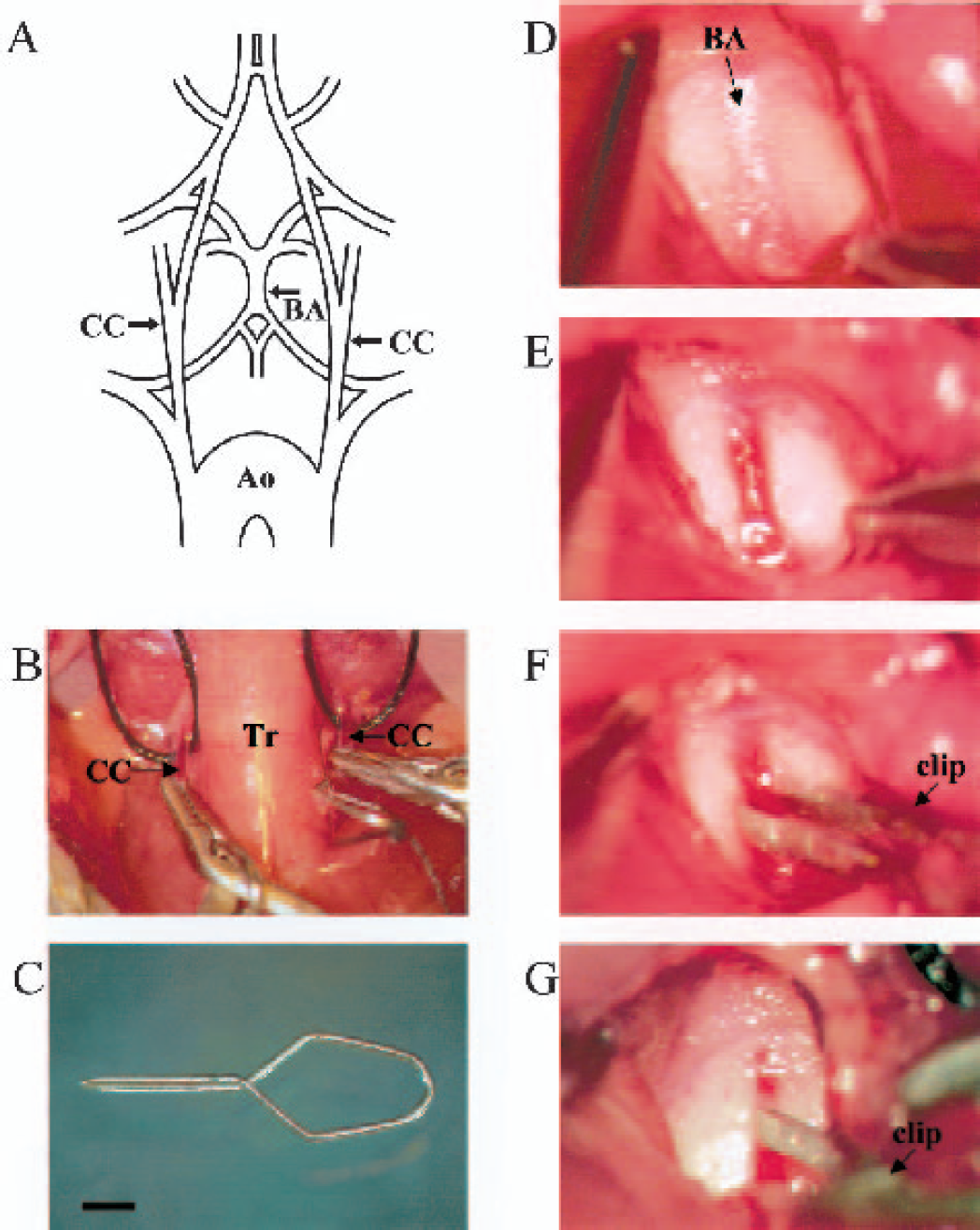
Surgical technique for inducing global cerebral ischemia.(A) Schematic illustration of arteries demonstrating the three points of clipping (arrows). (B) Surgical view of three-vessel occlusion. (C) A miniature clip for the basilar artery was made of stainless steel with a diameter of 0.2 mm. The tip of the blade was further tapered with a diamond drill under a microscope. Scale bar = 1 mm. (D) The dura and arachnoid membrane over the basilar artery was exposed from the left side by retraction of the trachea and esophagus to the right. (E) The dura mater and the underlying arachnoid membrane were removed to expose the basilar artery. (F) The miniature clip was applied to the basilar artery. (G) The cessation of blood flow was observed in the basilar artery. CC, common carotid artery; BA, basilar artery; Ao, aorta; Tr, trachea.
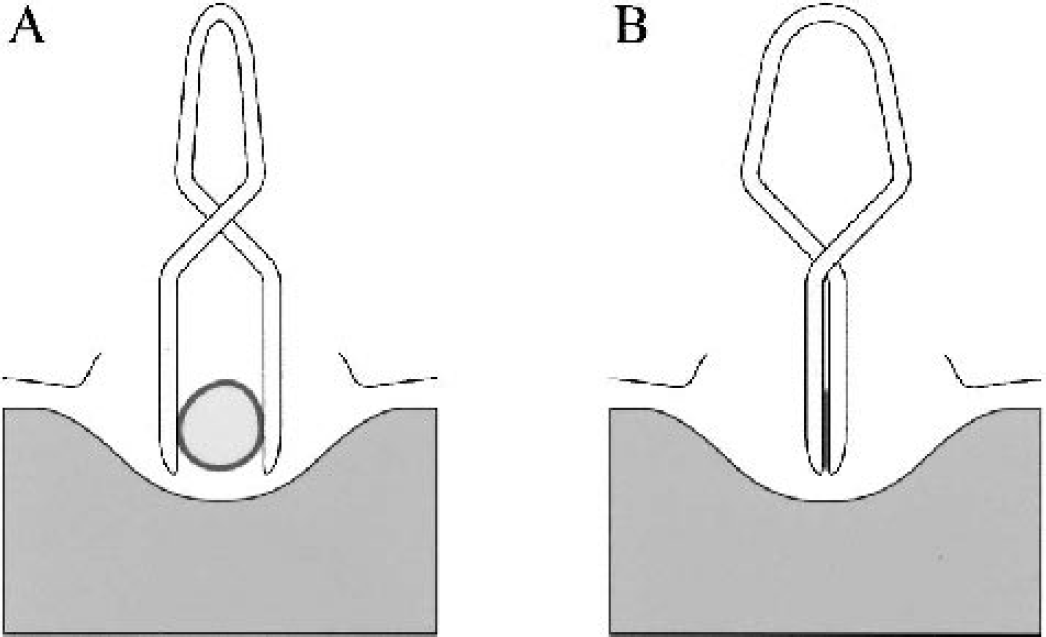
Schematic diagram depicting the clipping of the basilar artery in the coronal section. The miniature clip occluded the basilar artery without compressing the brain stem.
