
Abstract
Protein synthesis inhibition occurs in neurons immediately on reperfusion after ischemia and involves at least alterations in eukaryotic initiation factors 2 (eIF2) and 4 (eIF4). Phosphorylation of the α subunit of eIF2 [eIF2(αP)] by the endoplasmic reticulum transmembrane eIF2α kinase PERK occurs immediately on reperfusion and inhibits translation initiation. PERK activation, along with depletion of endoplasmic reticulum Ca2+ and inhibition of the endoplasmic reticulum Ca2+-ATPase, SERCA2b, indicate that an endoplasmic reticulum unfolded protein response occurs as a consequence of brain ischemia and reperfusion. In mammals, the upstream unfolded protein response components PERK, IRE1, and ATF6 activate prosurvivial mechanisms (e.g., transcription of GRP78, PDI, SERCA2b) and proapoptotic mechanisms (i.e., activation of Jun N-terminal kinases, caspase-12, and CHOP transcription). Sustained eIF2(αP) is proapoptotic by inducing the synthesis of ATF4, the CHOP transcription factor, through “bypass scanning” of 5‘ upstream open-reading frames in ATF4 messenger RNA; these upstream open-reading frames normally inhibit access to the ATF4 coding sequence. Brain ischemia and reperfusion also induce μ-calpain–mediated or caspase-3–mediated proteolysis of eIF4G, which shifts message selection to m7G-cap–independent translation initiation of messenger RNAs containing internal ribosome entry sites. This internal ribosome entry site–mediated translation initiation (i.e., for apoptosis-activating factor-1 and death-associated protein-5) can also promote apoptosis. Thus, alterations in eIF2 and eIF4 have major implications for which messenger RNAs are translated by residual protein synthesis in neurons during brain reperfusion, in turn constraining protein expression of changes in gene transcription induced by ischemia and reperfusion. Therefore, our current understanding shifts the focus from protein synthesis inhibition to the molecular pathways that underlie this inhibition, and the role that these pathways play in prosurvival and proapoptotic processes that may be differentially expressed in vulnerable and resistant regions of the reperfused brain.
Keywords
Significant new knowledge in the field of translation control has opened up fresh avenues for the understanding of neuronal PSI induced by brain I/R. Here we review (1) evidence that I/R induces inhibition of translation initiation by altering eIF2 and eIF4, (2) evidence that these eIF alterations are expected to cause major alterations in the repertoire of new peptides synthesized by residual translation in vulnerable neurons, (3) evidence that phosphorylation of the α-subunit of eIF2 [holoprotein eIF2(αP), subunit eIF2α(P)] during at least initial reperfusion is caused by the eIF2α kinase PERK, which is activated by the UPR elicited by ER stress, (4) the current understanding of the UPR and its role in prosurvival and proapoptotic signal transduction and transcription mechanisms, and (5) further evidence that the UPR is induced by brain I/R. The understanding that I/R-induced alterations in eIF2 and eIF4 impose translational constraints on gene expression (Read et al., 2001) shifts the focus for further investigation from the phenomena of PSI to the pathways mediating PSI and the profound consequences that differential translation may have on stress resolution or activation of proapoptotic processes in vulnerable and resistant neurons.
Brain ischemia and reperfusion induce inhibition of translation initiation through molecular alterations in eIF2 and eIF4
The characteristics of postischemic PSI have been amply documented (Hossmann, 1993; Krause and Tiffany, 1993). Immediately on reperfusion after global ischemia, protein synthesis is inhibited in neurons throughout the entire brain. Dependent on the duration of ischemia, protein synthesis will gradually recover in brain regions more resistant to ischemic damage, but remains suppressed in selectively vulnerable neurons. Ischemic preconditioning can attenuate postischemic PSI (Kato et al., 1995), and the use of cycloheximide to block protein synthesis during preconditioning abolishes the subsequent neuroprotective effects (Barone et al., 1998). In the most vulnerable brain region, the CA1 of the hippocampus, protein synthesis never recovers fully during reperfusion even after relatively brief ischemia (Araki et al., 1990; Thilmann et al., 1986). Thus, a number of investigators have suggested that persistent PSI is a marker for neurons destined to undergo delayed neuronal death in the reperfused brain (Hossmann, 1993; Krause and Tiffany, 1993; White et al., 2000; Paschen, 2000). As we will see, our current understanding of the UPR and the regulation of translation initiation provide potential causal pathways for this inference.
In their seminal description of postischemic PSI, Kleihues and Hossmann (1971) observed polysome disaggregation and accumulation of free monosomes and implicated inhibition of translation initiation as the mechanism of PSI. It is now accepted that PSI during brain reperfusion is caused by inhibited translation initiation that involves modifications to eIF2 and eIF4 (Krause and Tiffany, 1993; DeGracia et al., 1996).
Roles of eIF2 and eIF4 in translation initiation.
In eukaryotes, translational control at initiation is coordinated by eIFs that orchestrate the assembly of ribosomal subunits, the messenger RNA (mRNA) to be translated, and the methionine-charged initiator transfer RNA (Met-tRNAi) (Pain, 1996). A ternary complex comprising eIF2, Met-tRNAi, and GTP delivers the peptide-initiating methionine to the 40S ribosomal subunit (Fig. 1), and this step of translation initiation controls the gross rate of protein synthesis (Pain, 1996). At a later stage of initiation, hydrolysis of this GTP generates an eIF2-GDP complex, and eIF2B (guanine nucleotide exchange factor) must catalyze the removal of this GDP for eIF2 to bind another GTP and participate in another round of translation initiation (Rowlands et al., 1988). The eIF2 containing an α-subunit phosphorylated on Ser-51 is a competitive inhibitor of eIF2B (Rowlands et al., 1988); thus, increased levels of eIF2α(P) downregulate the rate of global protein synthesis (Pain, 1996). Indeed, because the ratio of eIF2 to eIF2B is approximately 5:1 in the brain (Alcàzar et al., 1995), translation initiation becomes substantially inhibited when approximately 20% of eIF2 is phosphorylated (Pain, 1996). The inhibition of eIF2B by eIF2(αP) is an equilibrium process, so a small amount of ternary complex will remain available for residual translation even when eIF2(αP) levels are high. Phosphorylation of eIF2α occurs in response to diverse cellular stresses, including the UPR (Ron and Harding, 2000), viral infection (Kaufman, 2000), heme depletion in reticulocytes (Chen, 2000), and nutrient deprivation (Kimball and Jefferson, 2000), and occurs rapidly during postischemic brain reperfusion.
