
Abstract
The authors are systematically exploring pharmacologic preservation for temporarily unresuscitable exsanguination cardiac arrest in dogs. They hypothesized that the antioxidant Tempol improves cerebral outcome when added to aortic saline flush at the start of cardiac arrest. In study A, no drug (n = 8), Tempol 150 mg/kg (n = 4), or Tempol 300 mg/kg (n = 4) was added to 25 mL/kg saline flush at 24°C (achieving mild cerebral hypothermia) at the start of 20-minute cardiac arrest. In study B, no drug (n = 8) or Tempol 300 mg/kg (n = 7) was added to 50 mL/kg saline flush at 2°C (achieving moderate cerebral hypothermia) at the start of 40-minute cardiac arrest. Cardiac arrest was reversed with cardiopulmonary bypass. Mild hypothermia lasted for 12 hours, controlled ventilation was sustained to 24 hours, and intensive care was provided for up to 72 hours. In study A, overall performance category 1 or 2 (good outcome) was achieved in all eight dogs treated with Tempol compared with three of eight dogs in the control group (P = 0.03). In study B, good outcome was achieved in all seven dogs treated with Tempol versus only two of 8 dogs in the control group (P = 0.007). In both studies, neurologic deficit scores were significantly better in the Tempol group, but not total histologic damage scores. At 72 hours, electron paramagnetic resonance spectroscopy of Tempol revealed direct evidence for its presence in the brain. Single- and double-strand DNA damage, nitrotyrosine immunostaining, total antioxidant reserve, and ascorbate acid levels were similar between groups, and thiol levels were decreased after Tempol in study B. The authors conclude that when added to aortic saline flush at the start of prolonged cardiac arrest, the antioxidant Tempol can enhance mild or moderate hypothermic cerebral preservation in terms of improved functional outcome. The mechanisms involved in this beneficial effect need further clarification.
Attempts to resuscitate patients from intrathoracic or intraabdominal exsanguination cardiac arrest (CA) before control of bleeding have failed (Bellamy et al., 1996). In the search for a needed new approach, Safar and Bellamy (1984) recommended research into “suspended animation for delayed resuscitation,” to preserve brain and organism during prolonged CA. This approach would enable transport and repair during pulselessness.
Hypothermia during CA induced and reversed by cardiopulmonary bypass (CPB) preserves brain and organism during up to 15-minute CA no-flow during mild hypothermia (33–36°C) (Safar, 1988), up to 20-minute CA during moderate hypothermia (28– 32°C) (Bigelow et al., 1950), up to 30-minute CA during deep hypothermia (16–27°C) (Livesay et al., 1983), and up to 60-minute CA during profound hypothermia (5–15°C) (Capone et al., 1996). In normothermic CA, the brain tolerates a no-flow time of only approximately 5 minutes (Safar, 1988). Therefore, to avoid the loss of viability of cerebral neurons, a suspended animation strategy must be induced within 5 minutes of CA onset. Because CPB is not immediately available in the field, we introduced and explored cold saline flush with large flush volumes into the aorta via a balloon tipped catheter to rapidly preserve cerebral viability in dogs during 15-minute (Woods et al., 1999), 20-minute (Behringer et al., 2000a), 30-minute (Behringer et al., 2000b), and 60-, 90-, or in some dogs even 120-minute CA (Behringer et al., 2001a). For suspended animation in the field, the aortic flush solution would need to be delivered using a portable small volume at ambient temperature. An aortic arch flush of approximately 25 mL/kg saline at 24°C at the start of 20-minute CA induced mild cerebral hypothermia (tympanic membrane ≈ 36°C) and resulted in survival with brain damage (Behringer et al., 2000a). We then hypothesized that adding a drug with cerebral preservation potential to this aortic arch saline flush would achieve functional and histologic normality. Using this 20-minute CA model, we systematically explored 14 pharmacologic cerebral preservation potentials (Behringer et al., 1999). Drugs were selected for the following six pharmacologic strategies: (1) delaying energy failure, (2) protecting membrane integrity, (3) preventing structural degradation, (4) regulating protein synthesis, (5) preventing re-oxygenation injury, and (6) preserving mitochondria. Concerning strategies one and two, adenosine (Woods et al., 2000), fructose-1,6-bisphosphate (Behringer et al., 2001b), the N-methyl- d -aspartate antagonist MK801 (Behringer et al., 2001b), and thiopental plus phenytoin (Behringer et al., 2001c) were not effective.
