
Abstract
A variety of endoplasmic reticulum (ER) stresses trigger the unfolded protein response (UPR), a compensatory response whose most proximal sensors are the ER membrane–bound proteins ATF6, IRE1α, and PERK. The authors simultaneously examined the activation of ATF6, IRE1α, and PERK, as well as components of downstream UPR pathways, in the rat brain after reperfusion after a 10-minute cardiac arrest. Although ATF6 was not activated, PERK was maximally activated at 10-minute reperfusion, which correlated with maximal eIF2α phosphorylation and protein synthesis inhibition. By 4-h reperfusion, there was 80% loss of PERK immunostaining in cortex and 50% loss in brain stem and hippocampus. PERK was degraded in vitro by μ-calpain. Although inactive IRE1α was maximally decreased by 90-minute reperfusion, there was no evidence that its substrate xbp-1 messenger RNA had been processed by removal of a 26-nt sequence. Similarly, there was no expression of the UPR effector proteins 55-kd XBP-1, CHOP, or ATF4. These data indicate that there is dysfunction in several key components of the UPR that abrogate the effects of ER stress. In other systems, failure to mount the UPR results in increased cell death. As other studies have shown evidence for ER stress after brain ischemia and reperfusion, the failure of the UPR may play a significant role in reperfusion neuronal death.
Global brain ischemia and reperfusion are associated with a profound inhibition of protein synthesis that is reversible in most brain regions but persists in selectively vulnerable brain regions (reviewed in Krause and Tiffany, 1993). Several laboratories have shown that protein synthesis inhibition at initial reperfusion is due to a large and rapid increase in phosphorylation of the α subunit of eIF2α (reviewed in DeGracia et al., 2002), most likely by the ER eIF2α kinase PERK (Kumar et al., 2001). PERK is activated only by the ER stress signaling system termed the unfolded protein response (Ron and Harding, 2000). Thus, protein synthesis inhibition after brain reperfusion is likely to be a part of a more comprehensive cellular response that contributes to the ultimate fate of reperfused neurons.
Several studies have presented evidence of ER dysfunction after ischemia and reperfusion (Hu et al., 2000; Kohno et al., 1997; Kumar et al., 2001; Parsons et al., 1997). Forms of ER stress include loss of ATP, depletion of ER luminal Ca2+, or inhibition of ER protein glycosylation (Kaufman, 1999). The UPR is activated to overcome ER stress by temporarily slowing accumulation of new proteins in the ER lumen and simultaneously up-regulating transcription of genes for ER-resident chaperones and enzymes that abate the effects of ER stress (Ron and Harding, 2000). In higher eukaryotes, prolonged activation of the UPR can also activate proapoptotic mechanisms (McCullough et al., 2001; Oyadomari et al., 2002). Thus, we have hypothesized that regional differences in brain UPR expression may contribute to selective vulnerability after ischemia and reperfusion (DeGracia et al., 2002).
To date, three upstream UPR components, all ER transmembrane proteins, have been discovered: PERK, IRE1α, and ATF6. The ER chaperone GRP78 binds to the ER luminal domains of both PERK and IRE1, which serves to repress activation of their cytosolic catalytic domains (Ma et al., 2002). On ER stress, GRP78 dissociates from PERK and IRE1α, allowing homooligomerization and activation by autophosphorylation (Bertolotti et al., 2000).
PERK contributes to a prosurvival response by attenuating the accumulation of new protein in the ER through phosphorylation of eIF2α [eIF2α(P)] leading to a generalized decrease of protein synthesis (Ron and Harding, 2000). Under conditions of high eIF2α(P), however, messenger RNAs (mRNAs) whose translation is normally obstructed by 5′ upstream open-reading frames can be translated by the process of bypass scanning (reviewed in DeGracia et al., 2002; Dever et al., 1995). Important examples of higher-eukaryote mRNAs that are synthesized under conditions of elevated eIF2α(P) are ATF4 (Harding et al., 2000a) and CHOP (GADD153) (Jousse et al., 2001). ATF4 is member of the CREB-2 family of transcription factors (Hai et al., 1989) and up-regulates transcription of chop mRNA (Harding et al., 2000a). CHOP is a proapoptotic member of the C/EBP family of transcription factors (Wang et al., 1996). Thus, eIF2α phosphorylation by PERK contributes in parallel to both prosurvival and proapoptotic mechanisms during the UPR.
IRE1α contains both autokinase and endoribonuclease activities. The latter IRE1α activity removes a 26-nt region from xbp-1 mRNA, which causes a shift in reading frame such that the xbp-1 translation product changes from a poorly translated 33-kd protein to a more efficiently translated 55-kd form [XBP-1(55)] (Calfon et al., 2002; Lee et al., 2002; Yoshida et al., 2001). XBP-1(55), in turn, is a transcription factor for the genes of a number of ER-resident chaperones and enzymes (Yoshida et al., 2001).
On ER stress, GRP78 dissociates from ATF6, which is translocated from the ER to the Golgi membrane (Shen et al., 2002) where it is sequentially cleaved by the proteases S1P and S2P, resulting in release of the cytosolic domain of ATF6 (Haze et al., 1999; Ye et al., 2000). The cytosolic domain of ATF6 translocates to the nucleus where it acts as a transcription factor and binds the ER stress response element on the genes of ER resident proteins (Yoshida et al., 1998, 2000). Hence, both IRE1α and ATF6 contribute to the transcriptional component of the UPR.
We investigated the PERK, IRE1α, and ATF6 pathways in brain stem, cerebral cortex, and hippocampus after 10-minute global brain ischemia and up to 4-h reperfusion in the rat. We found PERK activation that clearly correlated with eIF2α phosphorylation. Although IRE1α underwent a reperfusion-dependent alteration, there was no evidence of IRE1α activity in vivo. ATF6 was not activated, nor were the downstream effector proteins ATF4, CHOP, and XBP-1(55) translated. Moreover, there was a severe loss of immunostaining of PERK by 4-h reperfusion, and PERK was a substrate of μ-calpain in vitro. Our data suggest that other mechanisms of cellular damage known to occur during global ischemia and reperfusion could alter the integrated UPR response, thereby preventing cells from coping with ER stress and contributing to neuronal death after ischemia and reperfusion.
MATERIALS AND METHODS
Polyclonal anti-IRE1α and anti-PERK antisera were a gift from David Ron (New York University, NY, U.S.A.) and have been previously described (Harding et al., 1999; Wang et al., 1998). We previously characterized the antibody specific for serine-51 phosphorylated eIF2α (DeGracia et al., 1997), which for this study was purchased from Biosource International (Cammarillo, CA, U.S.A.). ATF6 antiserum has been previously characterized (Haze et al., 1999). CHOP (GADD153), XBP-1, and ATF4 antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, U.S.A.). All other chemicals were reagent grade.
Positive controls for CHOP, ATF4, and XBP-1(55) and for active forms of PERK, IRE1α, and ATF6 were obtained by exposing differentiated NB104 cells (Prasad, 1975) to 1-μmol/L thapsigargin for 2 h at 37°C. Thapsigargin causes ER stress by irreversible inactivation of the ER Ca2+-ATPase (Mengesdorf et al., 2001). Inactive PERK, IRE1α, and ATF6 were obtained from untreated NB104 cells, which do not produce CHOP, ATF4, or XBP-1(55).