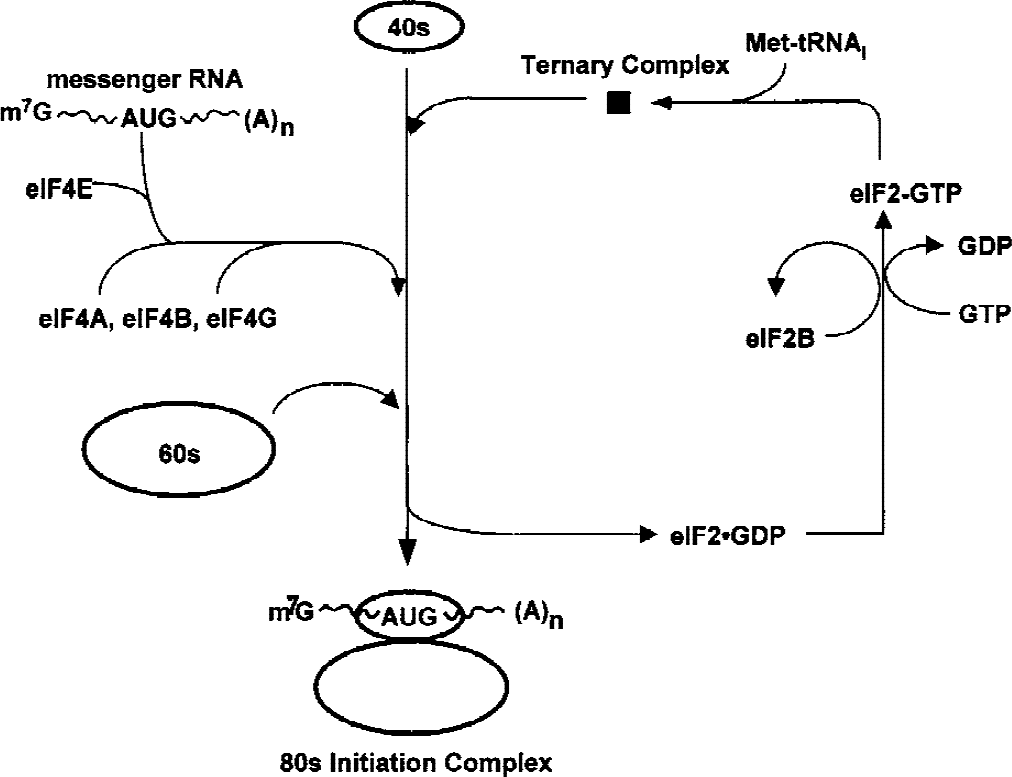
Abbreviated steps in translation initiation showing the roles of eIF2 and eIF4. The GTP-bound eIF2 delivers the methionyl-charged initiator transfer RNA to the 40S ribosomal subunit as a ternary complex. At the end of initiation, the GTP on eIF2 is hydrolyzed, producing inactive eIF2-GDP. Active eIF2 is recycled by the guanine nucleotide exchange factor, eIF2B. Phosphorylation of the α subunit of eIF2 leads to sequestration of eIF2B, accumulation of eIF2-GDP, and inhibition of protein synthesis by lack of delivery of the first amino acid. eIF4 is a tetramer, eIF4E recognizes the m7-G cap of capped messenger RNAs, eIF4A and eIF4B possess RNA helicase activity and allow ATP-dependent scanning along the 5′ untranslated region of the messenger RNA to the AUG start codon, and eIF4G is a large scaffolding protein mediating interactions of the various translation initiation components. Proteolytic fragments of eIF4G can initiate translation in the absence of eIF4E by binding IRES in the 5′ untranslated region.
The eIF4F complex that delivers the mRNA to the 40S ribosomal subunit (Fig. 1) also regulates translation initiation (Pain, 1996). The eIF4F consists of the following three subunits: (1) eIF4E, which binds the m7G-cap of the mRNA, (2) eIF4A, an mRNA helicase and ATPase, and (3) eIF4G, a scaffolding protein with binding sites for eIF4E, eIF3, and polyA binding protein. After binding the eIF4F complex with eIF4B, a cofactor of eIF4A, the small ribosomal subunit uses ATP to scan the 5′ untranslated region of the mRNA until it encounters the AUG start codon; the factors affecting recognition of the start codon are complex and can involve cis and trans factors at the 5′ and 3′ ends of the mRNA (Gray and Wickens, 1998). Phosphorylation of eIF4E on Ser-53 increases its binding to eIF4G (Lamphear and Panniers, 1990), and eIF4E activity is also controlled by the phosphorylation state of the eIF4E-binding proteins, which sequester eIF4E when unphosphorylated (Gingras et al., 1999). The eIF4G is also phosphorylated at multiple sites, but the function of this phosphorylation is unknown (Yan et al., 1992). Increasing the quantity or activity of eIF4F enhances the translation of mRNAs that are in low abundance (e.g., transcription factors, growth factor receptors) or have complex secondary structures (Gray and Wickens, 1998).
Translation control modifications during ischemia and reperfusion at steps other than translation initiation.
There is a strong general consensus that inhibition of translation initiation during at least the initial hours of brain reperfusion is caused by molecular alterations of eIF2 and eIF4, but recent studies have investigated other aspects of translation control. Althausen et al. (2001) observed that inhibitory phosphorylation of eukaryotic elongation factor 2 (eEF-2) moderately increased during 60-minute middle cerebral artery occlusion, but was normalized after 1 hour of reperfusion and decreased at 3 and 6 hours of reperfusion. These investigators took this modest and transient change to indicate that posttranslational modification of eEF-2 was not a substantial component of PSI induced by I/R. The phosphorylation state of p70S6K has also been recently examined during brain I/R. Hypophosphorylated p70S6K is associated with lowered protein synthesis during cell quiescence (Proud, 1996), and Althausen et al. (2001) also observed during 1-hour middle cerebral artery occlusion a marked reduction of p70S6K phosphorylation that only partially recovered after 6 hours of reperfusion. Janelidze et al. (2001) likewise found a persistent decrease in phosphorylation of p70S6K during reperfusion after 2-hour middle cerebral artery occlusion; however, Martin de la Vega et al. (2001a) found that although p70S6K kinase activity was decreased at 30-minute reperfusion after 30-minute ischemia by 4VO, it was normalized after 4 hours of reperfusion. Therefore, although these studies suggest that important alterations of translation control may occur at steps other than initiation during brain I/R, they do not provide an alternate causal basis for the immediate PSI, and they do not contradict the importance of the molecular alterations in eIF2 and eIF4 for the regulation of translation initiation.
Molecular alteration of eIF2 induced by brain ischemia and reperfusion.
Hu and Wieloch (1993) reported the first evidence that brain I/R induced substantial dysfunction in the eIF2 system. In rat brain homogenates obtained after 15-minute ischemia and 30-minute or 1-hour reperfusion, formation of the ternary complex was inhibited. The investigators found that this could be improved by the addition of purified eIF2B, and eIF2B activity (as measured by dissociation of preformed 3H-GDPeIF2) was inhibited. Burda et al. (1994) confirmed that 30-minute 4VO and 30-minute reperfusion caused reduction of eIF2B activity, and used isoelectric focusing to show that approximately 28% of total eIF2α comigrated with in vitro phosphorylated eIF2α(P). We confirmed an approximately 85% inhibition of translation initiation in rat brain homogenates obtained after 90-minute reperfusion after 10-minute cardiac arrest, and used isoelectric focusing to show that this was associated with approximately 23% of total eIF2α in the phosphorylated state (DeGracia et al., 1996). We then developed a phospho-specific antibody against Ser-51–phosphorylated eIF2α, and confirmed that it specifically recognized eIF2α(P) but not unphosphorylated eIF2α, and was monospecific for eIF2α(P) on Western blots of brain homogenates (DeGracia et al., 1997). We then showed that in postmitochondrial supernatant of control brain, eIF2α(P) is approximately 1% of total eIF2α, and that global I/R induces increases in eIF2α(P) of from 10-fold to 24-fold, depending on the duration of I/R (DeGracia et al., 1997, 1998). These large changes in the extent of eIF2α phosphorylation are not associated with any significant changes in the total amount of eIF2α (Burda et al., 1994; DeGracia et al., 1996, 1997; Althausen et al., 2001), and there is little increase in eIF2α(P) during ischemia in global (DeGracia et al., 1997) or focal models (Althausen et al., 2001). In focal ischemia, Althausen et al. (2001) showed an approximately 20-fold increase in eIF2α(P) in unfractionated mouse brain homogenates from the MCA territory after 1-hour reperfusion after 1-hour middle cerebral artery occlusion; in this study, the level of eIF2α(P) was substantially resolved approximately threefold that of normal levels after 6 hours of reperfusion. Our immunohistochemical studies with the anti-eIF2α(P) antibody after cardiac arrest show that by 5 to 10-minute reperfusion after global brain ischemia, eIF2α(P) is intensely stained in the perinuclear cytoplasm of principal neurons throughout the entire brain (DeGracia et al., 1997; Sullivan et al., 1999; Page et al., 2000), but by 60-minute or 4-hour reperfusion, there has been substantial clearing from injury-resistant neurons and a subcellular shift to intranuclear localization in vulnerable CA1 pyramidal neurons (DeGracia et al., 1997). In the focal ischemia model of Althausen et al. (2001), immunohistochemical staining also showed little eIF2α(P) in normal brain sections but intense staining of perinuclear cytoplasm and nuclei in cortical neurons after 1-hour reperfusion.