This report concerns strategy five. We added the cell-permeable antioxidant 4-hydroxy-2,2,6,6,-tetramethylpiperidine-1-oxyl (Tempol) to the saline flush. Tempol has been shown to benefit rats with hemorrhagic shock (Kentner et al., 2000) or splanchnic artery occlusion (Mota-Filipe et al., 1999), traumatic brain injury (Beit-Yannai et al., 1996; Zhang et al., 1998), or focal brain ischemia (Beaulieu et al., 1998; Rak et al., 2000), and gerbils with global brain ischemia (Cuzzocrea et al., 2000). Tempol was never investigated in a large (higher) animal species and a clinically relevant model of prolonged CA with evaluation of long-term outcome. In this study, adjunctive biochemical and immunohistologic observations were also made to explore suspected mechanisms of the action of Tempol. Using our exsanguination CA dog model, we hypothesized that Tempol added to aortic saline flush at 24°C at the start of a 20-minute no-flow CA enhances the documented benefit of mild cerebral hypothermia (ambient temperature flush for use in the field); and that Tempol added to aortic saline flush at 2°C with a larger volume at the start of 40-minute no-flow CA enhances the documented benefit of moderate cerebral hypothermia (feasible in hospitals). We expect that for both CA durations, Tempol by flush results in normal functional recovery without histologic brain damage.
MATERIALS AND METHODS
This study was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh and the Department of Defense, and followed national guidelines for the treatment of animals. All experiments were conducted by the same team. Study A was conducted within 9 months in mixed sequence and without randomization as part of a systematic exploration of 14 pharmacologic aortic flush-preservation potentials (Behringer et al., 1999), whereas study B was conducted within 3 months with randomization. Briefly, the protocol called for exsanguination to 20-minute no-flow CA (study A) or 40-minute no-flow CA (study B), resuscitation with CPB, mild hypothermia to 12 hours, controlled ventilation to 20 hours, intensive care to 72 hours, and outcome evaluation. In study A, after 2 minutes CA, the dogs received a flush over 1 minute into the thoracic aorta with 25 mL/kg saline at 24°C without the drug (control group, n = 8) or with 150 (n = 4) or 300 mg/kg Tempol (n = 4) added. In study B, after 2 minutes CA, the dogs received a flush over 2 minutes into the abdominal aorta (necessary to include preservation of the spinal cord when CA exceeds 20 minutes) with 50 mL/kg saline at 2°C, either without the drug (control group, n = 8) or with 300 mg/kg Tempol (n = 8).
Preparation
A detailed description of the model was published previously (Behringer et al., 2000a, 2000b). Thirty-two male custom-bred hunting dogs (body weight, 18–26 kg; age, 8–12 months) were premedicated with intramuscular ketamine 10 mg/kg, anesthetized with halothane (2–4%) and a 1:1 ratio of nitrous oxide to oxygen via a cone mask. After tracheal intubation, the dogs were mechanically ventilated (Harvard Piston Ventilator model 613, Harvard Apparatus, South Natick, MA) with tidal volumes of 15 mL/kg and a positive end-expiratory pressure of 5 cm H2 O without paralysis. Temperature probes were inserted for monitoring tympanic membrane, esophageal, and rectal temperatures. Fluid maintenance was with dextrose 5% in 0.45% sodium chloride at 5 mL · kg−1 · hour−1. A PE 90 arterial catheter was inserted for pressure monitoring and blood sampling, and a 7.5-Fr pulmonary artery catheter (Intellicath Continuous Cardiac Output thermodilution catheter, Baxter Co., Irvine, CA) was inserted for continuous monitoring of pulmonary artery pressure, temperature, and cardiac output (Baxter Vigilance Monitor, software 4.42). An 8-Fr prototype balloon catheter with one hole at the tip (Cardeon Corp., Cupertino, CA, U.S.A.) was inserted for arterial bleeding and aortic flush, and a multiple-hole 18-Fr transjugular vena cava catheter was inserted for venous bleeding and venous return to the CPB system. Tympanic membrane temperature was controlled at 37.5 ± 0.1°C with a heating blanket and lamp.