All animal experiments were approved by the Wayne State University Animal Investigation Committee and were conducted following the Guide for the Care and Use of Laboratory Animals (National Research Council, revised 1996). The thoracic compression method of cardiac arrest was used to induce global brain ischemia in male Long Evans rats (250 to 300 g) (Harlan, Indianapolis, IN, U.S.A.) by methods described previously (DeGracia et al., 1997). This model results in profound inhibition of protein synthesis, inhibited translation initiation, and rapid accumulation of eIF2α(P) (DeGracia et al., 1996, 1997) as well as lipid peroxidation products (White et al., 1993). All animals were maintained normothermic during both the ischemic and reperfusion periods by means of a homeostatic blanket control unit (Harvard Apparatus, Holliston, MA, U.S.A.). Experimental groups (n = 3 per group) included nonischemic controls (NIC), 10-minute ischemia and 10-minute reperfusion (10R), 10-minute ischemia and 90-minute reperfusion (90R), and 10-minute ischemia and 4-h reperfusion (4hrR).
Brains were carefully removed at the appropriate times and the brain stem, cerebral cortex, and hippocampus were rapidly dissected bilaterally. A midline incision followed by separation of the cerebral cortex via “peeling” at the internal capsule revealed the hippocampus, which was dissected in its entirety. The cerebral cortex was removed from all underlying structures. Brain stem was taken as that tissue caudal to the inferior colliculis and rostral to the base of the medulla.
Regions isolated from one side of the brain were used for immunoprecipitation and immunoblot studies and were immediately sonicated on ice in homogenization buffer containing 20-mmol/L HEPES (pH 7.5), 10% glycerol, 1-mmol/L ethylenediamine tetraacetic acid (EDTA), 10-mmol/L tetrasodium pyrophosphate, 100-mmol/L NaF, 17.5-mmol/L β-glycerophosphate, 1-mmol/L phenylmethylsulfonyl fluoride (PMSF), 4 μg/mL aprotinin, 2 μg/mL pepstatin A, and 5-μmol/L okadaic acid. Aliquots of unfractionated homogenates were used to determine protein concentrations by the Folin phenol reagent method, and the remainder were frozen (dry ice/ethanol) and stored at −80°C until further use.
IRE1α and PERK immunoprecipitation was performed using previously described methods (Kumar et al., 1999; Wang et al., 1998). To provide enough protein to successfully immunoprecipitate PERK from brain stem or hippocampus, samples were pooled. Homogenates of brain stem, cerebral cortex, and hippocampus containing 12, 4, and 8 mg protein, respectively, were used for PERK immunoprecipitation. For IRE1α, 4-mg protein samples were used from each region. All homogenates were taken to 1% Triton X-100 and then precleared with 1 μL of an unrelated antibody (lamin A/C; Santa Cruz Biotechnology) plus 20 μL Protein A-Sepharose beads (Zymed Laboratories, San Francisco, CA, U.S.A.). Precleared supernatants were incubated overnight at 4°C with end-over-end rotation with 15 μL Protein A-Sepharose beads prebound with 0.5 μL PERK or IRE1α antisera. Proteins were eluted from the beads in SDS-PAGE loading buffer, electrophoresed on 6% SDS-PAGE, and electroblot transferred to nitrocellulose. PERK and IRE1α immunoprecipates were then immunoblotted.
SDS-PAGE and immunoblotting were performed as previously described (DeGracia et al., 1996, 1998). For immunoblotting of unfractionated brain regions, 125 μg protein was loaded per lane. Primary antibody dilutions for immunoblotting were 1:750 anti-eIF2α(P), 1:10,000 anti-PERK, 1:2,000 anti-IRE1α, 1:400 anti-CHOP, 1:250 anti–XBP-1, 1:200 anti-ATF4, and 1:300 anti-ATF6. Relative band densities were determined using BioImage Intelligent Quantifier v3.0 (BioImage, Ann Arbor, MI, U.S.A.). Statistical analyses were performed using ANOVA with Fischer LSD post hoc where necessary.
In vivo protein synthesis was determined in nonischemic controls and in the 90-minute and 4-h reperfusion groups (n = 3 per group) by administering 1 mCi 35S-methionine/35S-cysteine (Trans 35S-Label; ICN Biomedicals, Inc., Irvine, CA, U.S.A.) 60 minutes before the animals were killed. Brain regions were dissected as previously described and sonicated in buffer containing 7-mol/L urea, 2-mol/L thiourea, 0.4% CHAPS, 10-mmol/L dithiothreitol (DTT), 1-mmol/L PMSF, 4-μg/mL aprotinin, 4-μg/mL leupeptin, and 2-μg/mL pepstatin A. Twenty-five microliters of each homogenate was spotted onto individual Whatmann GF-A glass fiber filters that had been prewet in 10% trichloroacetic acid (TCA), 1.6% unlabeled methionine, and 0.4% unlabeled cysteine. Filters were incubated in this same solution for 15 minutes at 4°C with rocking, washed twice with cold 5% TCA, and sequentially dried in 100% ethanol and 100% acetone. 35S incorporation into TCA-precipitable material was quantified by liquid scintillation counting and expressed as disintegrations per minute (dpm) · μg protein–1 · min–1.
To detect whether IRE1α cleaved the 26-nt segment from endogenous xbp-1 mRNA (processed xbp-1 mRNA), PCR was used to amplify a 600-bp complementary DNA (cDNA) encompassing nucleotides 571 to 1,144 in the mRNA sequence (Calfon et al., 2002). Digestion of the unprocessed 600-bp cDNA with the restriction enzyme Pst I normally yields two fragments of ∼300 bp on electrophoresis, but cleavage by active IRE1α of a 26-nt segment in the xbp-1 mRNA results in a 574-bp amplification product and removal of the Pst I restriction site (Calfon et al., 2002). Endogenous xbp-1 mRNA was amplified by sequential reverse transcription–polymerase chain reaction (RT-PCR) by the method of Calfon et al. (2002). First, total RNA was isolated by handheld homogenization of 50-mg wet weight tissue samples of brain stem, cerebral cortex, hippocampus, or 5 × 106 NB104 cells using the TRIzol reagent method as per vendor instructions (Invitrogen, Carlsbad, CA, U.S.A.). Five micrograms total RNA was used for reverse transcription of xbp-1 cDNA using the antisense primer described later and the C. Therm. Polymerase Reverse Transcription Kit (Roche Diagnostics, Indianapolis, IN, U.S.A.) per vendor instructions. Next, the 600-bp region of the xbp-1 cDNA was amplified with the Expand High Fidelity PCR Sytem (Roche Diagnostics). The following primers were used: sense (5′-AAACAGAGTAGCAGCGCAGACTGC-3′) and antisense (5′-GGATCTCTAAAACTAGAGGCTTGGTG-3′). Finally, 200 ng amplified xbp-1 cDNA was digested with 0.5 U Pst I for 60 minutes, 37°C and then detected on 2% agarose gels using 0.1% SYBR Gold (Molecular Probes, Eugene, OR, U.S.A.) in 1X TBE. To determine the sensitivity of the PCR assay, total RNA from thapsigargin-treated NB104 cells (which contain only processed xbp-1 mRNA) was mixed in varying proportions with total RNA from untreated NB104 cells (which contain only unprocessed xbp-1 mRNA) and subjected to sequential RT-PCR as described previously.
The effect of μ-calpain was determined using PERK immunoprecipitated from 4 mg protein from NIC and 10R cortex samples bound to Protein A-Sepharose beads. Immunoprecipitated PERK was equilibrated in 35 μL buffer containing 25-mmol/L Tris (pH 7.5), 50-mmol/L NaCl, 1-mmol/L DTT, 0.5-mmol/L EDTA, and 1-mmol/L CaCl2. Human μ-calpain (calpain I; Calbiochem-Novabiochem Corporation, San Diego, CA, U.S.A.) at doses of 25, 100, or 250 ng was added to start the reactions, which were run for 60 minutes at 37°C with gentle tap mixing every 10 minutes and stopped by boiling for 90 seconds in an equal volume of SDS-PAGE loading buffer. Reactions were electrophoresed on 6% SDS-PAGE gels, electroblot transferred, and immunoblotted with anti-PERK.