Measurement of physiologic parameters
In a separate group of BL/6 (n = 6, BL/6 BP group) and SV129 (n = 6, SV129 BP group) mice subjected to 14-minute ischemia, blood pressure was measured through a PE-10 cannula inserted into the left femoral artery by a carrier amplifier (Model AD-601G, Nihon Kohden, Tokyo, Japan). Arterial blood samples were analyzed before ischemia, 8 minutes after ischemia, and 15 minutes after reperfusion by a blood gas analyzer (Model 248, Chiron Diagnostics Ltd., Essex, U.K.) and a glucose analyzer (Glu-1, TOA Electronics Ltd., Tokyo, Japan).
Measurement of direct current potential
In another group of BL/6 (n = 6, BL/6 direct current [DC] group) and SV129 (n = 6, SV129 DC group) mice subjected to 14-minute ischemia, hippocampal DC potential was measured by a DC amplifier (Model AD-641G; Nihon Kohden, Tokyo, Japan) through silver chloride wires and a glass micropipette electrode (tip diameter 10 to 20 μm) inserted into the right hippocampus (Paxinos and Franklin, 2001) (1.9 mm posterior to the bregma, 1.2 mm lateral from the midline, 1.2 mm in depth from the dura) with a reference electrode placed in the neck muscle (Kawahara et al., 1995).
Blood pressure and blood gas analysis in C57BL/6 and SV129 mice subjected to 14-minute ischemia
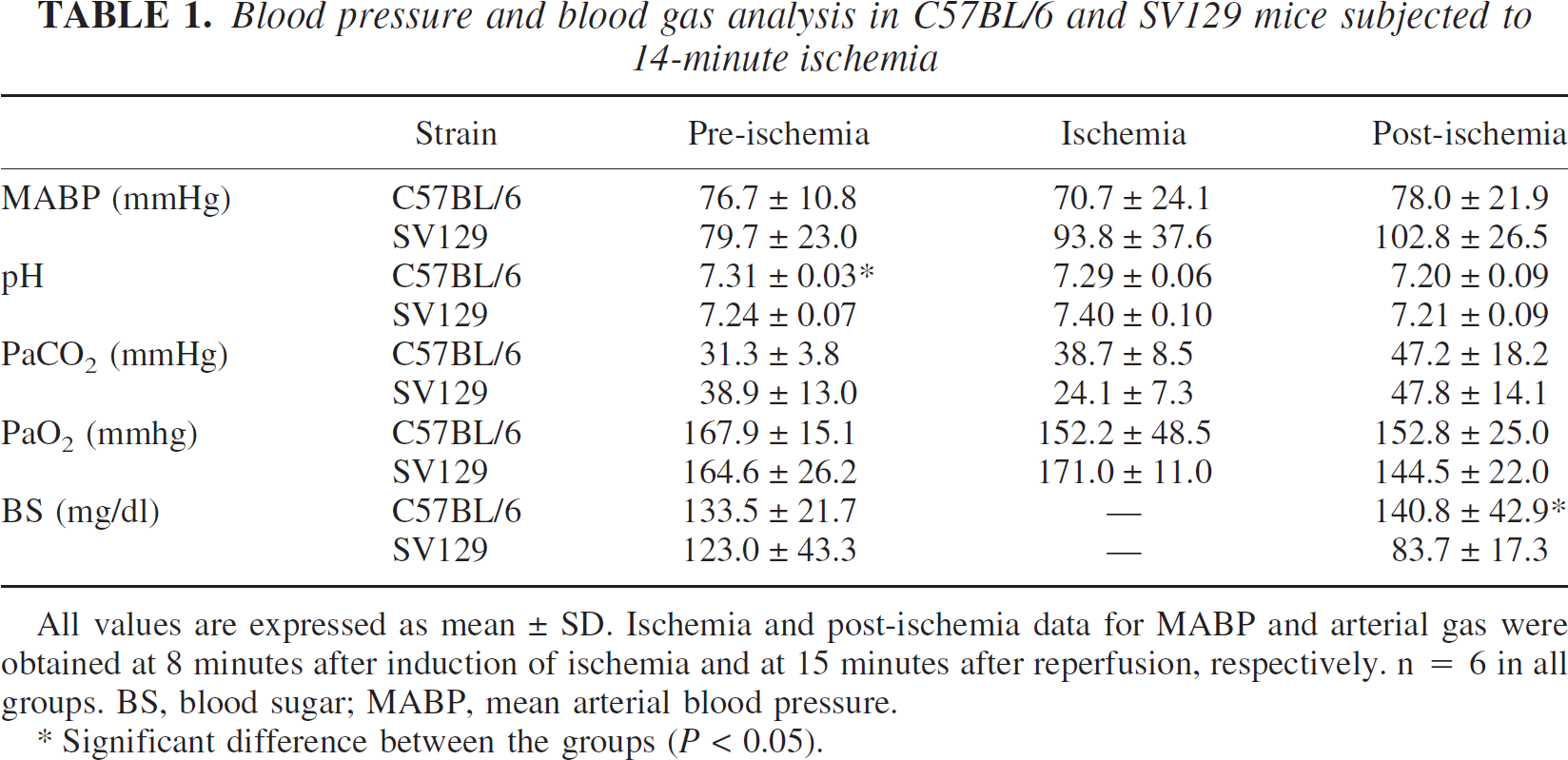
All values are expressed as mean±SD. Ischemia and post-ischemia data for MABP and arterial gas were obtained at 8 minutes after induction of ischemia and at 15 minutes after reperfusion, respectively. n = 6 in all groups. BS, blood sugar; MABP, mean arterial blood pressure.
Significant difference between the groups (P < 0.05).
Histologic evaluation of neuronal injury in hippocampal CA1 sector
BL/6 mice were subjected to 8 (n = 11, BL/6 8-minute group), 10 (n = 15, BL/6 10-minute group), 12 (n = 11, BL/6 12-minute group), or 14 (n = 18, BL/6 14-minute group) minutes of ischemia. SV129 mice were similarly subjected to 14 (n = 11, SV129 14-minute group), 16 (n = 9, SV129 16-minute group), or 18 (n = 14, SV129 18-minute group) minutes of ischemia. Four days later, the mice were anesthetized by 4% halothane exposure and transcardially perfused with 4% para***formaldehyde in 0.1 M phosphate buffer (pH = 7.4). After postfixation for several hours, the brains were removed and embedded in paraffin. Coronal sections (4 μm thickness) containing the dorsal hippocampi were stained with cresyl violet, and the number of intact neurons in the hippocampal CA1 sector was counted in a blind fashion. The average neuronal density calculated from both hemispheres was used to evaluate ischemic injury.
BL/6 mice were killed in a similar manner and evaluated at 24 hours (n = 6, BL/6 24 hour group) and 28 days (n = 7, 28 day group) after 14-minute ischemia to evaluate the temporal profile of neuronal death.
Animals subjected to the same surgery without vessel occlusion served as sham-operated controls in BL/6 (n = 6, BL/6 sham group) and SV129 (n = 6, SV129 sham group) mice.