Cardiac arrest and flush
After two baseline measurements at tympanic membrane temperature 37.5°C, heating devices, intravenous fluids, and halothane were discontinued while the dogs were weaned to spontaneous breathing of a 3:1 nitrous oxide to oxygen mixture via a T-tube. When the canthal reflex returned, hemorrhage was initiated to achieve a mean arterial pressure (MAP) of 40 mm Hg at 2 minutes, 30 mm Hg at 3 minutes, and 20 mm Hg at 4 minutes. At 5 minutes, to assure zero blood flow, ventricular fibrillation was induced with a 60-Hz transthoracic shock of 110 V (alternating current) for 2 seconds, and was repeated as needed. Total arrest time (no-flow) was 20 minutes in study A and 40 minutes in study B.
Two minutes after the onset of CA, the balloon of the aortic catheter was inflated to occlude the aorta and saline was flushed using a roller pump, as described previously. After the flush and during CA, the aortic catheter was replaced by a short arterial CPB cannula (7 or 8 Fr).
Resuscitation
Reperfusion after CA was achieved with CPB using a centrifugal pump (Biomedicus, Eden Prairie, MN, U.S.A.) and hollow-fiber membrane oxygenator with heat exchanger (Medtronic, Anaheim, CA, U.S.A.) primed with 400 mL dextran 40 (10% in saline) and Ringer solution (1:1) including sodium bicarbonate (2 mEq/kg) and heparin (1,500 U). Just before the start of CPB, additional heparin (1,500 U) and sodium bicarbonate (2 mEq/kg) were given intravenously. Cardiopulmonary bypass began with a flow of 100 mL/kg per minute, and reinfusion of the shed blood was titrated to achieve a central venous pressure of 10 to 15 mmHg and a MAP greater than 100 mm Hg. If necessary, epinephrine (boluses of 0.01 mg/kg) was administered intravenously. For defibrillation we used external direct-current countershocks of 150 J, and repeated shocks were increased by 50 J. Controlled ventilation was resumed with 100% oxygen at a rate of 8 to 10 inflations per minute. A base deficit of greater than 6.0 mEq/L was corrected with intravenous sodium bicarbonate. When restoration of spontaneous circulation was established, a norepinephrine infusion was titrated intravenously to achieve a brief hypertensive bout of MAP greater than 150 mm Hg, after which MAP was maintained at 90 to 150 mm Hg (Sterz et al., 1990). Shed blood was gradually reinfused into the CPB system to maintain a central venous pressure of 8 to 15 mm Hg and a hematocrit greater than 30%. During CPB, the activated clotting time was maintained at more than 300 seconds with heparin. Cardiopulmonary bypass flow for assisted circulation was reduced to 75 mL · kg−1 · minute−1 at 60 minutes and 50 mL·kg−1 ·minute−1 at 90 minutes, and was stopped at 120 minutes.
Intensive care
After weaning dogs from CPB assist at 2 hours, controlled ventilation was continued to 20 hours with a 1:1 mixture of nitric oxide and oxygen. Paralysis was maintained with doses of intravenous pancuronium (0.1 mg/kg, repeated as needed). To prevent “stress” (mydriasis, hypertension), fentanyl was titrated intravenously in boluses of 5 to 10 μg/kg. Hypotension (MAP < 90 mm Hg) was managed with intravenous titrated Ringer solution or norepinephrine. Severe hypertension (MAP > 150 mmHg) was controlled with intravenous boluses of labetalol (0.25–0.5 mg/kg) or hydralazine (0.1–0.2 mg/kg). At 20 to 24 hours, paralysis was reversed to spontaneous breathing with neostigmine (50 μg/kg) plus atropine (25 μg/kg) and the dogs were extubated. Thereafter, seizures, running movements, opisthotonos, or spontaneous tachypnea were controlled with titrated doses of diazepam (0.2–0.3 mg/kg intravenously) as needed. Tympanic membrane temperature was controlled at 34°C with external cooling and warming for the first 12 hours after the start of CPB, and at 37.5°C until 72 hours.