It is not surprising that there is some variation in reports of the relative I/R-induced increase in eIF2α(P). In addition to the effect of different I/R models, some of these differences probably reflect the different methods used to process brain homogenates. We have generally used the postmitochondrial supernatant obtained after 20,000 g centrifugation of forebrain homogenates, and observed similar 20-fold to 25-fold I/R-induced increases in eIF2α(P) on Western blots of completely unfractionated brain homogenates (DeGracia et al., unpublished data, 2000). Similarly, Althausen et al. (2001) observed an approximately 20-fold increase in eIF2α(P) in unfractionated brain homogenates obtained after 1-hour reperfusion. In contrast, Salinas's laboratory has used salt-washed postnuclear supernatants obtained after 200,000 g centrifugation (SK100 fraction) (Burda et al., 1994). These investigators observed that in this preparation, approximately 28% of eIF2α is phosphorylated after 30-minute 4VO and 30-minute reperfusion in rats (Burda et al., 1994; Martin de la Vega, 2001a, b ), a finding that is in general agreement with results from other laboratories. However, they reported that 7% to 10% of total eIF2α obtained from control animals is in the form of eIF2α(P) (Burda et al., 1994; Martin de la Vega, 2001a, b ), which suggests that the SK100 fractionation procedure either is concentrating eIF2(αP) or is resulting in a loss of unphosphorylated eIF2 from the control samples. Given the immunohistochemical data showing nuclear-localized eIF2α (Lobo et al., 2000), which also occurs in vulnerable neurons during reperfusion for longer than 1 hour (DeGracia et al., 1997), homogenate fractionation procedures that eliminate the nucleus will underestimate total eIF2α(P) levels. Although these caveats suggest the need to interpret relative eIF2α(P) levels with some caution, they do not weaken the strong consensus that during early postischemic brain reperfusion there is a rapid and large increase in levels of eIF2α(P) associated with the inhibition of eIF2B activity and inhibition of translation initiation. Moreover, the observation of Althausen et al. (2001) that PSI persists after substantial resolution of eIF2α(P) by 6-hour reperfusion suggests that additional mechanisms are involved at these later reperfusion times.
Molecular alteration of eIF4 induced by brain ischemia and reperfusion.
Several investigators have examined the effects of brain I/R on eIF4E and eIF4G. We reported that total eIF4E decreased approximately 35% during 20-minute decapitation ischemia in rats and showed that eIF4E is a μ-calpain substrate in vitro (Neumar et al. 1995). In contrast, neither 10 minutes of cardiac arrest nor 90 minutes of subsequent reperfusion changed the quantity or phosphoserine immunoreactivity of eIF4E (DeGracia et al. 1996). Burda et al. (1998) found no loss of eIF4E to 30-minute 4VO, but did show a 50% decrease in its phosphorylation during ischemia with a return to control values after 30-minute reperfusion. Martin de la Vega et al. (2001a) revisited this issue and presented evidence of a 20% decrease in cortical and hippocampal eIF4E levels during 30-minute 4VO that persisted for at least 4 hours of reperfusion. In addition, this study found that the phosphorylation status of eIF4E-binding protein-1 (4EBP-1) decreased during ischemia but returned to control levels by 30-minute reperfusion. Therefore, the extent of degradation or modified phosphorylation of eIF4E induced by brain I/R is modest, and it appears unlikely that eIF4E modifications play a major role in postischemic PSI.
There is a more compelling case for the involvement of eIF4G. We reported the presence of multiple eIF4G fragments by 90-minute reperfusion after a 10-minute cardiac arrest (DeGracia et al., 1996). Proteolysis of eIF4G was confirmed by Neumar et al. (1998), who showed, an approximately 70% loss of eIF4G that was blocked in vivo by inhibition of μ-calpain after 20-minute global ischemia; this same study showed that brain homogenates exhibit in vitro Ca2+-dependent eIF4G degradation that is blocked by calpastatin. Martin de la Vega et al. (2001a) have recently confirmed proteolysis of eIF4G during ischemia and showed a persistent reduction in eIF4G of approximately 35% at 4 hours of reperfusion after 30-minute 4VO. At this longer reperfusion time, it may be important that eIF4G is also a target of caspase-3 (Marissen and Lloyd, 1998). Moreover, although the eIF4G antibodies have not yet been sufficiently specific to allow immunohistochemical mapping of the localization of the loss of this protein, the localization of calpain (Siman et al., 1989; Neumar et al., 2001) and caspase-3 (Chen et al., 1998) activation to vulnerable neurons during I/R suggests that the loss of eIF4G is also localized to neurons. Finally, it is also intriguing that according to microarray examination (Jin et al., 2001), eIF4G transcripts containing an IRES (Hellen and Sarnow, 2001) are among those most increased in the hippocampus during the initial hours of reperfusion.
I/R-induced alterations of eIF2 and eIF4 are expected to cause major changes in the repertoire of new peptides synthesized by residual translation in vulnerable neurons
In simultaneous immunohistochemical examination of eIF2α(P) and quantitative autoradiographic assessment of protein synthesis in CA1 neurons at 90-minute reperfusion, approximately 15% of the control level of protein synthesis and intense eIF2α(P) immunostaining were observed (Sullivan et al., 1999). There has been tremendous focus on altered gene expression during I/R, and it is clearly important to determine if these transcripts are being translated (Read et al., 2001). However, protein expression levels generally cannot be determined from quantitative mRNA data (Gygi et al., 1999). Moreover, during cerebral I/R, residual protein synthesis occurs in an environment of altered translation initiation, which poses significant constraints on the repertoire of proteins synthesized.
The levels of eIF2α(P) seen during the first hours of reperfusion not only greatly reduce the overall rate of translation, but can also cause paradoxically enhanced translation of some proteins by the process of “bypass-scanning” (Dever et al., 1995) (Fig. 2). In yeast, the mRNA for the transcription factor GCN4 is polycistronic with a long 5′ leader containing three short ORFs followed by the actual code for GCN4 in the last ORF. Normally, the ready availability of eIF2-GTP–Met-tRNAi complexes allows the translation initiation at the ORF nearest the 5′ terminal followed by the disassembly of the translation complex at the stop codon of this ORF, thereby precluding the synthesis of GCN4. However, with increased levels of eIF2(αP), the ribosome is less likely to find a competent ternary complex in time to initiate translation at the upstream ORFs (Dever et al., 1995). The probability of a ternary complex binding to the small ribosomal subunit is enhanced over longer scanning times; thus, GCN4 is translated because initiation does not occur on the ORFs nearer the 5′ end of the message (Dever et al., 1995). An important mammalian transcript translated by eIF2α(P)-mediated bypass scanning is ATF4 (Harding et al., 2000a), the transcription factor for apoptosis-inducing CHOP (GADD153) (Zinszner et al., 1998).