TUNEL staining for ischemic injury
Paraffin sections of BL/6 mice subjected to 8 (n = 2) or 14 (n = 2) minutes of ischemia were processed for histologic evaluation of neuronal injury in hippocampal CA1 sector. After deparaffinization, the sections were treated with proteinase K, and the fragmented DNA was visualized with biotinylated***dUTP and terminal transferase (TUNEL method) according to the manufacturer's protocol (Roche Diagnostics, Mannheim, Germany). For evaluation of the thalamic reticular nucleus, BL/6 mice subjected to 14-minute ischemia and killed 24 hours thereafter (n = 2) were used in a similar fashion (Kawai et al., 1992).
Analysis
All values were expressed as mean ± SD. The cell counts (neuronal density) in the CA1 sector and CBF data were compared using one-way analysis of variance (ANOVA) followed by Bonferroni's post-hoc comparison with significance set at P < 0.05. Other physiologic parameters were compared using an unpaired two-tailed t-test.
RESULTS
Physiologic parameters
Physiologic parameters measured in BL/6 and SV129 BP groups subjected to 14-minute ischemia are listed in Table 1. In general, there was a large standard deviation, indicating difficulty in confining these parameters within a narrow range. Nonetheless, the mean values fell mostly within the physiologic range under our experimental conditions, and there were no significant differences between BL/6 and SV129 mice except for preischemic pH and postischemic blood glucose levels.
In the animals subjected to histologic analysis, rectal and temporal muscle temperatures were strictly controlled at 37.0 ± 0.4°C, as shown in Table 2. No hypothermic phase was noted in either strain during the time the mice were maintained in the humidified incubator, up to 96 hours after ischemia (Table 3). Although the mean rectal temperature was significantly lower in BL/6 mice at 2 and 6 hours after ischemia, and higher in BL/6 mice at 96 hours after ischemia (P < 0.05), all temperatures of both strains were within the physiologic range (36.6 to 37.8°C).
Temperature and CBF profile after 14-minute ischemia in C57BL/6 and SV129 mice
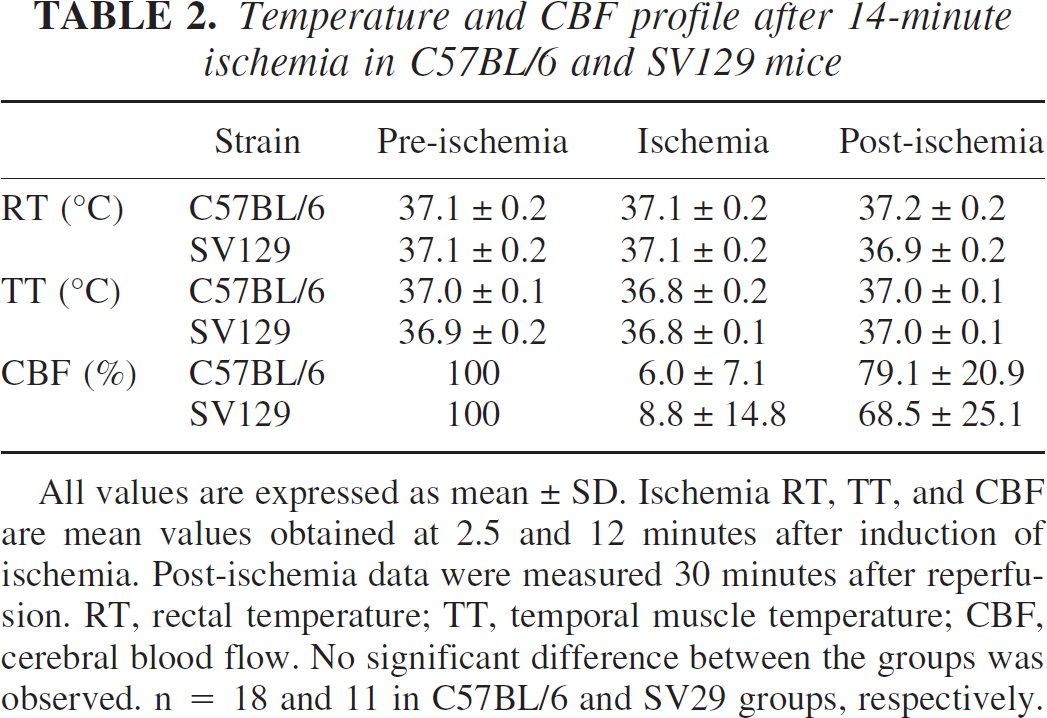
All values are expressed as mean ± SD. Ischemia RT, TT, and CBF are mean values obtained at 2.5 and 12 minutes after induction of ischemia. Post-ischemia data were measured 30 minutes after reperfusion. RT, rectal temperature; TT, temporal muscle temperature; CBF, cerebral blood flow. No significant difference between the groups was observed. n = 18 and 11 in C57BL/6 and SV29 groups, respectively.
All values are expressed as mean ± SD. Ischemia RT, TT, and CBF are mean values obtained at 2.5 and 12 minutes after induction of ischemia. Post-ischemia data were measured 30 minutes after reperfusion. RT, rectal temperature; TT, temporal muscle temperature; CBF, cerebral blood flow. No significant difference between the groups was observed. n = 18 and 11 in C57BL/6 and SV29 groups, respectively.
Rectal temperature after 14-minute ischemia in C57BL/6 and SV129 mice

All values are expressed as mean ± SD. Rectal temperature (RT) was measured at 2, 6, 24, 48, and 96 hours (h) after 14-minute ischemia. n = 18 and 11 in C57BL/6 and SV129 groups, respectively.
Significant difference between the groups at the same time point (P < 0.05).
Measurement of CBF
Cortical CBF was immediately reduced after the three vessels were clipped and remained constant during the ischemic period in all animals (Fig. 3). The mean CBF value before recirculation, at 2.5 minutes after ischemia, in BL/6 and SV129 groups was less than 10% of the preischemic control value in both groups, although the individual variation was greater in SV129 mice (Table 2). Statistically, there were no significant group or side differences (P > 0.05), indicating bilateral uniform reduction of CBF in both groups.