Preservation with Tempol
Tempol was purchased from Sigma-Aldrich (Milwaukee, WI, U.S.A.) and dissolved in isotonic saline solution. The Tempol solutions were filtered with a 0.22-μm filter (Fisherbrand; Fisher Scientific, Pittsburgh, PA, U.S.A.) before the aortic flush. In study A, we explored the addition of Tempol 150 (n = 4) or 300 mg/kg (n = 4) to the aortic flush after 2 minutes of cardiac arrest. These doses were chosen based on the literature (Beit-Yannai et al., 1996; Zhang et al., 1998; Mota-Filipe et al., 1999) and pilot experiments. In study B, we chose the addition of a larger dose of Tempol (300 mg/kg) (n = 8) to the aortic saline flush after 2 minutes of cardiac arrest because of the longer duration of ischemia.
General outcome evaluation
Performance was evaluated according to overall performance categories (OPC) (1 = normal, 2 = moderate disability, 3 = severe disability, 4 = coma, and 5 = death) (Leonov et al., 1990). Neurologic function was evaluated as neurologic deficit scores (NDS) (0–10% = normal, 100% = brain death) (Radovsky et al., 1995). Scores were evaluated every 8 hours after extubation until final evaluations at 72 hours. Attempts were made to discontinue any sedation at least 4 hours before final evaluations. If necessary, sedation was reversed with flumazenil (0.1 mg intravenously, repeated if needed).
Brain histopathology
For morphologic studies, the dogs were reanesthetized after the final outcome evaluation with ketamine 10 mg/kg intramuscularly, followed by 0.5 to 1.5% halothane with a 1:1 mixture of nitric oxide and oxygen via tracheal tube and controlled ventilation. After left thoracotomy, the dogs were killed by infusing paraformaldehyde (4%, pH 7.4) into the aortic arch using a roller pump at a pressure of approximately 100 mm Hg, with the right atrium opened, until clear fluid returned (usually 2 L).
Light microscopic scoring.
The brain was removed after 1 hour of fixation. After cutting 3-mm thick slices, the same six slices of each brain were embedded in paraffin, cut into 4-μm thick sections, and stained with hematoxylin-eosin-phloxine. Using light microscopy, the same pathologist (AR), unaware of treatment assignments, scored 19 distinct anatomic brain regions according to severity and the extent of ischemic neuronal changes, infarcts, and edema, as described previously (Radovsky et al., 1995). The total brain histologic damage score (HDS) was the sum of all area scores. A score of more than 40 represents significant damage, and more than 100 represents severe damage.
Nitrotyrosine immunohistochemistry (study B).
Nitrotyrosine was detected in the hippocampus immunohistochemically at 72 hours, indicating of the presence of peroxynitrite and other nitrosating agents (Whalen et al., 1999). Tissue sections were removed of paraffin, rehydrated, and processed by a high-temperature antigen-retrieval technique. Sections were incubated in a 1:200 dilution of antinitrotyrosine antibody (Upstate Biotechnology, Lake Placid, NY, U.S.A.) diluted in phosphate-buffered saline, followed by incubation in appropriate secondary antibody. Sections were then washed in phosphate-buffered saline and incubated with an avidin-biotin complex (ABC Standard kit; Vector Labs, Burlingame, CA, U.S.A.) and then reacted with diaminobenzidine (DAB, Vector Labs). Nitrotyrosine immunoreactivity was evaluated by one observer (RSBC) who was blinded to experimental groups as follows: 0 = no increase in immunoreactivity above background, 1+ = few immunoreactive cells, 2+ = moderate immunoreactivity in cells in < 4 200X fields, 3+ = moderate immunoreactivity in cells in ≥ 4 200X fields, and 4+ = marked immunoreactivity in cells and prominent dendritic labeling in ≥ 4 200X fields.
Double-strand DNA damage (study B).