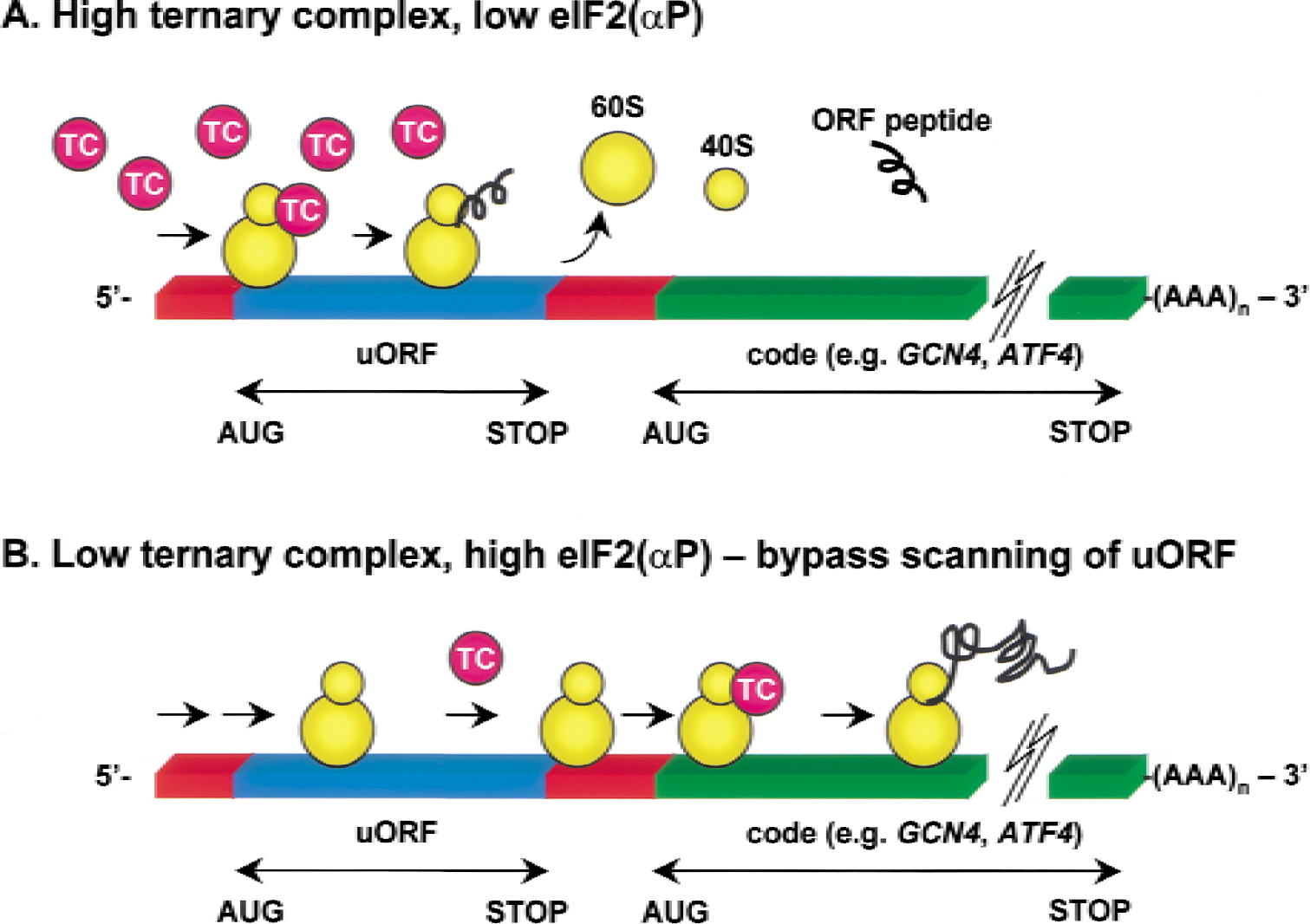
Schematic representation showing how high levels of eIF2α(P) mediate bypass scanning on messenger RNAs with 5′ uORFs. (A) When eIF2α(P) levels are low, ternary complex (TC) availability is high, allowing a scanning ribosome (yellow) to initiate at an uORF (blue), synthesize a short ORF peptide, and terminate translation, thereby precluding translation of the actual protein-coding sequence (green). (B) When eIF2α(P) levels are high and TC levels are greatly reduced, the ribosome without a TC scans through uORFs, and prolonged scanning of the rest of the 5′ leader increases the probability of the ribosome binding a TC before reaching the authentic AUG start codon (green). Examples of such messenger RNAs include yeast GCN4 and mammalian ATF4.
Fragmentation of eIF4G can also induce large changes in which mRNAs are translated. Loss of intact eIF4G can cause failed docking of eIF4E-bound m7G-capped mRNAs to the small ribosomal subunit during translation initiation. This extensively alters mRNA selection by favoring only those messages that contain an IRES (Gingras et al., 1999), which do not require the cap-binding mechanism to load onto ribosomes. The issue of IRES-sequence characteristics is complex and few of the known IRES sequences exhibit consensus; furthermore, there is an unpredictable involvement of idiosyncratic cofactors that affect IRES function (Hellen and Sarnow, 2001). For example, the ER chaperone GRP78 is vital for UPR resolution. Although its transcript has an IRES, a polypyrimidine-binding protein known to upregulate IRES-dependent translation of several other transcripts downregulates GRP78 translation (Hellen and Sarnow, 2001). Nevertheless, poliovirus research has shown the truly massive effect of eIF4G degradation on mRNA selection. The poliovirus uses proteolysis of eIF4G to shut off translation of most constitutive host mRNAs while IRES-containing viral mRNAs are translated (Johannes et al., 1999). According to complimentary DNA microarray assays, there is little change in the total levels of host mRNAs when eIF4G is fragmented, but approximately 97% of host mRNAs no longer appear on ribosomes whereas there is a substantial enrichment of approximately 3% of host mRNAs (Johannes et al., 1999).
Involvement in IRES-mediated translation by truncated eIF4G isoforms lacking the eIF4E binding domain may be greater than just the exclusion of capped messages (Henis-Korenblit et al., 2000). Caspase-3 cleaves eIF4G at multiple sites but leaves a stable 76-kd “middle fragment” that may continue to play a significant role in message selection during apoptosis (Hellen and Sarnow, 2001). Truncated eIF4G isoforms can also be generated by transcription of separate genes. For example, death-associated protein-5 (DAP5) is an eIF4G isoform that lacks the eIF4E binding domain, and on caspase-mediated removal of 11 kd of its C-terminal, DAP5 is activated to facilitate IRES-initiated translation of mRNAs, including its own (Henis-Korenblit et al., 2000). This mechanism maintains a high level of DAP5 during caspase-mediated cell death. Another important example of mammalian IRES-mediated translation is the synthesis of apoptosis-activating factor-1, which occurs only by an IRES-mediated mechanism, again serving to maintain sufficient levels of this protein even when cap-dependent translation initiation mechanisms are compromised (Coldwell et al., 2000). Indeed, PSI involving both initial eIF2α phosphorylation and proteolytic fragmentation of eIF4G now appears to be stereotypical for apoptosis induced by a variety of mechanisms (Marissen and Lloyd, 1998; Morley et al., 2000).
Although there have been numerous studies of transcripts induced by I/R, the predicted large shift in the repertoire of peptides synthesized by residual translation in reperfused vulnerable neurons necessitates substantial additional proteomic investigation because the real message is indeed in the translation (Richter and Theurkauf, 2001). In this regard, some experiments have examined the whole hippocampus (Kiessling et al., 1986; Wengenack et al., 1998). The work of Kiessling et al. (1986) found enhanced synthesis of proteins with mobilities of 110, 70, 65, 50, and 27 kd at 3-hour reperfusion after 30-minute 4VO. Although these studies asked prescient questions, inclusion of the entire hippocampus introduced resistant regions (DG, CA2, and much of CA3). Compared with the CA1, these regions (1) have initially less eIF2α(P) and substantially more clearance of this isoform by 90-minute to 4-hour reperfusion (DeGracia et al., 1997), (2) display less evidence of activation of μ-calpain (Roberts-Lewis et al., 1994) and caspase-3 (Chen et al., 1998) and are thus likely to have more limited or even absent eIF4G degradation, and (3) have better preservation of global protein synthesis (Thilman et al., 1986). Currently available tools are adequate for the identification of pulse-labeled proteins from a single brain ventral median nucleus (Mobbs et al., 1989), and the use of this technology to identify the repertoire of proteins produced in the reperfused CA1 by residual translation is now urgent.
Phosphorylation of eIF2α during initial reperfusion is caused by the UPR elicited by stress to the ER
With recognition of the rapidity and extent of I/R-induced eIF2α phosphorylation, it became important to understand the mechanism leading to eIF2α(P) formation because this would likely provide insight into a stress pathway leading to a major component of PSI. The eIF2α can become phosphorylated by the following three mechanisms: (1) deglycosylation of the eIF2-binding protein p67 that is permissive for eIF2α phosphorylation (Gupta et al., 1997) (2) inhibition of an eIF2α(P) phosphatase, or (3) activation of an eIF2α kinase.