RESULTS
Consistent with our previous work (DeGracia et al., 1996; Page et al., 2000), there is a rapid increase of eIF2α(P) in both vulnerable and nonvulnerable brain regions shortly after reperfusion (Fig. 1A). Mean eIF2α(P) levels peaked at 10-minute reperfusion and were 5.8-fold (P = 0.05), 28.5-fold (P = 4 × 10–5), and 15.7-fold (P = 3 × 10–5) higher than nonischemic controls in brain stem, cortex, and hippocampus, respectively (Fig. 1B). Recovery of eIF2α(P) to basal levels was seen by 4-h reperfusion in brain stem. A slower recovery was observed in cortex and hippocampus: eIF2α(P) levels were indistinguishable between 10 and 90 minutes, but decreased to 5.4-fold control and 3.3-fold control, respectively, at 4 h of reperfusion.
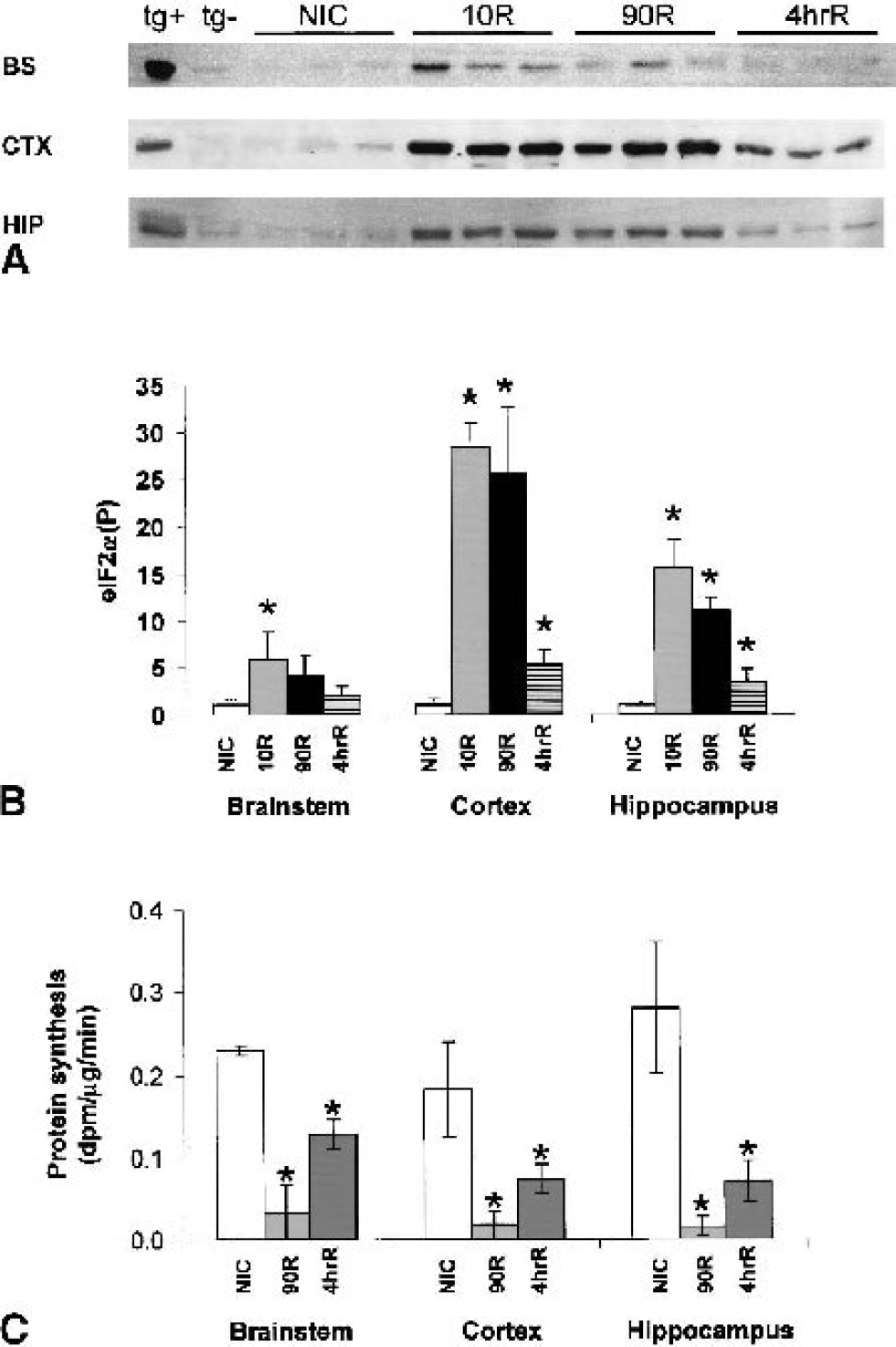
Also consistent with our previous measurements (DeGracia et al., 1996; Sullivan et al., 1999), there was an ∼85% inhibition of protein synthesis in all brain regions at 90-minute reperfusion (Fig. 1C). Some recovery of translation occurred at 4 h of reperfusion; protein synthesis was inhibited 45% in brain stem (P = 0.008), 60% in cortex (P = 0.036), and 75% in hippocampus (P = 0.024) as compared to controls. Interestingly, protein synthesis in the brain stem continued to be inhibited, despite near-normal levels of eIF2α(P).