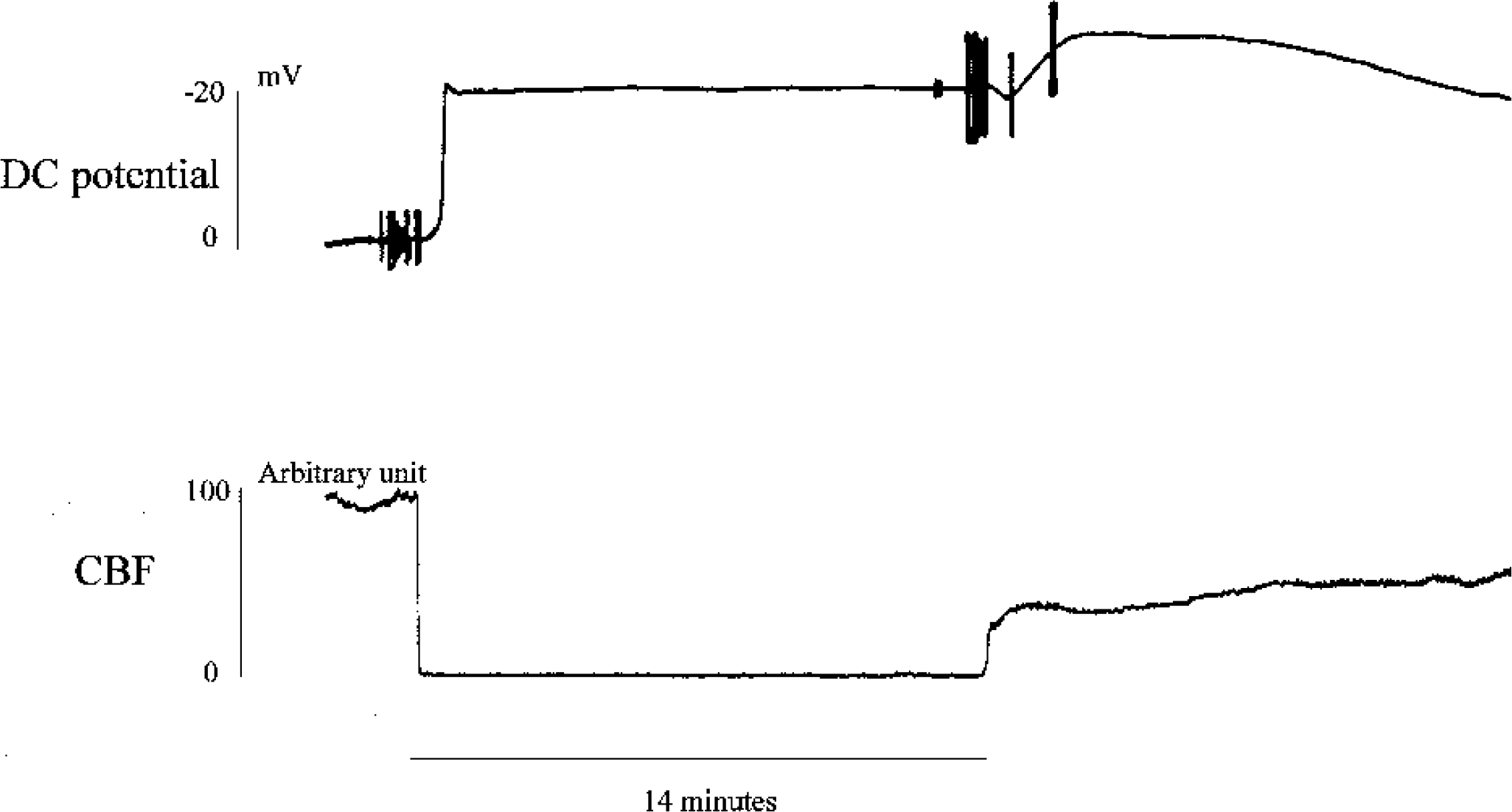
The changes of DC potential and CBF in C57BL/6 mice. CBF was reduced to less than 10% of the control value immediately after ischemia and remained constant during 14-minute ischemic period. The negative shift of DC potential was observed at 1 minute after induction of ischemia and showed further negative shift following reperfusion. DC, direct current; CBF, cerebral blood flow.
Anoxic depolarizaton
After the three-vessel occlusion, an abrupt negative shift of DC potentials was noted with a delay of 57.0 ± 14.6 seconds in the BL/6 DC group (Fig. 3) and 57.4 ± 11.9 seconds in the SV129 DC group; this observation was not statistically significant (unpaired two-tailed t-test). DC potential showed a further negative shift, followed by slow recovery after recirculation.
Survival rate and surgical failure
Surgical success and survival rates in this small rodent model are critical for the assessment of histologic outcome days after ischemia. We encountered technical difficulties, such as failure of ventilatory support, basilar artery bleeding, and airway obstruction, which resulted in death of the animals during or immediately after recirculation. However, we finally achieved an overall success rate of 94.0% in our series (n = 116, calculated from all animals in SV129 and BL/6), independent of ischemic duration and animal strain.
With regard to survival after successful recirculation, a reasonable overall survival rate of 85.9% (n = 69) at 4 days was obtained in the BL/6 group irrespective of ischemic duration. Although the survival rate after 14-minute ischemia was 100% at 24 hours, it decreased to 58.3% at 28 days. In contrast, the survival rate at 4 days in SV129 groups decreased according to the duration of ischemia, resulting in a survival rate of 51.9% after 18-minute ischemia, compared with 100% following a 14- or 16-minute period of ischemia.
Neuronal injury in the hippocampal CA1 sector
Consistent neuronal injury in the CA1 sector was observed in BL/6 mice as the ischemic duration increased (Fig. 4). Although few neurons showed degeneration after 8-minute ischemia (Figs. 4C and 4D), most of the pyramidal neurons exhibited pyknotic, shrunken nuclei after 14-minute ischemia at 4 days (Figs. 4E and 4F). Neuronal death or loss was confirmed by the complete disappearance of these degenerated neurons at 28 days after 14-minute ischemia (Figs. 4G and 4H). DNA fragmentation detected by TUNEL staining was demonstrated only in this ischemic group at 4 days but not at 24 hours or after 8-minute ischemia (Figs. 5A–C). These observations indicate that, using our model, a 14-minute period of ischemia in BL/6 mice leads to severe, irreversible neuronal injury by 4 days.