TUNEL (terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling) was performed at 72 hours (Clark et al., 2001). Briefly, 5-mm thick paraffin-embedded coronal sections containing hippocampus were removed of paraffin and incubated in 1 μM proteinase K (Boeringer Mannheim, Indianapolis, IN, U.S.A.). Sections were then incubated in 300 U/mL terminal deoxynucleotidyl transferase and 20 nmol/mL biotin-16-deoxyuridine (Boeringer Mannheim). TUNEL was viewed using ABC and DAB. The extent of DNA damage in the hippocampus for each brain section was evaluated by one observer (RSBC) who was blinded to experimental groups as follows: 0 = no TUNEL-positive cells; 1 = 1 to 10 TUNEL-positive cells in > 3 400X fields, 2 = 11 to 20 TUNEL-positive cells in > 3 400X fields, 3 = 21 to 30 TUNEL-positive cells in > 3 400X fields, 4 = 31 to 40 TUNEL-positive cells in > 3 400X fields, and 5 = > 41 TUNEL-positive cells in > 3 400X fields.
Single-strand DNA damage (study B).
The DNA PANT (polymerase I-mediated biotin deoxyadenosine triphosphate nick translation) labeling was also performed at 72 hours (Clark et al., 2001), and was optimized for the in situ detection of cells with increased single-strand DNA breaks after cerebral ischemia. Briefly, deparaffinized sections were incubated in 1 μmol/L proteinase K (Boeringer Mannheim). Sections were then incubated in 40 U/mL DNA polymerase I and 29 μmol/mL biotin-14-deoxyadenosine triphosphate (both from Life Technologies, Gaithersburg, MD, U.S.A.) in buffer. The slides were incubated in ABC and DNA strand breaks viewed with DAB. Slides were coverslipped for light-microscopic analysis and evaluated by one observer (RSBC) who was blinded to experimental groups.
Biochemical mechanisms
Ascorbate radicals and Tempol.
Electron paramagnetic resonance (EPR) spectroscopy was used to measure ascorbate radical and Tempol in brain homogenates at 72 hours, and in plasma samples. We used a JEOL-RE1X spectrometer (Kyoto, Japan) at 25°C in gas-permeable Teflon tubing (0.8-mm internal diameter and 0.013-mm thickness from Alpha Wire Corp (Elizabeth, NJ, U.S.A.). The tube (approximately 8 cm long) was filled with 60 μL mixed sample, folded into quarters, and placed in an open 3-mm internal diameter EPR quartz tube in such a way that all of the sample was within the effective microwave irradiation area. The spectra were recorded at a 3,355-G center field, 20-mW power; 0.79-G field modulation, 50-G sweep width, 4000 receiver gain, and 0.1-second time constant. Spectra were collected using EPRware software (Scientific Software Services, Bloomington, IL, U.S.A.).
Total antioxidant reserve.
The total antioxidant reserve in brain homogenates at 72 hours in both studies was assayed by chemiluminescence produced in the presence of luminol and peroxyl radicals, as described previously (Tyurina et al., 1995). A water-soluble azoinitiator (AAPH) was used to produce peroxyl radicals at a constant rate. Oxidation of luminol (400 μmol/L) by AAPH-derived peroxyl radicals in 50 mmol/L disodium phosphate buffer (pH 7.4) at 37°C was started by the addition of AAPH (50 mmol/L). A delay in the chemiluminescence response, which is caused by interaction of endogenous antioxidants with AAPH-derived peroxyl radicals, is observed upon addition of brain homogenate (0.1 mg protein/mL). Based on the known rate of peroxyl radical generation by AAPH, the amount of peroxyl radicals scavenged by endogenous antioxidants was determined. A luminescent plate reader (ML 1000; Dynatech Laboratories, Billingshurt, U.K.) was used for determinations.
Ascorbic acid.