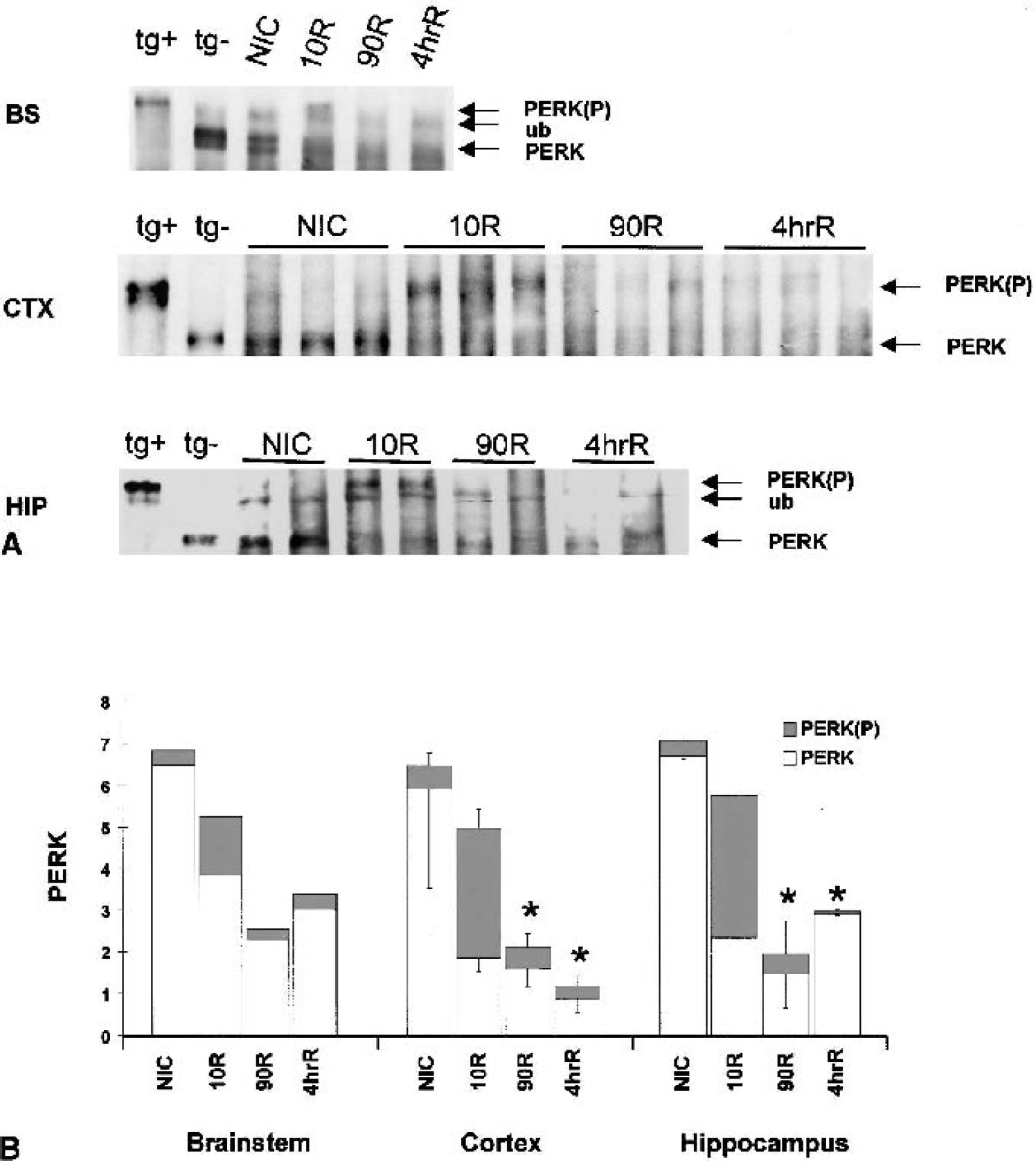
PERK activity is indicated by its phosphorylation state, which can be determined by its reduced mobility on SDS-PAGE after immunoprecipitation (Harding et al., 1999). PERK from all three brain regions behaved in a similar fashion (Fig. 2A). At 10-minute reperfusion, PERK showed a large mobility reduction that comigrated with PERK from positive controls (Fig. 2A), indicative of active, phosphorylated PERK (Harding et al., 1999). Densitometry of total PERK signal revealed a loss of signal at 90-minute and 4-h reperfusion; by 4 h, PERK was decreased by 80% in cortex, and ∼50% in brain stem and hippocampus (Fig. 2B).
We previously showed that μ-calpain is activated during global brain ischemia (Neumar et al., 1996). Therefore, we assessed the effects of in vitro treatment of immunoprecipitated PERK with μ-calpain. As shown in Fig. 3, there is almost complete loss of PERK signal at the lowest dose of μ-calpain (25 ng) and total loss at the highest dose (250 ng), regardless if PERK was in the inactive (NIC) or active (10R) state.
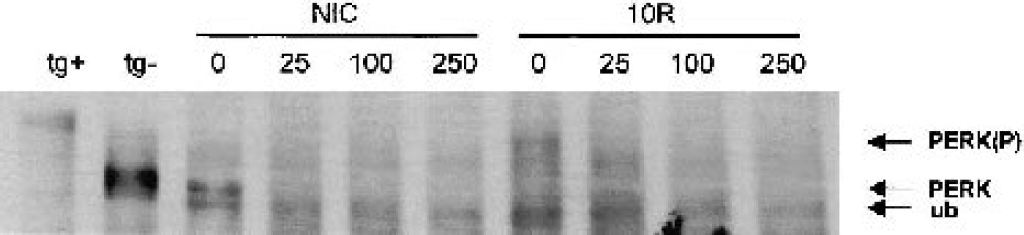
In vitro treatment of immunoprecipitated PERK with μ-calpain. PERK was immunoprecipitated from 4 mg protein of a nonischemic control (NIC) or 10-minute ischemia/10-minute reperfusion (10R) cerebral cortex. Amounts of μ-calpain (ng) added to each reaction are indicated above the respective lanes. There was almost complete loss of PERK signal whether in the inactive (NIC) or active (10R) state at the 25-ng dose of μ-calpain, and total loss of PERK signal at higher doses. Mobility controls for PERK(P) and PERK were thapsigargin-treated (tg+) and untreated (tg–) NB104 cells, respectively. This blot shows an unidentified band (ub) that consistently immunoprecipitates with PERK in all brain samples and migrates slightly below inactive PERK.
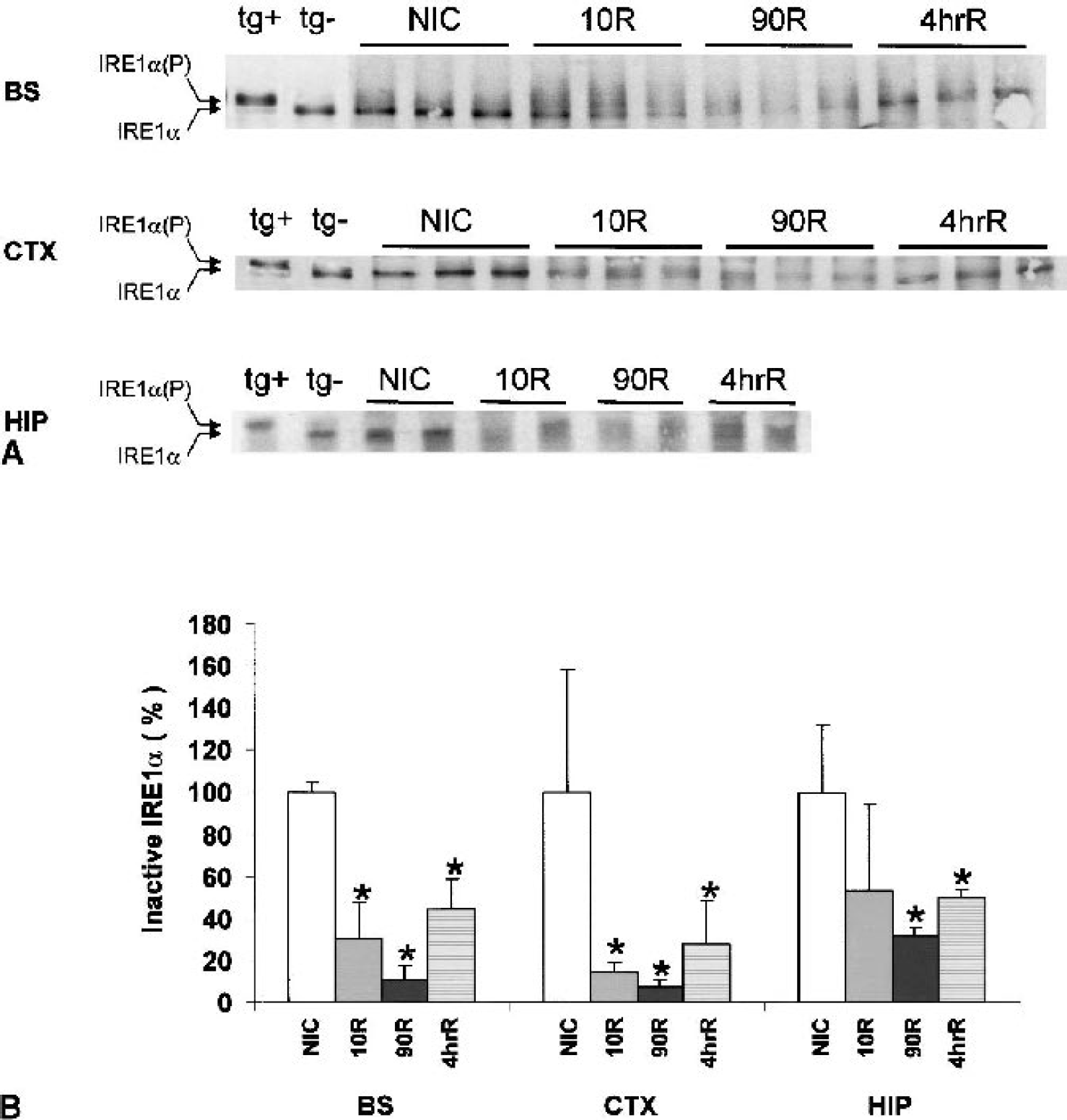
IRE1α activity also correlates with its phosphorylation status, showing reduced mobility on SDS-PAGE after immunoprecipitation (Bertolotti et al., 2000). For all three brain regions, IRE1α from nonischemic control animals was detectable and comigrated with inactive NB104 IRE1α with a mobility of ∼110 kd as expected (Fig. 4A). We did not see a distinct shift to IRE1α(P) in brain, as was observed in the thapsigargin-treated NB104 cells. There is, however, evidence of multiple bands of IRE1α in the 10R brain stem and cortex samples that comigrate in the region of NB104 IRE1α(P). We did see a consistent loss of inactive IRE1α in the experimental groups (Fig. 4B). A similar pattern is observed in all three brain regions: maximal loss of the inactive IRE1α band by 90 minutes of reperfusion with a return toward control levels at 4 h (Fig. 4B). The 85% loss of inactive IRE1α in brain stem and 90% loss in cortex were statistically significant, but the 40% loss in hippocampus was not (ANOVA P = 0.01, 0.02, and 0.2, respectively).