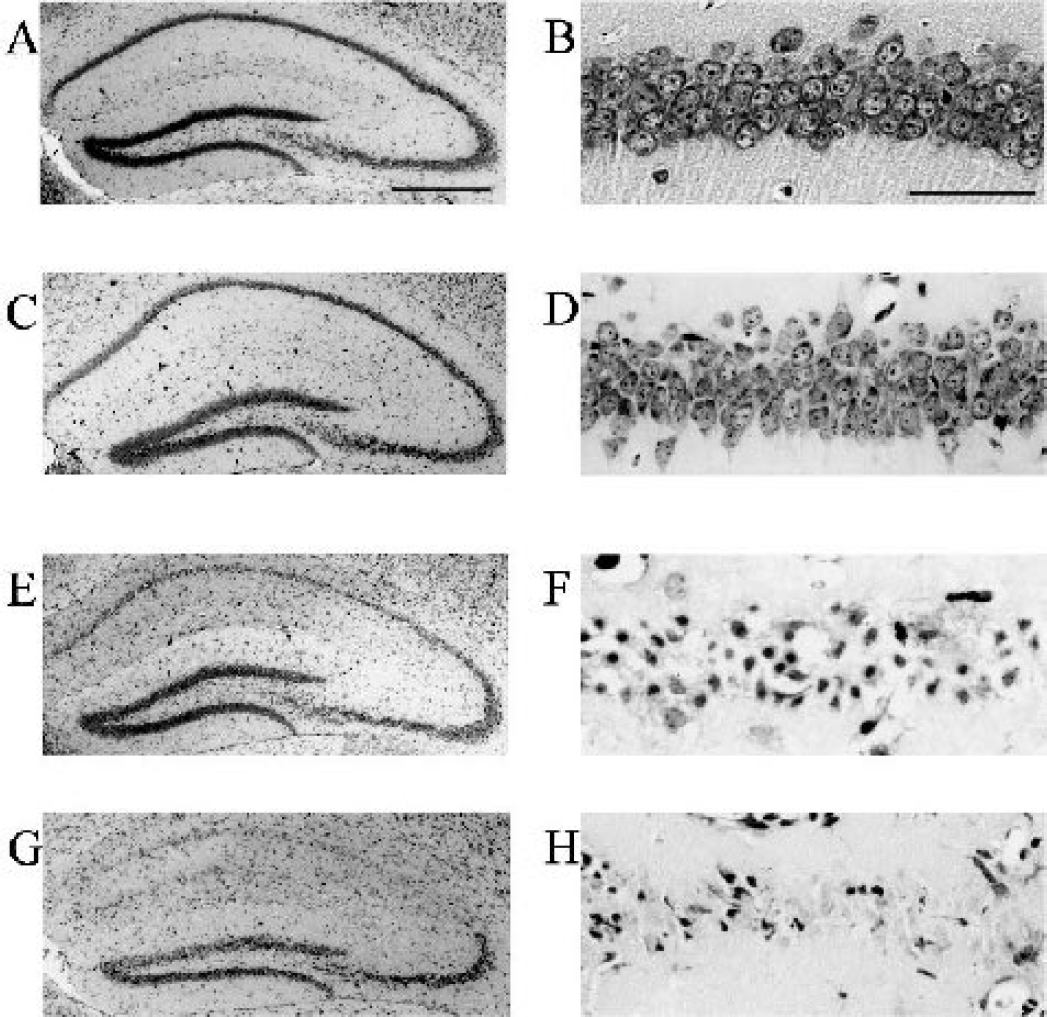
Histologic evaluation with cresyl violet staining of the hippocampal CA1 sector in C57BL/6 mice. (A, B) Normal control sections. After 8-minute ischemia, only a few degenerated neurons were noted in CA1 (C and D). After 14-minute ischemia, however, CA1 neurons were uniformly degenerated at 4 days (E and F). This was more clearly observed at 28 days (G and F). Scale bar = 50 μm (left column) and 50 μm (right column).
These CA1 neuronal injuries were quantitatively compared according to ischemic duration and survival time in BL/6 groups (Fig. 6A). Although the number of intact neurons was slightly reduced after 8-minute ischemia (78.5 ± 8.5% of normal, P > 0.05), there was a significant duration-dependent reduction after ischemia lasting longer than 10 minutes, reaching 8.4 ± 12.7% of normal control numbers after 14-minute ischemia, with consistent injury and minimal individual variation. There was no difference in CA1 neuronal injury at 4 and 28 days after 14-minute ischemia (Fig. 6B).
In contrast to these observations in BL/6 mice, severe neuronal injury was not observed in SV129 mice (Fig. 6C). The neuronal count remained at 67.8 ± 30.7% after 14-minute ischemia. When ischemic duration was increased to 16 or 18 minutes, the neuronal count did not decrease further. This result indicates that the profile of ischemic injury differs in BL/6 and SV129 mice.
Ischemic injury in other regions
Similarly, ischemic injury was observed in the hippocampus of BL/6 mice at 4 days after ischemia as shown by cresyl violet and TUNEL staining. In the CA2 sector, neuronal degeneration was noted in 32% of animals after 8-minute ischemia, reaching 100% after 12-minute ischemia. Similarly, in the CA3 sector, neuronal degeneration was observed in 14% of animals after 8-minute ischemia and in 46 to 75% after 10 to 14 minutes of ischemia, as depicted by TUNEL-positive neurons in this region (Fig. 5E). In addition, dentate granule cells displayed scattered neuronal degeneration (Fig. 5D) in 27 to 33% of animals after 10 and 12 minutes of ischemia and 86% of animals after 14-minute ischemia.
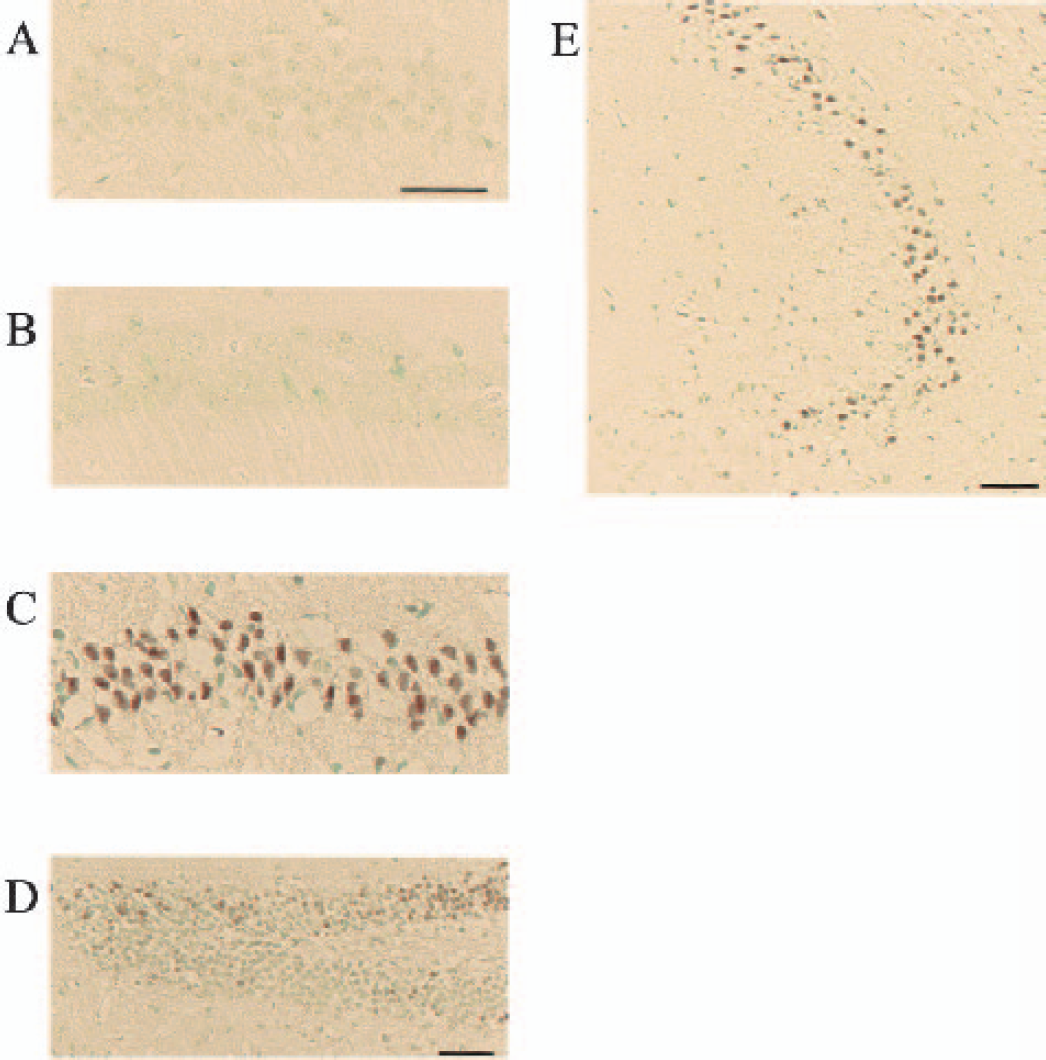
TUNEL staining in the hippocampi of C57BL/6 mice. DNA fragmentation as depicted by TUNEL staining was not observed in the CA1 sector 4 days after 8-minute ischemia (A) or 24 hours after 14-minute ischemia (B). However, 4 days after 14-minute ischemia, TUNEL-positive cells were observed in the CA1 sector (C). TUNEL positive cells were also observed in the dentate gyrus (D) and CA3 sector (E) in some animals 4 days after 14-minute ischemia. Scale bar = 50 μm.