Using Western blot analysis, we did not observe the loss or deglycosylation of p67 associated with global brain I/R, and using immunohistochemistry we also found no loss of glycosylated p67 from vulnerable neurons (Owen et al., 2001). Althausen et al. (2001) recently confirmed no change in p67 glycosylation in the focal ischemia model.
The in vivo eIF2α(P) phosphatase is protein phosphatase 1 (Redpath and Proud, 1990). We found in our global ischemia model no difference (based on multipoint time courses) in the rate of in vitro dephosphorylation of exogenous eIF2(αP) by rat forebrain homogenates from control, ischemic, and reperfused animals (DeGracia et al., 1998). In a reperfusion time course obtained after 30-minute 4VO, Martin de la Vega et al. (2001b) reported a modest and transient I/R-induced reduction of exogenous eIF2α(P) dephosphorylation during in vitro reactions assayed at a single 15-minute reaction time point. This latter study failed to show a correlation between eIF2α(P) phosphatase activity and endogenous levels of eIF2α(P). Our more detailed kinetic study suggests that loss of eIF2α(P) phosphatase activity may not have a major causal role in the accumulation of eIF2α(P) during reperfusion.
There is, however, a fundamental methodologic concern regarding these studies that may account for their inconsistent results. The protein phosphatase 1 catalytic subunit activity against any given substrate is regulated primarily through targeting subunits specific to the substrates (Lee et al., 1999). The aforementioned studies necessarily used brain homogenates with the attendant disruption of subcellular molecular localization in all of the different neuronal, glial, and vascular components of the tissue. This fact, taken together with the targeting subunit issue, argues that without confirmation of distinct I/R-induced posttranslational modification of an eIF2α(P) phosphatase or a targeting subunit in neurons displaying eIF2α(P), the results of gross activity assays must be viewed with substantial caution. Indeed, the in vivo activities of phosphatases or kinases could easily be masked during in vitro activity assays in gross homogenates.
Assessment of the effect of I/R on gross eIF2α kinase activity in brain homogenates has not been more compelling than that of the eIF2α(P) phosphatase assays. In general, the effect of I/R has been seen as a marginal increase (Burda et al., 1998) or an actual decrease (DeGracia et al., 1998; Martin de la Vega et al., 2001b) in in vitro eIF2α kinase activity of gross brain homogenates at reperfusion times when eIF2α(P) levels are increasing severalfold in vivo. Therefore, it is essential to directly inspect the individual eIF2α kinases.
Genetic and biochemical analyses indicate that there are only four homologous mammalian eIF2α kinases: GCN2 (Kimball and Jefferson, 2000), HRI (Chen, 2000), PKR (Kaufman, 2000), and PERK (Ron and Harding, 2000). The amino acid-regulated eIF2α kinase GCN2 is expressed in mammalian brain (Sood et al., 2000), and potential accumulation of uncharged transfer RNAs consequent to lowered ATP levels in ischemic neurons might activate GCN2. HRI coordinates globin synthesis with heme availability in erythroid cells. Although there appears to be little HRI in the brain (Pal et al., 1991), its activation by nitric oxide (Uma et al., 2001), heavy metals such as iron (Hurst et al., 1987), oxidized glutathione, and polyunsaturated fatty acids and lipoperoxides (De Herreros et al., 1985) rendered HRI an inviting candidate for mediating I/R-induced eIF2α phosphorylation. PKR is activated not only by viral nucleotides but also by heparin and is widely distributed in various tissues, including the brain (Haines et al., 1993). PERK (or PEK-pancreatic eIF2α kinase) spans the ER membrane, and recognition of unfolded luminal peptides by GRP78 dissociation from the PERK N-terminal domain causes activation of the eIF2α kinase in the cytoplasmic C-terminal domain (Bertolotti et al., 2000). PERK is most highly expressed in pancreas, but is also expressed in brain (Shi et al., 1998).
When the effect of homozygous eIF2α kinase functional knockouts on I/R-induced eIF2α(P) was studied, PKR-, HRI-, and GCN2-knockout mice showed no reduction of reperfusion-induced eIF2α(P) (DeGracia et al., 1998; Kumar et al., 2001). It was not possible to analyze PERK knockouts because these mice die from type I-like diabetes within 6 to 8 postnatal weeks due to apoptosis of pancreatic β and acinar cells (Harding et al., 2001). However, PERK activation by autophosphorylation induces a characteristic sodium dodecyl sulfate-polyacrylamide gel electrophoresis mobility shift (Harding et al., 1999). We found that brain I/R always induced a shift in PERK mobility from the inactive to the activated isoform (Kumar et al., 2001). Martin de la Vega et al. (2001b) reported no change in the eIF2α kinase activity of PERK immunoprecipitated from control and reperfused samples; however, Western blot analysis was not used to confirm that PERK had been immunoprecipitated, or to examine the effect of I/R on its electrophoretic mobility. In fact, assays of immunoprecipitated PERK activity are unlikely to be a useful approach to its in vivo activation state because the in vitro kinase reaction conditions will themselves activate PERK, even if it is immunoprecipitated in the inactive state (Bertolotti et al., 2000).
Brain I/R does not affect p67 glycosylation or levels, has little or no effect on eIF2α(P) phosphatase activity, and PKR, HRI, and GCN2 are not needed for phosphorylation of eIF2α during reperfusion. However, brain I/R consistently induces transformation of PERK to its active isoform, and PERK activation together with the immunolocalization of eIF2α(P) provide direct molecular evidence that an ER UPR is occurring in neurons as a consequence of brain I/R.
Current understanding of the UPR
The ER is the site of lipid synthesis and of maturation and processing of secreted and transmembrane proteins (Stevens and Argon, 1999). The UPR occurs when cells are subjected to ER stress, and can be thought of as a stimulus-effector system. The stimuli triggering the UPR are those that upset the functions occurring in the ER lumen related to protein folding, maturation, posttran-scriptional processing, or lipid synthesis (Kaufman, 1999). The effectors of the UPR include both immediate and long-term transcriptional, translational, and posttranslational modification components that operate in parallel. Even in yeast, the overall UPR transcriptional response is complex and involves approximately 5% of the genome, including genes for proteins involved at all stages of secreted and membrane-bound protein processing (Travers et al., 2000). It is expected that a similarly large change in gene transcription occurs during the mammalian UPR. The biologic objective of the UPR is to reduce the requirement for ER protein processing by temporary PSI while simultaneously upregulating the transcription of genes or processes that will abate the effects of the ER insult or push the cell into apoptosis if ER dysfunction persists (Patil and Walter, 2001). The primary effectors of the mammalian UPR are three ER-transmembrane proteins: PERK, IRE1, and ATF6.
Several disruptions of ER biochemistry activate the UPR. These changes include depletion of ER Ca2+ stores (e.g., by the ER Ca2+-ATPase inhibitor thapsigargin), shift of the lumen from an oxidizing to a reducing environment (e.g., by dithiothreitol or β-mercaptoethanol), inhibition of protein glycosylation (e.g., with tunicamy-cin), glucose or oxygen deprivation (Kaufman, 1999; Ron and Harding, 2000), and disruption of ER-associated protein degradation (Patil and Walter, 2001).
Both PERK (Shi et al., 1998; Harding et al., 2001) and IRE1(Miyoshi et al., 2000) are present in the brain and expressed in neurons. Activation of PERK and IRE1α is mediated by their homologous ER-luminal N-terminal sequences (Fig. 3). Normally, the luminal domains of the inactive forms of PERK and IRE1α form 1:1 complexes with the chaperone GRP78 (also know as BiP or KAR2) (Bertolotti et al., 2000). In the presence of unfolded ER-luminal proteins, GRP78 dissociates from PERK and IRE1α to bind the unfolded proteins to prevent their aggregation (Ron and Harding, 2001). We have found that immunoprecipitation of PERK from homogenates of nonischemic brains coimmunoprecipitates GRP78, but PERK coimmunoprecipitation of GRP78 is lost after a 10-minute cardiac arrest and 10-minute reperfusion (DeGracia et al, 2000). Once dissociated from GRP78, IRE1α and PERK homooligomerize and activate by transautophosphorylation (Bertolotti et al., 2000; Welihinda and Kaufman, 1996).