To determine whether the alterations seen in IRE1α by immunoprecipitation correlated with its endoribonuclease activity, we assessed the processing of endogenous xbp-1 mRNA. Active IRE1α cleaves a 26-nt sequence from xbp-1 mRNA resulting in processed xbp-1. Because removal of the 26-nt sequence results in loss of a Pst I restriction site, the PCR amplification product is refractory to Pst I cleavage if xbp-1 is in the processed form. As expected, thapsigargin treatment of NB104 cells resulted in a 574-bp xbp-1 product that is refractory to Pst I cleavage (Fig. 5A). In contrast, there was no evidence of xbp-1 mRNA processing in the brain during reperfusion (Fig. 6). The 574-bp processed xbp-1 product was not observed, and the 600-bp fragment that we observed was cleaved by Pst I. Based on the sensitivity of our assay (Figure 5B), this result suggests that, if xbp-1 processing occurs during reperfusion, less than 5% of endogenous xbp-1 is processed. In some brain samples, and in all NB104 samples, we observed a ∼650-bp unknown amplification product (uap, Figs. 5 and 6) that was unresponsive to Pst I cleavage.
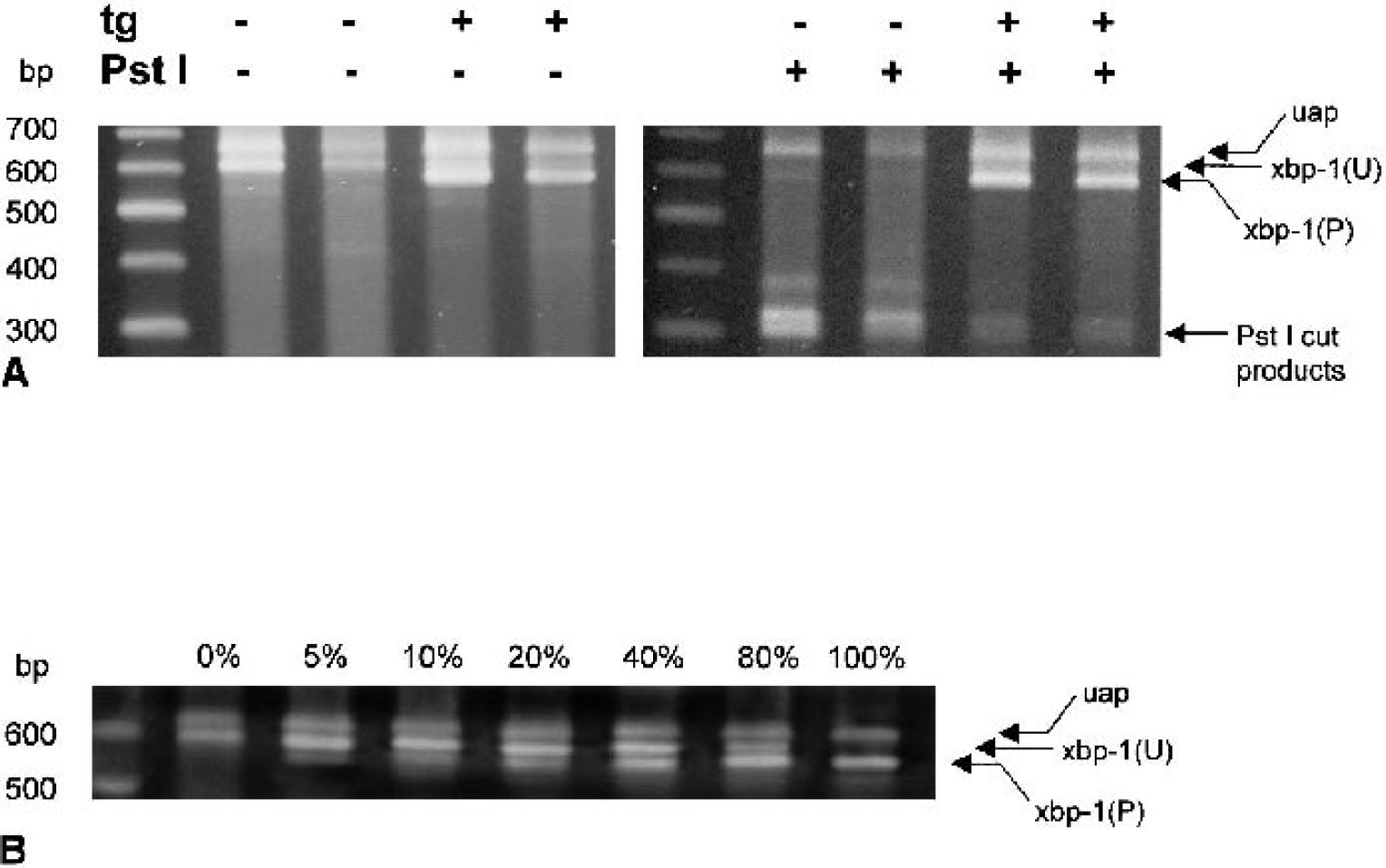
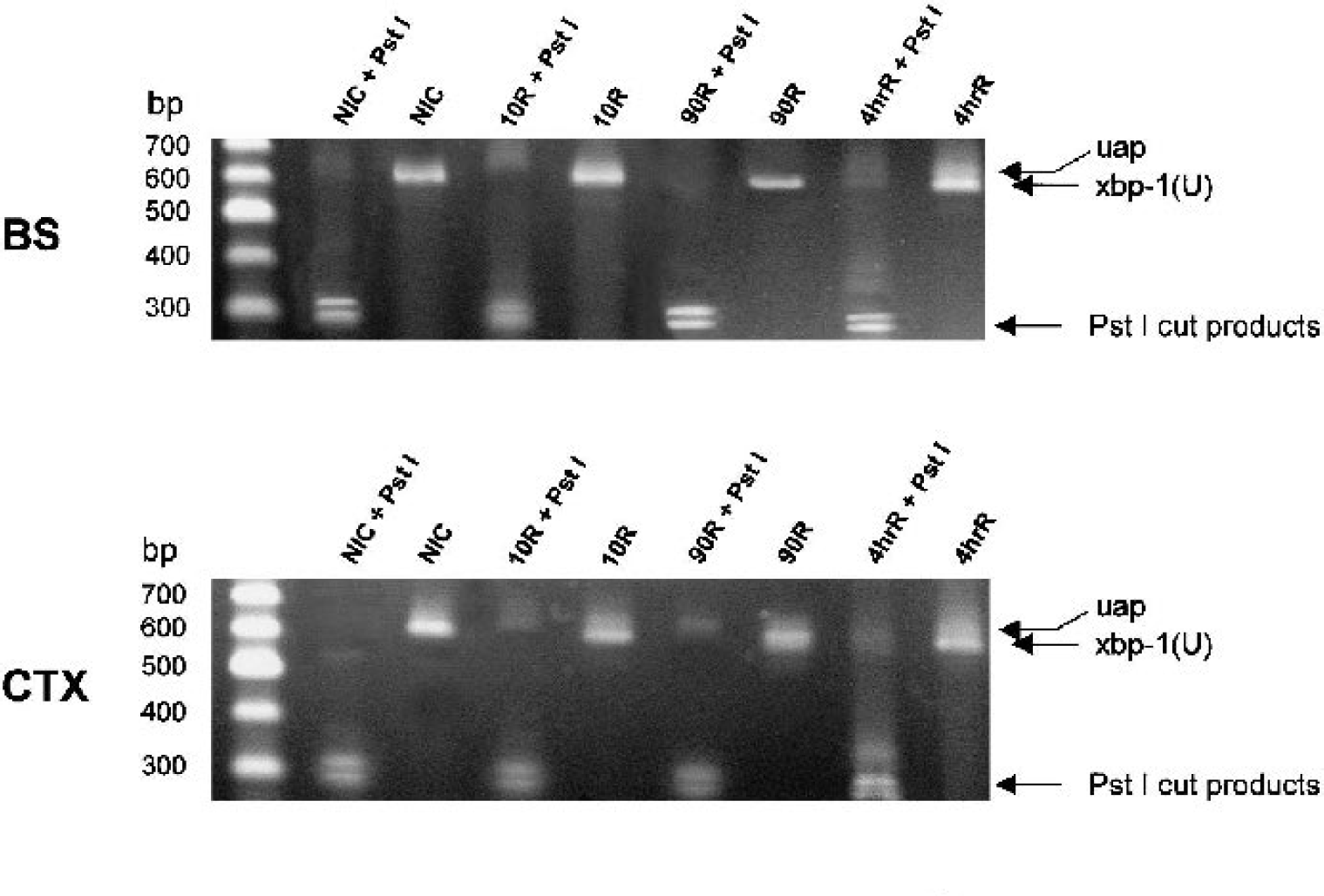
The effect of Pst I cleavage on a 600-bp fragment [xbp-1(U)] amplified from endogenous xbp-1 of brain stem (BS), cerebral cortex (CTX), or hippocampus (HIP) of nonischemic controls (NIC), or after 10-minute (10R), 90-minute (90R), or 4-h reperfusion (4hrR) after a 10-minute cardiac arrest. Representative 2% agarose gels with and without Pst I treatment. There was no evidence of the 574-bp processed xbp-1 complementary DNA, and all samples were cleaved by Pst I.