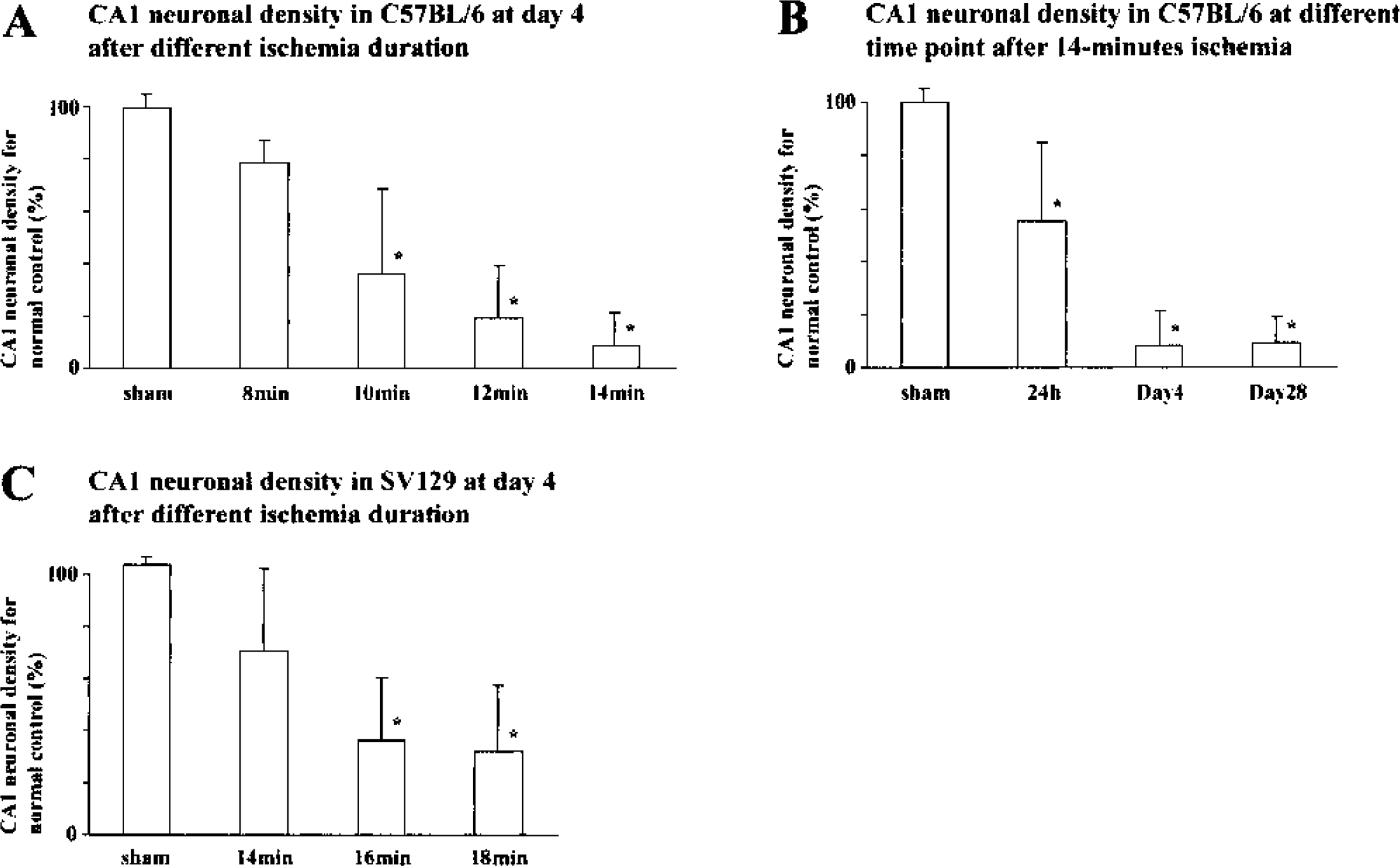
Quantitative evaluation of CA1 neuronal injury in C57BL/6 and SV129 mice. (A) CA1 neuronal count was compared between sham-operated control groups and groups subjected to 8- to 14-minute ischemia at 4 days after ischemia in C57BL/6 mice. A significant reduction in surviving neurons was observed after 10-, 12-, and 14-minute ischemia when compared with the sham-operated control groups. (B) CA1 neuronal count after 14-minute ischemia was evaluated at different time points after ischemia in C57BL/6 mice. A severe and significant reduction in the neuronal count was not observed at 24 hours but was observed at 4 and 28 days. There was complete degeneration at 4 days. (C) Similar analysis in SV129 mice subjected to 14- to 18-minute ischemia at 4 days. Mild but significant ischemic injury was observed after 14-minute ischemia compared with that in C57BL/6 mice. Although the ischemic duration was increased up to 18 minutes, no severe injury was attained in this strain. All values are expressed as mean ± SD. * Significant difference when compared with sham-operated control groups (P < 0.0001).
Outside of the hippocampus, TUNEL-positive cells were also observed after 14-minute ischemia in BL/6 mice (Fig. 7). These cells were noted mainly in layers 3 to 5 of the cortex, the ventrolateral part and the reticular nucleus of the thalamus, and in the dorsal-lateral part of the striatum. However, neither the severity nor intensity of these injuries was consistent among the animals studied.