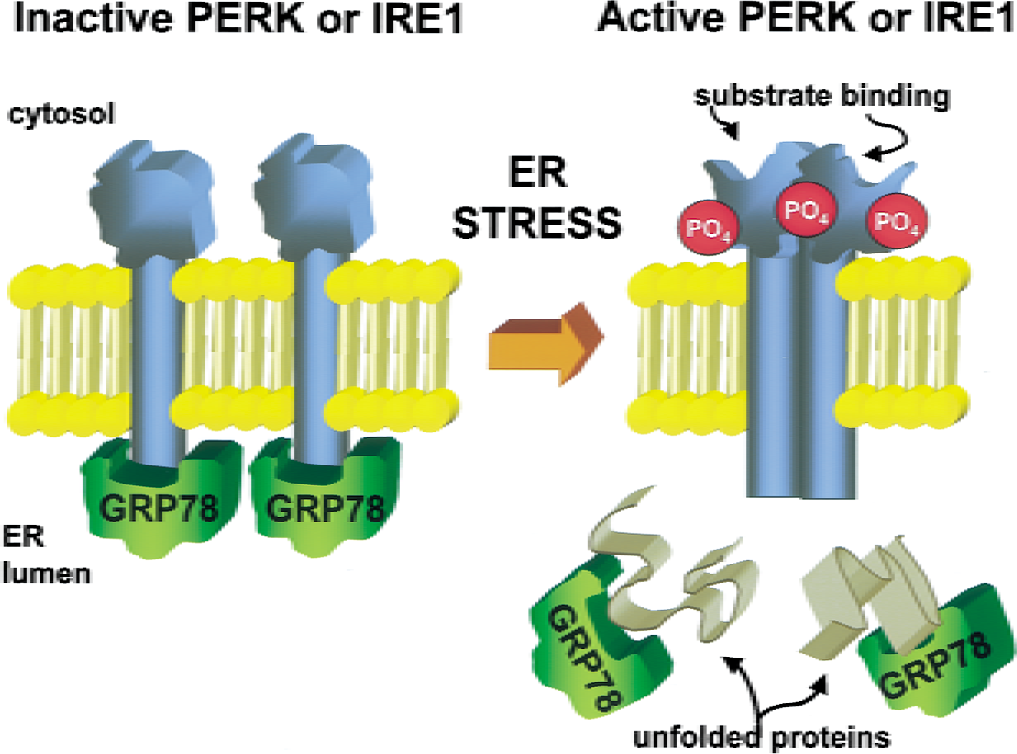
Schematic representation showing how PERK and IRE1 are activated by the dissociation of GRP78, homooligomerization, and autophosphorylation. (Left) In unstressed cells, PERK and IRE1 are ER transmembrane proteins maintained in an inactive state by binding GRP78 on their ER-lumenal domains. (Right) On ER stress, GRP78 dissociates and binds unfolded or misfolded proteins, allowing homooligomerization of PERK or IRE1 or both, leading to autophosphorylation of these enzymes and activation toward their substrates.
Activated PERK phosphorylates eIF2α, thereby inhibiting most protein synthesis. However, David Ron's group has shown that at least the mammalian mRNA for ATF4 undergoes translation initiation during the UPR by “bypass-scanning” similar to that seen for yeast GCN4 mRNA (Harding et al., 2000a) (Fig. 2). Like the mRNA for GCN2, ATF4 mRNA has a long 5′ leader with multiple uORFs. This mRNA is constitutively present in mammalian cells, including neurons (Yukawa et al., 1999), but is not efficiently translated until there is increased eIF2α phosphorylation (Harding et al., 2000a). ATF4 is a transcription factor, originally identified as CREB-2 in the family of cyclic AMP-responsive genes (Hai et al., 1989). ATF4 is responsible for CHOP transcription (Fawcett et al., 1999). Thus, PERK activation, by way of increased eIF2α(P), is responsible not only for general PSI but also for at least one important change in gene expression (i.e., the upregulation of CHOP) through bypass scanning-mediated enhanced translation of ATF4 during the UPR (Fig. 2).
The functions of mammalian IRE1 are more complex and less completely understood. The cytoplasmic domains of all IRE1 isoforms also include an endonucleolytic RNAse domain (Sidrauski and Walter, 1997) that is activated by IRE1 transautophosphorylation. In yeast, the activated IRE1 RNAse removes a 252-nucleotide translation-attenuator sequence from the constitutively expressed but inefficiently translated HAC1 mRNA (Kawahara et al., 1997), which is then ligated by transfer RNA ligase (Sidrauski and Walter, 1997) and translated efficiently (Kawahara et al., 1997). HAC1 protein is a member of the basic leucine zipper (bZIP) family of transcription factors; in yeast it binds to the UPR element (Kohno et al., 1993) in the promoter of UPR-responsive yeast genes (including that for GRP78) and thereby up-regulates their transcription. Although a sequence analogue of yeast HAC1 does not appear to exist in mammalian genomes, the mammalian UPR induces accurate intron splicing of vector-introduced HAC1 mRNA by IRE1 isoforms (Niwa et al., 1999), and expression of the yeast HAC1 protein in mammalian cells induces the promoter activity of the GRP78 gene on UPR induction (Foti et al., 1999). Cross-linking experiments have recently shown that UPR induction in mammalian cells is associated with complex formation between IRE1α and an unidentified constitutively expressed RNA (Bertolotti and Ron, 2001), and several investigators have shown that overexpression of mammalian IRE1 isoforms induces enhanced GRP78 transcription. It is not yet clear whether this UPR-responsive transcriptional effect of mammalian IRE1 involves its processing of an mRNA (as in yeast) or a role in upstream activation of ATF6 (Wang et al., 2000), but dominant negative mutants of IRE1 inhibit UPR-induced transcription dependent on an ATF6-binding minimum consensus sequence TGACGT (Wang et al., 2000).
As PERK can modulate apoptosis via CHOP expression, both IRE1α and IRE1β (expressed mainly in the gut) are also connected to mammalian apoptotic mechanisms (Fig. 4). Recently, Iwawaki et al. (2001) showed that overexpression of IRE1β can induce apoptosis by a mechanism involving cleavage of the 28S ribosomal RNA by the IRE1 β RNAse, and that a chimeric protein with the ER luminal domain of IRE1β and the cytoplasmic domain of IRE1α also cleaves the 28S ribosomal RNA. Although it is not yet known whether native IRE1α can process ribosomal RNA, such a mechanism has the potential to explain persistent PSI in an unresolved UPR, even after levels of eIF2α(P) are reduced. Moreover, activation of IRE1α leads to activation of the apoptosis-associated stress-induced protein Ser/Thr kinases known as SAPKs (stress-activated protein kinases) or JNKs via interaction of the IRE1α cytoplasmic domain with the TRAF2 (tumor necrosis factor receptor-associated factor-2) adaptor protein (Urano et al., 2000) and JIK (Jun inhibitor kinase) (Yoneda et al., 2001). The same complex of activated IRE1α with JIK and TRAF2 is also implicated in inducing activation of ER-localized procaspase-12 (Yoneda et al., 2001).