High-pressure liquid chromatography was used to determine ascorbic acid levels in brain homogenates at 72 hours in both studies. We used an ODS Hypersil column (200 × 4.6 mm, 5 μm) (Hewlett Packard). The supernatant obtained by precipitation of proteins by 10% ethanol acid and sedimentation (10,000 g × 10 minutes) was used. The Shimadzu high-pressure liquid chromatography system (Kyoto, Japan) was used with an LC-600 pump and SPD-M10A diode array detector (detection by absorbance at 264 nm). The eluant was methanol:water (1:24 by volume) adjusted to pH 3.0 by ethanol acid at a flow rate of 1 mL/minute. Under these conditions, the retention time for ascorbic acid was 3 minutes. Acquired data were exported from detectors using Shimadzu EZChrom software.
Thiols.
The concentrations of low molecular weight thiols and protein thiols were determined in brain homogenates at 72 hours in both studies. We used ThioGlo-1 (CalBiochem), a maleimide reagent, which produces a highly fluorescent product upon reaction with sulfhydryl groups. To homogenates of brain tissue containing 15 to 30 μg protein/mL, ThioGlo-1 was added to a final concentration of 10 μmol/L (in dimethyl sulfoxide solution). Low molecular weight thiol content was estimated by an immediate fluorescence response observed upon addition of ThioGlo-1 to the brain homogenate. A standard curve was established by addition of low molecular weight thiols (glutathione 0.04–4.0 μmol/L) to 50 mmol/L disodium phosphate buffer (pH 7.4) containing 10 μmol/L ThioGlo-1. Total protein thiols were determined as an additional fluorescence response after addition of sodium dodecyl sulfate (4 mmol/L) to the same homogenate. A Cytofluor 2350 fluorescence plate reader (Millipore, Bedford, MA, U.S.A.) was used in the assay using excitation filter 360 ± 40 nm and emission filter 530 ± 25 nm.
Determination of proteins.
Protein concentration were determined with the Bio-Rad Protein Assay kit. A standard curve was established by addition of bovine serum albumin to the Bio-Rad assay kit, and protein content was calculated.
Statistical analysis
Dogs that did not meet protocol criteria or died from extracerebral causes were excluded. Brain death as an outcome was included if the experiment was performed according to protocol. Data are given as mean and SD, if normally distributed, or as median and interquartile range (IQR; difference between the 25th and 75th percentiles). We used the independent-samples t-test or the Mann-Whitney Test for the comparison of continuous variables (physiologic variables: NDS, HDS, ascorbate, thiols, and antioxidant reserve in brain), and the Fisher exact test for differences in proportions between groups (OPC 1, 2 = good outcome vs. 3–5 = bad outcome). To quantify the change of temperature over time during arrest, we calculated the area under the temperature curve. All data were computed with SPSS for Windows, release 8.0 (Chicago, IL, U.S.A.), or NCSS for Windows (Keysville, UT, U.S.A.). A P value < 0.05 was considered statistically significant.
RESULTS
For both studies, a total of 32 dogs were exsanguinated to CA. In study A, all 16 dogs survived to 72 hours in protocol (control group, n = 8; Tempol group, n = 8). In study B, one dog in the Tempol group was excluded because of a technical mistake in the aortic flush. A total of 15 dogs survived to 72 hours in protocol (control group, n = 8; Tempol group, n = 7).
Resuscitation
In studies A and B, there were no group differences in extracerebral variables important for cerebral recovery at baseline (Table 1) and 6 hours after resuscitation (Table 2). In both studies, there were no differences in the required number and energy of defibrillating countershocks, in the time needed to achieve restoration of spontaneous circulation, and in bicarbonate requirement. Total epinephrine doses required were significantly higher with Tempol in both studies (Table 3). The total norepinephrine requirements were the same for both groups in study A, but significantly higher in the Tempol group in study B (Table 3). The induced brief hypertension had the same duration, with a MAP greater than 150 mm Hg and the same peak MAP in both groups of both studies, but the beginning of hypertension was significantly delayed in the Tempol groups in both studies (Table 3). Cardiac index varied greatly between dogs, and postarrest values were often doubled compared with baseline values, but there were no differences between groups.