As further confirmation that endogenous xbp-1 mRNA was in the unprocessed form, there was no accumulation of the 55-kd form of XBP-1, which was readily detected in thapsigargin-treated NB104 cells (Fig. 7C). Similarly, we observed no increase in CHOP protein (Fig. 7A) or ATF4 (Fig. 7B) in rat brain regional homogenates of any experimental group, both of which were readily detectable in thapsigargin-treated NB104 cells. Finally, there was no evidence of proteolysis of the 90-kd ATF6 band (p90ATF6) in all three brain regions of all experimental groups (Fig. 8), although we detected in thapsigargin-treated NB104 cells 85-kd (p85ATF6) and 50-kd (p50ATF6) proteolytic cleavage products associated with translocation of the cytosolic domain of ATF6 to the nucleus (Haze et al., 1999).
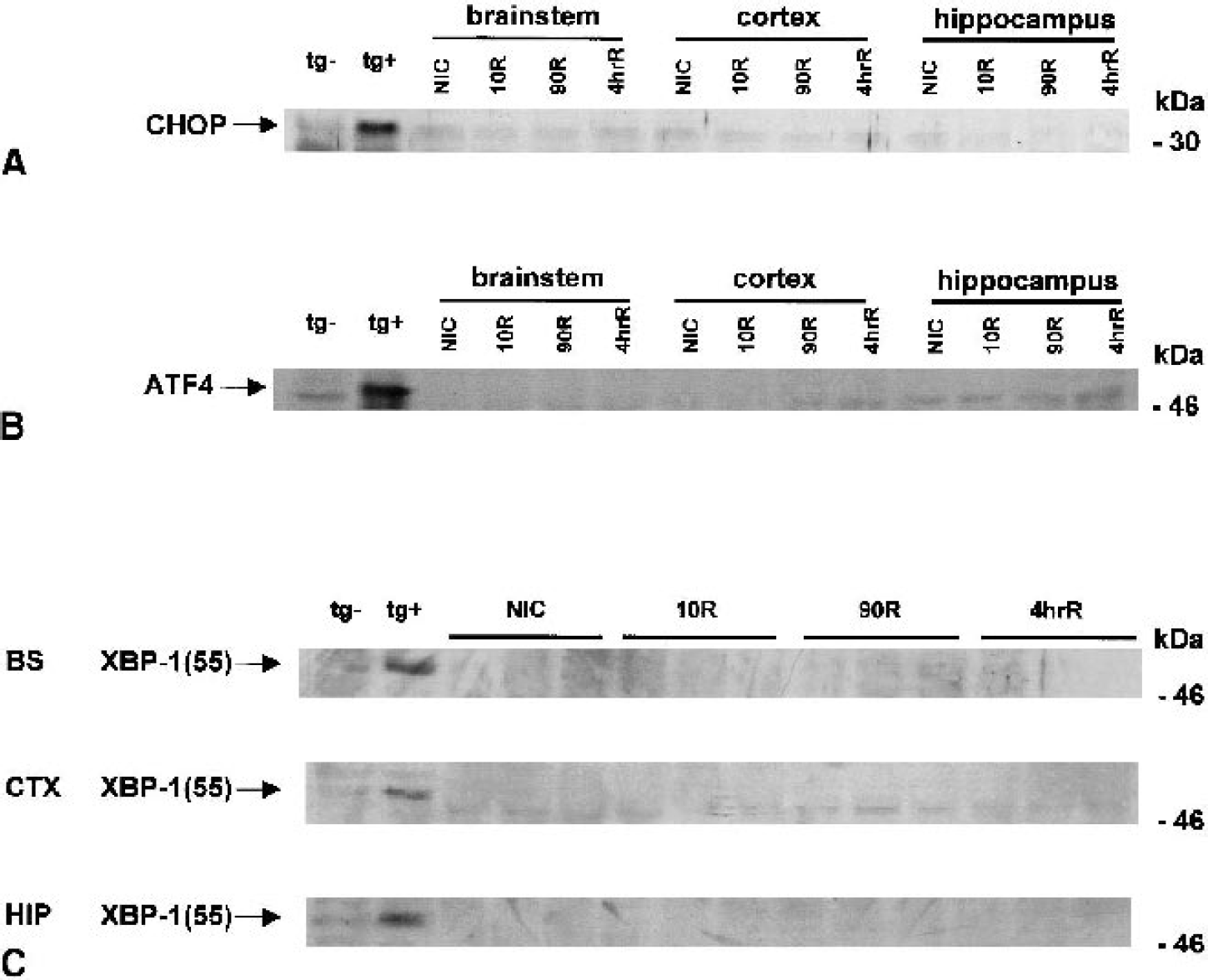
Immunoblots of
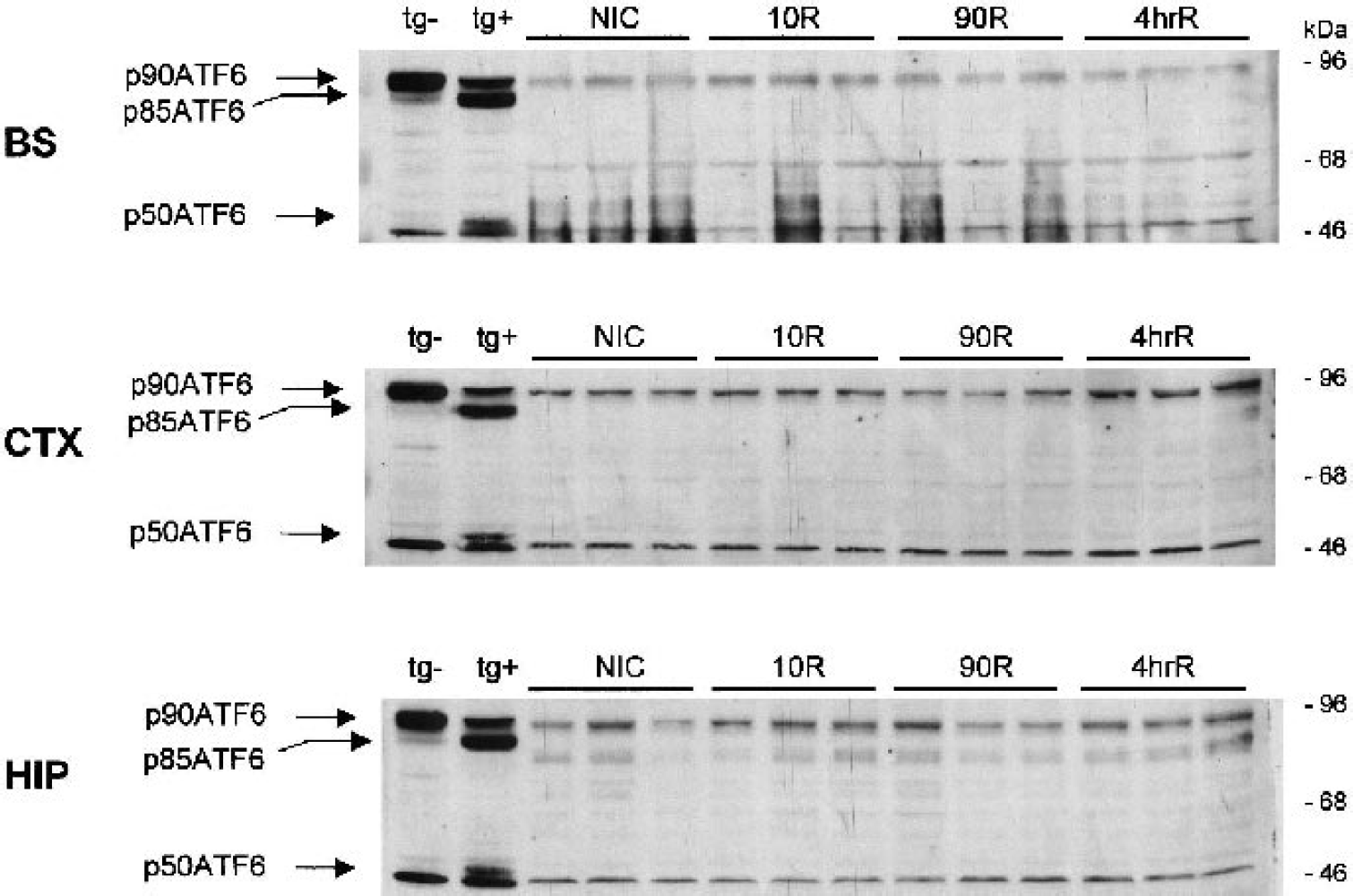
ATF6 does not undergo proteolytic activation by 4-h brain reperfusion after a 10-minute cardiac arrest. Immunoblotting of 125 μg of unfractionated homogenate proteins of brain stem (BS), cerebral cortex (CTX), and hippocampus of nonischemic controls (NIC), or after 10-minute (10R), 90-minute (90R), or 4-h reperfusion (4hrR) after a 10-minute cardiac arrest. In all groups, the 90-kd ATF6 band (p90ATF6) remains intact. After thapsigargin treatment in NB104 cells (tg+), ATF6 undergoes transformation to the 85-kd (p85ATF6) and 50-kd (p50ATF6) forms, which are not present in the untreated NB104 cells (tg–). There is an unidentified band at ∼46 kd in all brain samples and the NB104 cells, but this does not comigrate with the p50ATF6 band seen in thapsigargin-treated NB104 cells.
DISCUSSION