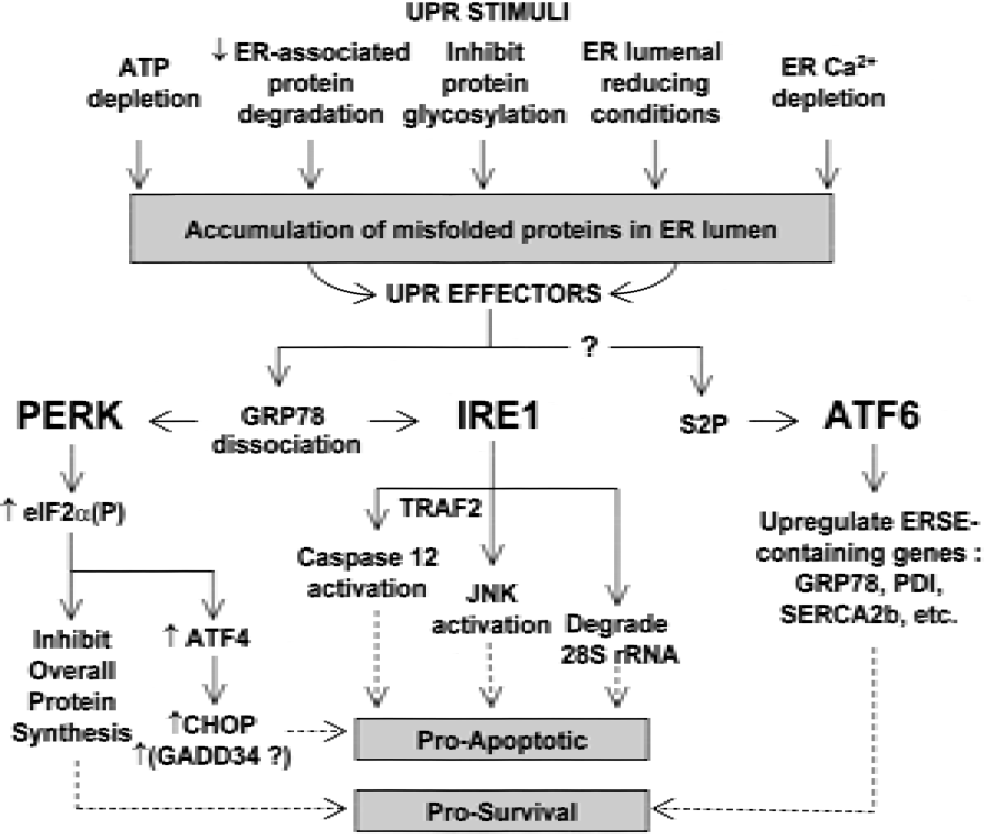
The mammalian UPR is a stimulus-effector system. Several mechanisms, including depletion of ER Ca2+, can lead to misfolding of proteins being processed in the ER lumen. Malfolded lumenal proteins result in activation of three known effectors (PERK, IRE1, and ATF6). These effectors cause reduction of the processing-requiring peptide load and induce transcription for resolution of the ER stress, but two of these effectors (PERK and IRE1) also initiate apoptotic pathways that will result in the death of the cell if ER stress resolution is unsuccessful. PERK-mediated eIF2α phosphorylation generally inhibits protein synthesis, but also paradoxically increases bypass scanning-mediated translation of at least proapoptotic ATF4. IRE1 may be involved in induction of transcription for ER chaperones through mechanisms involving RNA processing or the activation of ATF6 or both; however, IRE1 also induces proapoptotic activation of JNKs and caspase-12, and its cytoplasmic RNAse domain can degrade 28S ribosomal RNA. After site-2 metalloprotease releases ATF6 from the ER membrane, it is translocated into the nucleus and promotes transcription for ER-resident proteins needed for resolution of the UPR; ATF6 has no known proapoptotic effects.
ATF6 is a key transcription factor in the resolution of the mammalian UPR (Patil and Walter, 2001), and unlike the situation with IRE1 and PERK, there is no evidence that ATF6 is also involved in proapoptotic mechanisms (Fig. 4). Normally resident as an ER transmembrane protein, on UPR induction ATF6 is proteolyzed by the ER-localized site-2 metalloproteinase (S2P) (Ye et al., 2000). As noted previously, there is no consensus whether IRE1 activation is upstream of ATF6 proteolysis (Patil and Walter, 2001). The liberated cytoplasmic domain of ATF6 translocates to the nucleus and collaborates with other transcription factors (NF-Y and YY1) (Li et al., 2000) to induce transcription of genes containing the mammalian ER stress-response element (Roy and Lee, 1999).
Overexpression of dominant negative mutants of the mammalian IRE1s (Wang et al., 1998), S2P (Ye et al., 2000), or ATF6 (Wang et al., 2000) attenuate the mammalian UPR, and PERK-knockout cells exhibit enhanced susceptibility to UPR-induced death (Harding et al., 2000b). Therefore, at least in the initial stage of UPR induction, all three ER-transmembrane upstream effectors are important for an effective UPR response.
Resolution of the UPR.
Successful resolution of the UPR would be expected to involve overcoming the initiating insult, restoring the ER peptide-folding functions, silencing IRE1α and PERK by their reassociation with GRP78, and dephosphorylating eIF2α(P), IRE1α, and PERK. Although a PP2C-class protein phosphatase, Ptc2, has been shown to dephosphorylate and inactivate IRE1 in yeast (Welihinda et al., 1998), nothing is known about dephosphorylation of IRE1 and PERK in higher eukaryotes. More is known about the role of the UPR-responsive gene products GRP78 in deactivating IRE1α and PERK, CHOP in induction of apoptosis, and GADD34 in directing dephosphorylation of eIF2α(P), and thus downregulating translation of ATF4 and consequent CHOP transcription. The promoter region of the neuronal ER-Ca2+-ATPase SERCA2b (Baba-Aissa et al., 1998) also contains three ER stress-response element sequences, and SERCA2b expression may be an integral part of the resolution of the UPR (Caspersen et al., 2000). We will briefly review the role of each of these proteins in UPR resolution or apoptosis after ER Ca2+ depletion, and then we will summarize a recent study by Paschen's group (Mengesdorf et al., 2001) that has provided the first integrated examination of their transcription and translation in response to in vitro ER Ca2+ depletion in cultured neurons.
The GRP78 promoter contains the ER stress-response element, and overexpression of GRP78 inhibits activation of IRE1 isoforms and PERK in response to ER stress (Bertolotti et al., 2000). Resolution of the ER stress is associated with GRP78 binding both IRE1 and PERK, even if they remain in phosphorylated states (Bertolotti et al., 2000). These results argue that translation of the UPR-induced transcripts for GRP78 leads to downregulation of the activities of PERK and IRE1. Indeed, preferential translation of GRP78 has been found to invariably precede resumption of general translational competence after ER Ca2+ depletion (Brostrom et al., 1990). In contrast, failure to synthesize GRP78 in response to ER Ca2+ depletion is directly associated with cell death. Thapsigargin inactivation of the SERCA pump causes death in cultured cells when the GRP78 transcriptional response to ER Ca2+ depletion is constitutively downregulated (McCormick et al., 1997) or translation of GRP78 is blocked by an antisense oligonucleotide (Li et al., 1992). Therefore, it is not surprising that in the case of cultured hippocampal neurons, antisense-mediated depletion of GRP78 substantially enhances cell death induced by glutamate or Fe2+-mediated radical damage (Yu et al., 1999), both of which are associated with mechanisms that induce ER Ca2+ depletion and eIF2α phosphorylation in cultured neurons (O'Neil et al., 1999).
Normally, CHOP is expressed at low or undetectable levels (Fornace et al., 1988). CHOP forms mainly functionally negative heterodimers with members of the C/EBP and FOS-JUN families of transcription factors (Ubeda et al., 1999), and induces apoptosis (Matsumoto et al., 1996; Zinsner et al., 1998) by mechanisms that do not involve p53 (Matsumoto et al., 1996) but do induce downregulation of Bcl2 protein expression and overproduction of oxygen radicals (McCullough et al., 2001). A transpositional rearrangement of the CHOP gene that inactivates the apoptosis-inducing function is found in most sarcomas (Wang et al., 1996). Although apoptosis-associated CHOP transcription was first recognized in association with DNA damage (Fornace et al., 1988), its transcription is actually caused by the ER UPR (Wang et al., 1996). As described previously, CHOP transcription is induced by eIF2α(P) through translation of ATF4 (Harding et al., 2000a), and the nonphosphorylatable S51A eIF2α mutant blocks UPR-induced CHOP transcription (Scheuner et al., 2001). Therefore, it is not surprising that the overexpression of GRP78 attenuates UPR-induced expression of CHOP (Wang et al., 1996). Transcription and translation of CHOP represent a pathway to apoptosis if the UPR is not resolved.