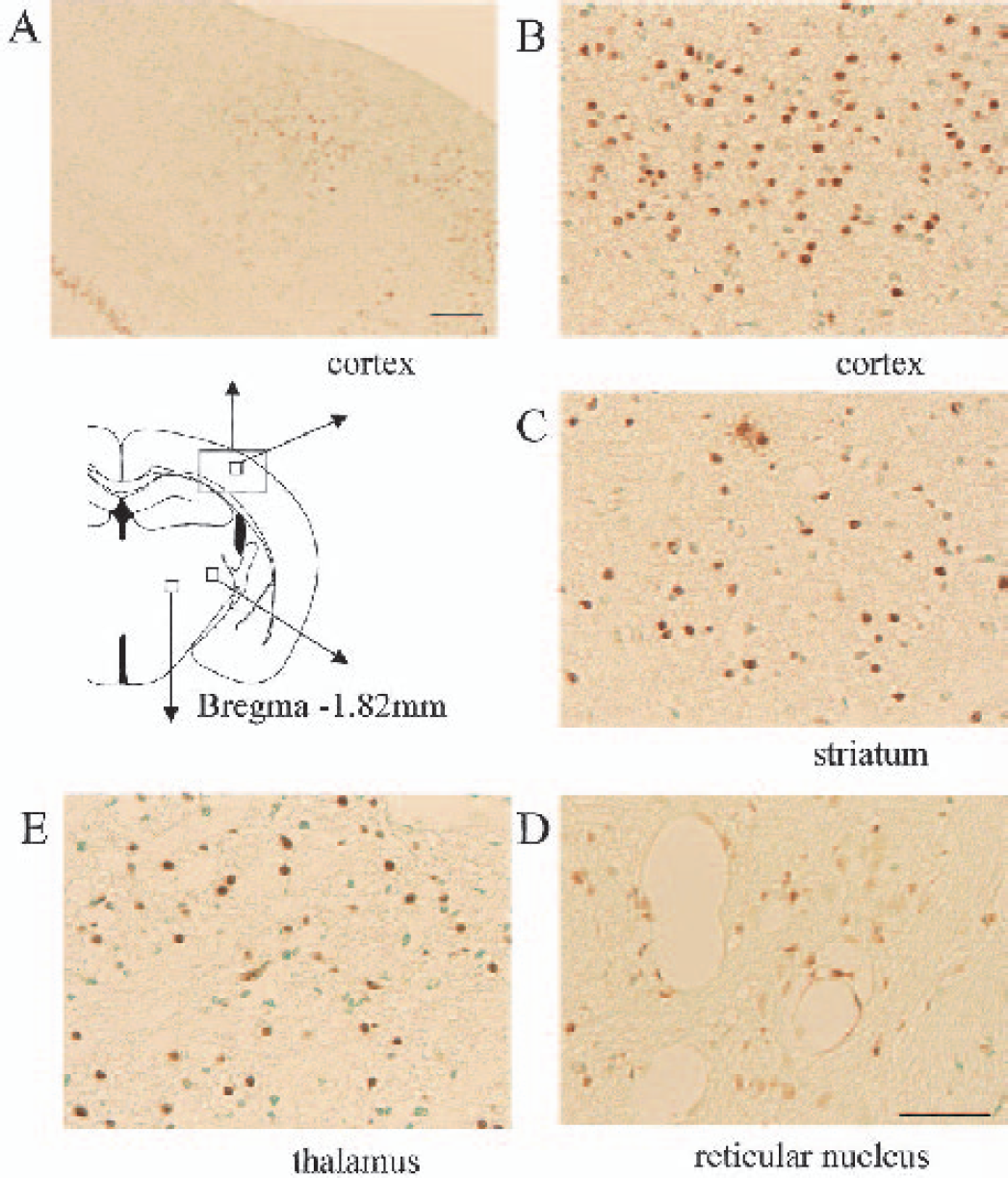
TUNEL staining in C57BL/6 mice subjected to 14-minute ischemia. TUNEL-positive cells were observed outside of the hippocampus and cortex 4 days after ischemia (A and B), striatum 4 days after ischemia (C), reticular nucleus 24 hours after ischemia (D), ventrolateral thalamus 4 days after ischemia (E). Scale bar = 250 μm (A) and 50 μm (B–E).
DISCUSSION
In this study, we developed a global ischemia model with consistent and uniform neuronal degeneration in the hippocampal CA1 sector in BL/6 mice. In this model, cortical CBF was sufficiently reduced and the anoxic depolarization was obtained within 1 minute, showing near-complete ischemia in the hippocampus in all the animals studied.
Several previous authors have attempted to establish global ischemia models with quantitatively uniform injury in CA1, using techniques including two-vessel occlusion and two-vessel occlusion with hypotension (Fujii et al., 1997; Kitagawa et al., 1998; Sheng et al., 1999; Wellons et al., 2000). However, because of the variability of collateral flow via the posterior communicating artery, it has been difficult to obtain uniform injury in CA1 while retaining high survival and success rates. Although we attempted a cardiac arrest model independent of collaterals, we could not produce CA1 neuronal injury within the ischemic time window required for animal survival (Kawahara et al., 2002). Thus we tried basilar artery occlusion combined with bilateral common carotid artery occlusion (three-vessel occlusion), originally described by Panahian et al. (1996). However, the animal survival rate was not high as reported later by the same group (Fujii et al., 1997), and CA1 neuronal injury was inconsistent. The basilar artery of mice actually runs through a narrow groove in the brain stem and is attached to the pia mater and arachnoid membrane with arachnoid trabeculae (Figs. 8A and 8B). For this reason, the Zen clip originally used by Panahian et al. (1996) is too large to obtain complete clipping of the basilar artery without compression injury to the brain stem. Thus we developed a new tapered miniature clip that fits to the groove and applied it after the complete untethering of the artery. This technique minimized brain stem injury at the clipping site (Figs. 8C and 8D) and contributed to relatively high survival rate (85.9%, n = 69, at 4 days). This survival rate is comparable with that obtained by Kitagawa et al. (1997) in a two-vessel occlusion model.