Physiologic variables at baseline
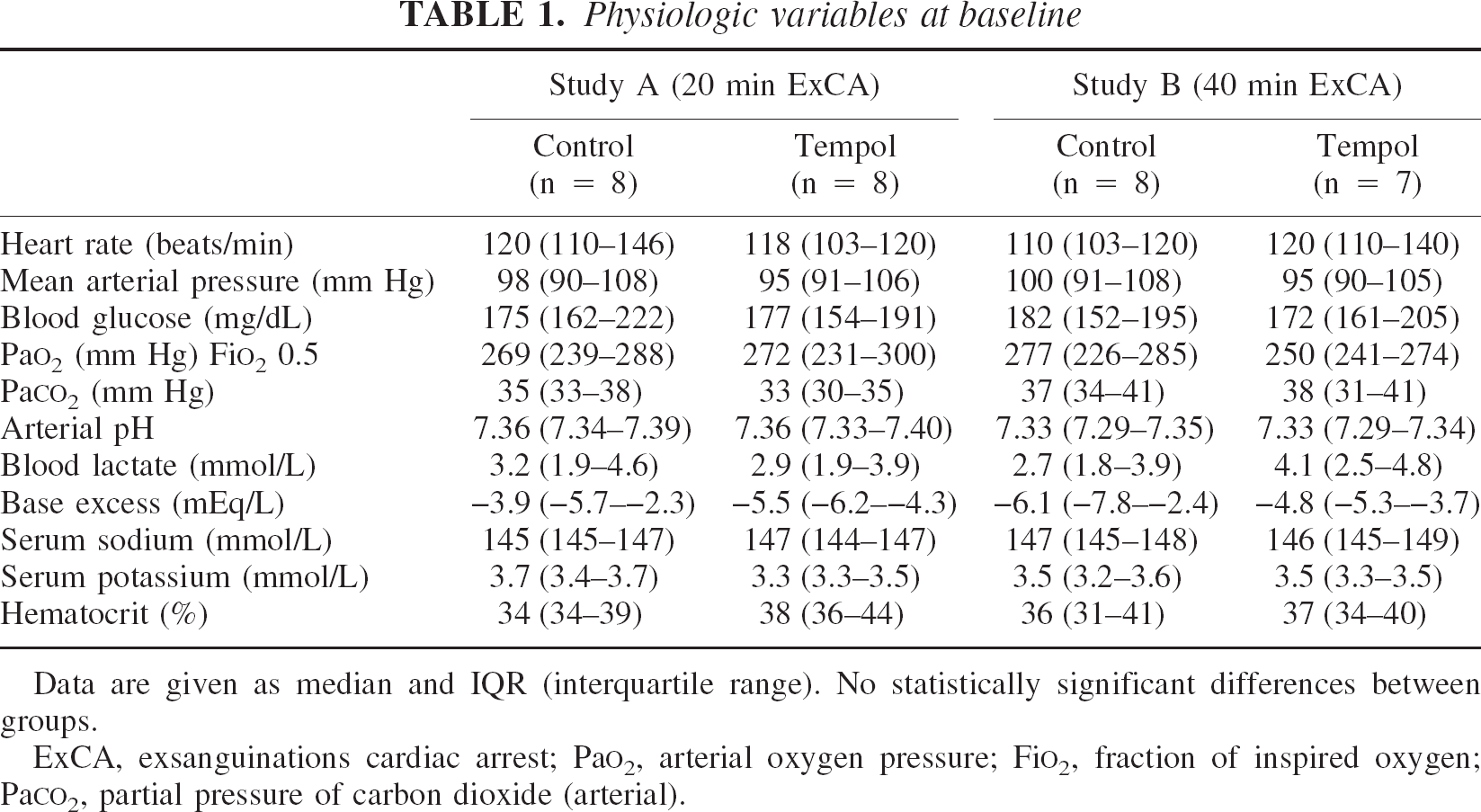
Data are given as median and IQR (interquartile range). No statistically significant differences between groups.
ExCA, exsanguinations cardiac arrest; PaO2, arterial oxygen pressure; FiO2, fraction of inspired oxygen; PaCO2, partial pressure of carbon dioxide (arterial).