GADD34 appears to be part of the machinery to resolve the UPR by localizing protein phosphatase 1 to the ER for dephosphorylation of eIF2α(P) (Novoa et al., 2001). The GADD34 promoter does not include a classical ER stress-response element (Hollander et al., 1997), but its transcription may be activated by ATF4 (Novoa et al., 2001). Although GADD34 induces the dephosphorylation of UPR-induced eIF2α(P), an N-terminal–truncated form of GADD34 is even more active in this regard (Novoa et al., 2001). The C-terminus of GADD34 is approximately 50% identical with the herpes simplex protein γ134.5 (Hollander et al., 1997) and the protein phosphatase 1 binding motif of γ134.5 (He et al., 1997), which targets protein phosphatase 1 to eIF2α(P) and causes an approximately 3,000-fold increase in the rate of eIF2α(P) dephosphorylation, thereby abrogating the effect of PKR activation by viral-derived nucleotides (He et al., 1997). Although GADD34 was also discovered in association with DNA damage, its association with apoptosis is variable (Hollander et al., 1997). Stable expression of GADD34 in highly differentiated cells appears to prevent apoptosis (Chou and Roizman, 1994).
Prolonged depletion of ER Ca2+ induced in vulnerable neurons by I/R (Kohno et al., 1997) may be caused by covalent modifications of SERCA2b (Doutheil et al., 2000; Parsons et al., 1999), and SERCA2b transcription is induced as part of the UPR (Caspersen et al., 2000). Compared with other SERCA isoforms, SERCA2b displays enhanced susceptibility to damage by peroxide (Grover et al., 1997), superoxide (Barnes et al., 2000), and nitric oxide (Doutheil et al., 2000), all of which are precursors or intermediates in radical damage during postischemic brain reperfusion (White et al., 2000). Indeed, our observations (Kumar et al., 2000) that with brief durations of brain I/R eIF2α phosphorylation is attenuated in homozygous eNOS- or nNOS-knockout mice and in wild-type mice treated with
The importance of the issue of whether these proteins are translated in vulnerable neurons during postischemic reperfusion has become apparent in the recent study from Paschen's group of the UPR in primary cultures of cortical neurons (Mengesdorf et al., 2001). After UPR induction by thapsigargin, both quantitative polymerase chain reaction and Western blot analysis were used to characterize transcription and translation of GRP78, CHOP, GADD34, SERCA2b, and several other proteins. In this study, the levels of total and phosphorylated eIF2α were also determined by Western blot analysis. The expected enhancement of eIF2α(P) persisted for 3 to 6 hours. During this time, GRP78 and CHOP transcripts increased approximately 20-fold, GADD34 transcripts increased approximately 40-fold, and SERCA2b transcripts doubled. However, only CHOP, which was absent in control cells, displayed strong translation.
The ER UPR is induced in vulnerable neurons by brain ischemia and reperfusion
Paschen has been a seminal advocate of the concept that ER stress represents an important fundamental mechanism of I/R-induced brain injury (Paschen, 1996, 2000). His insight grew in part from the foundation developed by Brostrom and colleagues, who showed that depletion of ER Ca2+ induced PSI through inhibited translation initiation (Brostrom and Brostrom, 1998). It is now clear that low levels of ER [Ca2+] inhibit ER-luminal protein-disulfide isomerases, causing malfolding in the ER lumen of newly synthesized peptides (Corbett et al., 1995; Oliver et al., 1999).
Depletion of ER Ca2+ is caused by brain I/R. Kohno et al. (1997) have shown that after 5-minute ischemia in the gerbil, ER Ca2+ in vulnerable neurons is lost by 15-minute reperfusion and is not recovered after 3 hours of reperfusion. Free arachidonic acid, which is produced during ischemia (Avaldano and Bazan, 1975), induces depletion of ER Ca2+ stores, phosphorylation of eIF2α, and inhibition of protein synthesis (Fleming and Mellow, 1995; O'Neil et al., 1999). Furthermore, microsomes from postischemic brains display decreased Ca2+ uptake (Parsons et al., 1997; Racay et al, 2000) that may be related to inactivation of SERCA2b by a nitric oxide-mediated mechanism (Doutheil et al., 2000) and prolonged uncoupling of Ca2+ transport and ATPase activity (Parsons et al., 1999). In this regard, Paschen's group has shown that in primary neuronal cultures, the nitric oxide donor SNAP induces inhibition of SERCA2b and depletion of ER Ca2+ stores (Doutheil et al., 2000). We have recently found that in eNOS- and nNOS-knockout mice, a greater duration of ischemia is needed to induce PERK activation (Kumar et al., 2001) and eIF2α phosphorylation (Kumar et al., 2000) during reperfusion, suggesting that nitric oxide production exacerbates ER stress.
Hu et al. (2000, 2001) recently provided ultrastructural evidence that brain I/R induces abnormal protein aggregates in several neuronal subcellular compartments, including the ER. These aggregates clear from injury-resistant neurons but persist in vulnerable CA1 neurons that progress to cell death. The persistence of these aggregates is associated with increased ubiquination of detergent-insoluble proteins, suggesting that they are being targeted for degradation but not in fact being cleared.
Further support for the I/R-induced UPR in vulnerable neurons derives from studies of individual UPR pathways. As discussed previously, PERK activation and greatly increased levels of eIF2α(P) are evident within 5 minutes of reperfusion. Furthermore, N-methyl-
A general pathophysiologic view of I/R-induced PSI
As Lipton (1999) has recently noted, it is surprisingly difficult to answer to the seemingly straightforward question of whether PSI during brain reperfusion is protective or pathologic. Given the complexity of the relations between the system components reviewed here, this question may not in fact be helpful. Indeed, the data reviewed suggest the need to shift the emphasis from PSI to the underlying pathways that mediate PSI and to the issue of residual translation products. Such a transformation of viewpoint suggests that reperfusion-induced PSI may be both protective and pathologic.
During initial reperfusion, eIF2α(P)-mediated PSI may be a protective response to ischemia-induced ER stress. Downregulation of the UPR through the expression of dominant negative IREα (Miyoshi et al., 2000) or PERK (Harding et al., 2000b) enhances cell death. However, persistence of eIF2α(P) coupled with eIF4G degradation may result in a pathologic transition involving major alterations of message selection that lead to disproportionate expression of proapoptotic genes by residual translation. In fact, the molecular evidence we have reviewed shows that UPR activation (essentially immediately on reperfusion) includes components that may be the earliest proapoptotic pathway activated by brain I/R. This finding suggests that inducing early resolution of the UPR and reducing eIF4G degradation are likely to be important targets for therapeutic interventions for the survival of selectively vulnerable neurons. A proteomic comparison of vulnerable and resistant neurons regarding their translation products during the first several hours of reperfusion should provide more insight into the validity of this view.
The evidence we have reviewed strongly supports the arguments of Richter and Theurkauf (2001) that “the message is in the translation,” of Read et al. (2001) that translation assessment is essential to understanding brain ischemia-induced gene expression, and of Niwa and Walter (2000) that it is at the level of translational control that cells decide their fate when they “pause” in an UPR. Therefore, the issue of injury-resistant versus vulnerable neurons now seems to involve which messages are on the ribosomes, and whether during early reperfusion residual translation produces proteins involved in the resolution of the UPR and other cell stresses or those mediating apoptosis.
Footnotes
Acknowledgments:
The authors thank Douglas Cavener, Jane Jane Chen, Michael Clemens, Heather Harding, Randy Kaufman, Scot Kimball, Robert Matts, Barb McGrath, Christopher Proud, David Ron, Donalyn Schuener, and Ron Wek for their ongoing support and interest in the problem of PSI during brain reperfusion.