Other studies have shown that in ER-stressed cells, the inability to express all components of the UPR via expression of dominant-negative IRE1α (Miyoshi et al., 2000) or PERK (Harding et al., 2000b) or downregulation of GRP78 (McCormick, 1997) increases cell death. Evidence of ER stress during brain ischemia and reperfusion (reviewed in DeGracia et al., 2002) necessitates investigation of the mechanisms responsible for coping with ER stress. Here we have characterized the major UPR pathways (summarized in Fig. 9) to 4 h of reperfusion in three different brain regions after cardiac arrest–induced global brain ischemia.
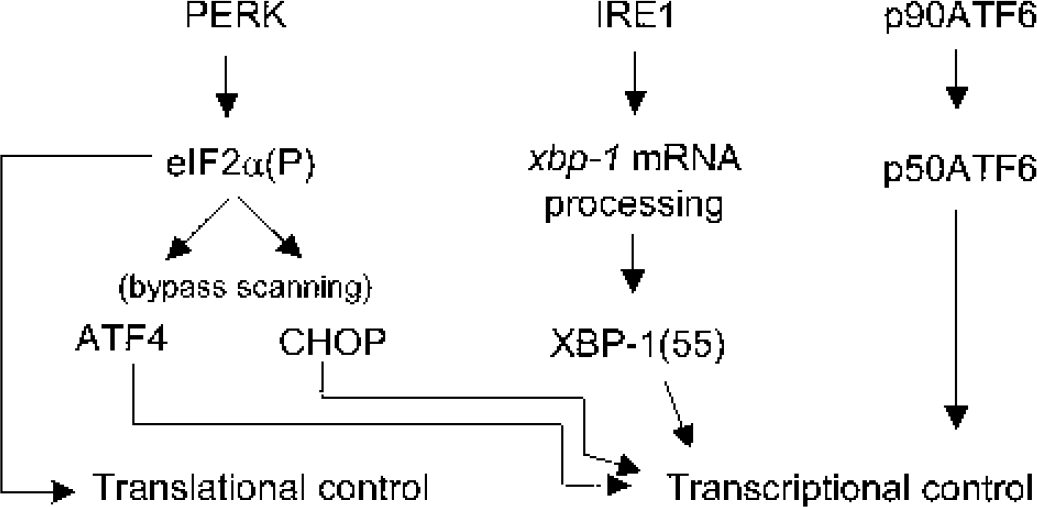
Diagram of the three major unfolded protein response (UPR) pathways. PERK, IRE1α, and ATF6 are endoplasmic reticulum (ER) transmembrane proteins that are activated in response to ER stress. Activated PERK phosphorylates the α subunit of eIF2 leading to a general suppression of protein synthesis and to increased translation of ATF4 and CHOP via the bypass scanning translation mechanism. Activated IRE1α possesses endoribonuclease activity specific for xbp-1 mRNA, which cleaves a 26-nt sequence (xbp-1 messenger RNA [mRNA] processing) and results in an efficiently translated xbp-1 mRNA, whose translation product is a 55-kd protein, XBP-1(55). ATF6 activation occurs by sequential proteolysis, resulting in the 50-kd p50ATF6 that translocates to the nucleus; the 85-kd form of ATF6 (p85ATF6) is thought to be a deglycosylated intermediate. The proteins ATF4, p50ATF6, CHOP, and XBP-1 are all transcription factors that alter gene expression to affect the UPR program. Together, these three pathways represent an integrated response by the cell to abate and repair the effects of ER stress through control of translation and transcription mechanisms.