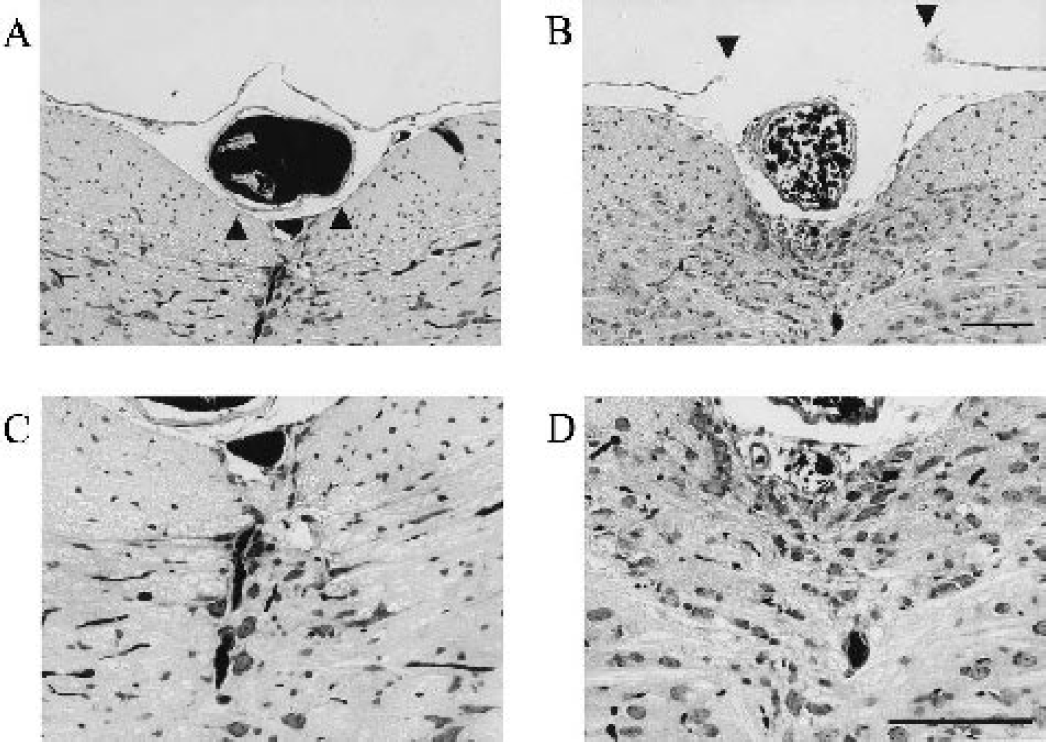
Histologic demonstration of the basilar artery and brain stem in C57BL/6 mice. (A) In an intact animal, the basilar artery is covered by the arachnoid membrane and embedded in a narrow groove, which is further tethered to the pia mater by arachnoid trabeculae (triangle pointing up). (B) In an animal subjected to ischemia, the arachnoid membrane was removed (triangle pointing down) and the basilar artery separated from the arachnoid trabeculae, thus enabling complete clipping by a tapered clip without compression injury to the brain stem. C and D are higher magnification images of A and B, respectively. (C) The ventral part of brain stem around the basilar artery is shown in an intact animal. (D) In animals subjected to ischemia, the injury to the ventral part of brain stem was minimal, although a slight glial reaction was noted beneath the basilar artery. The basilar artery is filled with carbon dye. Scale bar = 100 μm.
Confirmation of CBF reduction in ischemia models is important, particularly in murine models, for the exclusion of nonischemic animals. In our model, we measured cortical CBF bilaterally from the border zone of the middle and posterior cerebral arteries, and obtained severe reduction of CBF in all animals, with a mean CBF value of less than 10%. The CBF values were slightly high compared with those advocated as exclusion criteria for the two-vessel occlusion model by Kitagawa et al. (1998). However, anoxic depolarization by DC potential measurement was obtained at approximately 60 seconds in all animals, confirming near-complete ischemia in the hippocampus. Similar DC potential measurement results were obtained in a previous study of the cardiac arrest model (Kawahara et al., 2002). The resultant neuronal injury in the CA1 sector was increased in a duration-dependent manner, such that near-complete neuronal degeneration was observed after 14-minute ischemia, but only minimal degeneration was observed after 8-minute ischemia. This observation indicates that, in our model, it was possible to induce reproducible neuronal injury bilaterally by adjusting ischemic duration in all animals. In contrast, in a previous study using the two-vessel occlusion model, only 50% of BL/6 mice showed bilateral CA1 injury even after severe cortical CBF reduction criteria were applied; however, these criteria were not yet amenable to quantitative analysis (Kitagawa et al., 1997). The uniformity and consistency of our model of forebrain ischemia should make it more reliable than existing models for the evaluation of various interventions in mice.
We evaluated neuronal injury in the CA1 sector at 4 days after ischemia because the survival rate decreased throughout the postischemic period. We showed that neuronal death at 4 days after 14-minute ischemia is almost complete and irreversible in BL/6 mice. The survival rate at 4 days (85.9%, n = 69) would be acceptable for various quantitative evaluations, but the survival rate at 28 days (58.3%) is less satisfactory. Nonetheless, this model could be used as a long-term survival model in certain situations, such as the neuronal regeneration study of CA1 pyramidal neurons we reported previously (Nakatomi et al., 2002).
The ischemic duration required for complete CA1 neuronal injury was longer in BL/6 mice than has been previously reported for rats and gerbils (Kirino, 1982; Pulsinelli et al., 1982; Smith et al., 1984). Because ischemic injury is dependent upon many factors including severity of ischemia, residual flow, and temperature, this variation in ischemic duration cannot be directly attributed to species differences. In the current study, however, we maintained normothermia during and after ischemia and showed almost complete ischemia comparable with that in cardiac arrest by anoxic depolarization. Of particular interest is our DC recording finding of very slow repolarization after reperfusion. This result was not found after nonlethal ischemia in a cardiac arrest model (Kawahara et al., 2002). This discrepancy might indicate that severe metabolic insult is required for neuronal injury in mice, compared with other rodent global ischemia model. Vulnerability to ischemia may therefore be species-specific, although the mechanisms for such differences are unknown.
In line with these observations, the results obtained in SV129 mice, a strain commonly used as a genetic background for many mutant mice (Gerlai, 1996), should be evaluated carefully. Although severe ischemia, as demonstrated by cortical CBF measurement, was achieved in SV129 mice in our model, neuronal injury after 14-minute ischemia was much less severe in SV129 than in BL/6 mice. Because subtle differences in residual flow could influence the degree of neuronal damage, we compared cortical CBF reduction and histologic outcome in both strains after 14-minute ischemia. We did not observe a relationship between CBF and histologic outcome in either strain in this low-flow range (data not shown). However, anoxic depolarization showed comparable near-complete ischemia in both strains. Because temperature is critical to ischemia in small rodents, we maintained intra- and postischemic temperature in both strains within the physiologic range using a humidified incubator set at 33°C (Table 3). A slightly higher temperature (less than 1°C) was noted in strain SV129 during the early reperfusion phase. However, this difference did not appear to influence our results, as it was the BL/6 mice, not the SV129 mice, that had the more severe neuronal damage. These findings, when considered with the species-specific vulnerability of rodents, suggest that strain influences ischemic vulnerability in mice independent of CBF and temperature. In fact, strain differences for kainate-induced neuronal degeneration have already been demonstrated in mice (Schauwecker and Steward, 1997). Although the reasons for this discrepancy are currently unknown, it must be considered when designing or interpreting studies using animals with different genetic backgrounds. Backcrossing for several generations can infer the benefits of the genetic background of BL/6 without causing changes in the mutated gene.
In this study, we established a global ischemia model in BL/6 mice using a modified three-vessel occlusion technique and a 14-minute ischemic duration. The model consistently results in quantitatively uniform neuronal injury in CA1. This model could greatly contribute to research on the in vivo molecular mechanisms of neuronal death in genetically engineered mice. We also observed similar ischemic injuries in other regions, including CA3, dentate gyrus, cortex, striatum, and thalamus, but the frequency and intensity of these injuries were inconsistent. Additional methodologic improvement in the quantitative evaluation of these regions would further increase the usefulness of this model for ischemia research.
Footnotes
Acknowledgements
The authors thank Ms. Reiko Matsuura and Ms. Kaori Hasegawa for their technical assistance.