In the reperfused brain, PERK is activated and phosphorylates its substrate eIF2α, and this is accompanied by inhibited protein synthesis. Elevated levels of eIF2α(P), however, should result in an increase of the bypass scanning translation products ATF4 and CHOP, but this was not seen at any time point. Additionally, there was a substantial loss of PERK by 4 h of reperfusion. Inactive IRE1α decreased substantially by 90 minutes of reperfusion, but the lack of both xbp-1 processing and appearance of XBP-1(55) indicates that this alteration occurred in the absence of in vivo IRE1α functional activity. ATF6 was not activated by 4-h reperfusion.
The UPR components from all three brain regions generally followed similar patterns. One difference observed among brain regions, however, was the more rapid clearance of eIF2α(P) and a greater return of protein synthesis in brain stem as compared with cerebral cortex or hippocampus. One could speculate that the stimulus responsible for UPR activation may be less persistent in brain stem than in the other regions and may, in part, account for the relatively greater resistance of brain stem to ischemia–reperfusion injury.
Two recent studies measured brain eIF2α(P) levels during reperfusion and showed that eIF2α(P) levels decreased to near-control levels by ∼6 h of reperfusion (Althausen et al., 2001; Martin de la Vega et al., 2001), but protein synthesis continued to be inhibited. These authors suggested there is further dysfunction to the translation machinery independent of eIF2α(P) levels. Our data support this view. Despite near-normal levels of eIF2α(P), protein synthesis in the brain stem is only 55% of control, and there is no synthesis of ATF4 or CHOP protein in the first 4 h of brain reperfusion.
These PERK data confirm our previous finding of forebrain PERK activation during reperfusion after bilateral carotid artery occlusion in the mouse (Kumar et al., 2001) and extend the finding in terms of regional activation and reperfusion duration after cardiac arrest in the rat. There is evidence of a prominent PERK mobility reduction, which indicates activation of PERK (Harding et al., 1999), at 10-minute reperfusion in all three brain regions. A surprising finding of the present study is that there is an absolute loss of PERK signal in all three brain regions. The in vivo half-life of PERK is 13 h in dividing cell cultures (Bertolotti et al., 2000) but has not been determined in brain. If the half-life in brain is similar, we would expect only a 15% loss of PERK in 4 h, under conditions of inhibited protein synthesis. Thus, our data indicate that PERK is undergoing proteolysis. In support of this, we have shown that PERK is an in vitro substrate of μ-calpain (Fig. 3), and our laboratory previously showed direct activation of μ-calpain during global brain ischemia (Neumar, 1996). Because other proteases may be responsible in whole or part for the PERK loss reported here, proof of μ-calpain involvement in vivo will require pharmacologic inhibition of μ-calpain and assessment of PERK levels.
Unlike PERK, IRE1α appears to undergo a reversible alteration during reperfusion as determined by loss and then partial return of the inactive form of IRE1α (Fig. 4). This does not seem to reflect an in vivo functional activation, however, because we did not observe processing of endogenous xbp-1 mRNA or detect synthesis of XBP-1(55). Because IRE1α presumably requires the concerted activity of tRNA ligase to join the cleaved ends of processed xbp-1 mRNA (Kaufman, 1999), it is possible that the IRE1α alteration we observed reflects IRE1α activation, and the lack of detection of processed xbp-1 possibly indicates the inability to ligate the cleaved mRNA. To distinguish these possibilities, future studies need to directly measure the functional activity of endogenous IRE1α in the first hours of reperfusion.
Damage mechanisms known to occur during reperfusion may account for why ATF6 was not activated by 4-h reperfusion. Electron microscopy studies have shown that the ER and Golgi apparatus of reperfused neurons loose their “flattened pancake” appearance and become rounded vesicles (Petito and Pulsinelli, 1984). We have shown accumulation of lipid peroxidation products in these structures during reperfusion (White et al., 1993). It is possible that the necessary stimulus is present for ATF6 activation, but alterations of ER and Golgi morphology may reflect an altered biochemical environment that precludes ATF6 activation.
Normally, the PERK, IRE1α, and ATF6 pathways respond in concert to abate the effects of ER stress. Because there is strong evidence for ER stress during brain reperfusion, including depletion of Ca2+ from the ER (Kohno et al., 1997), decreased ER Ca2+-ATPase activity (Parsons et al., 1997), and accumulation of protein aggregates in the ER (Hu et al., 2000), it is unexpected that the three UPR pathways are not activated concurrently in the reperfused brain. In contrast, in the thapsigargin-treated NB104 cells, PERK, IRE1α, and ATF6 were rapidly activated and, within a 2-h time frame, XBP-1(55), ATF4, and CHOP proteins were synthesized. Because the UPR can be induced pharmacologically in cell culture with 2-μg/mL tunicamycin, 200-nmol/L thapsigargin, or 5-mmol/L DTT, we attempted to activate the UPR in intact nonischemic control brain by intracerebroven-tricular injection of 20-μg/mL tunicamycin (10X), 100-μmol/L thapsigargin (500X), or 2-mol/L DTT (400X). No evidence of in vivo UPR induction was observed 2 h after any treatment (data not shown). Clearly, there are substantial differences between the in vivo and in vitro responses of the UPR components to pharmacologic insults, but the basis for these differences is completely unknown and further studies are needed.
Given the differences between pharmacologic induction of the UPR in cell culture and the in vivo results presented here, the present results can be interpreted in at least two ways: either the UPR does not occur during brain reperfusion and PERK is activated by a currently unknown mechanism, or ER stress induced by brain ischemia and reperfusion activates the UPR but there is disruption in its expression in vivo. The present study did not allow us to distinguish between these possibilities. Previous evidence, however, suggests the latter possibility may be correct. Paschen et al. (2001) found that sequential treatment of primary cortical neuronal cultures with H2O2 followed by thapsigargin led to an attenuated UPR, as measured by grp78, grp94, and chop mRNA synthesis, compared with thapsigargin treatment alone. This result indicates that oxidative stress can attenuate the UPR. There is some evidence that such a mechanism may also be operative in vivo. Whereas there is a modest increase in grp78 mRNA after focal brain ischemia (Wang et al., 1993), grp78 mRNA expression after global ischemia and normothermic reperfusion is significantly suppressed in hippocampal pyramidal cells (Aoki et al., 2001). Free radicals are generated after global ischemia (reviewed in White et al., 2000), but there appears to be an absence of free radical generation in the core of focal ischemia (Solenski et al., 1997), which may account for the difference in these two studies.
In summary, our present results indicate that there is a lack of an integrated expression of the UPR in the early period after transient brain ischemia. Other damage mechanisms including oxidative reactions, unregulated proteolysis, and translation inhibition may contribute to this dysfunction. Regardless of the precise mechanism, it is expected that the inability to mount an effective response against ER stress will contribute to the death of neurons.
Footnotes
Acknowledgments:
The authors thank Heather Monti and Salman Azam for their important technical assistance, and David Ron and Heather Harding for providing PERK and IRE1α antisera.