Physiologic variables at 6 hours after start of resuscitation
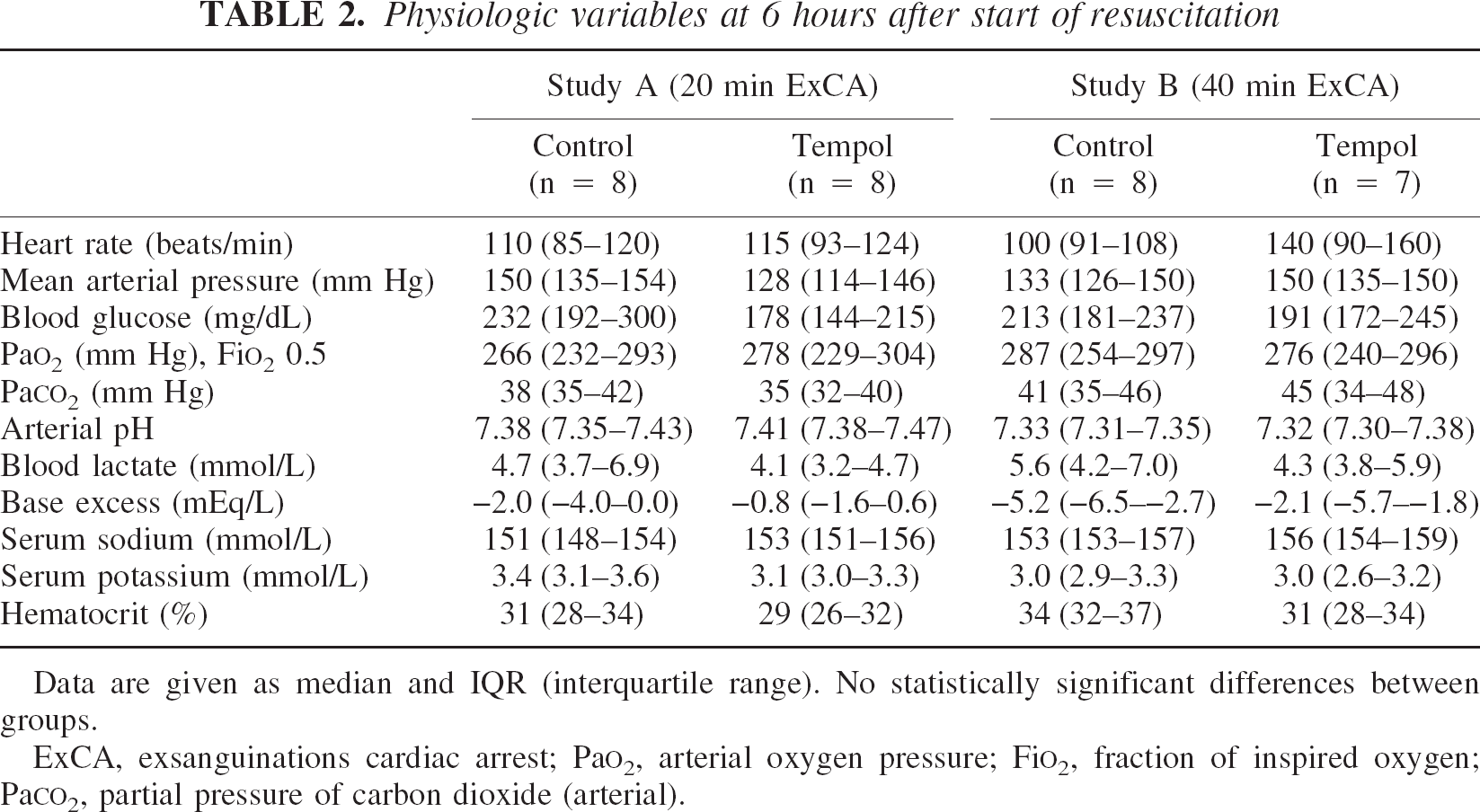
Data are given as median and IQR (interquartile range). No statistically significant differences between groups.
ExCA, exsanguinations cardiac arrest; PaO2, arterial oxygen pressure; FiO2, fraction of inspired oxygen; PaCO2, partial pressure of carbon dioxide (arterial).
Requirements for restoration of spontaneous circulation (ROSC)
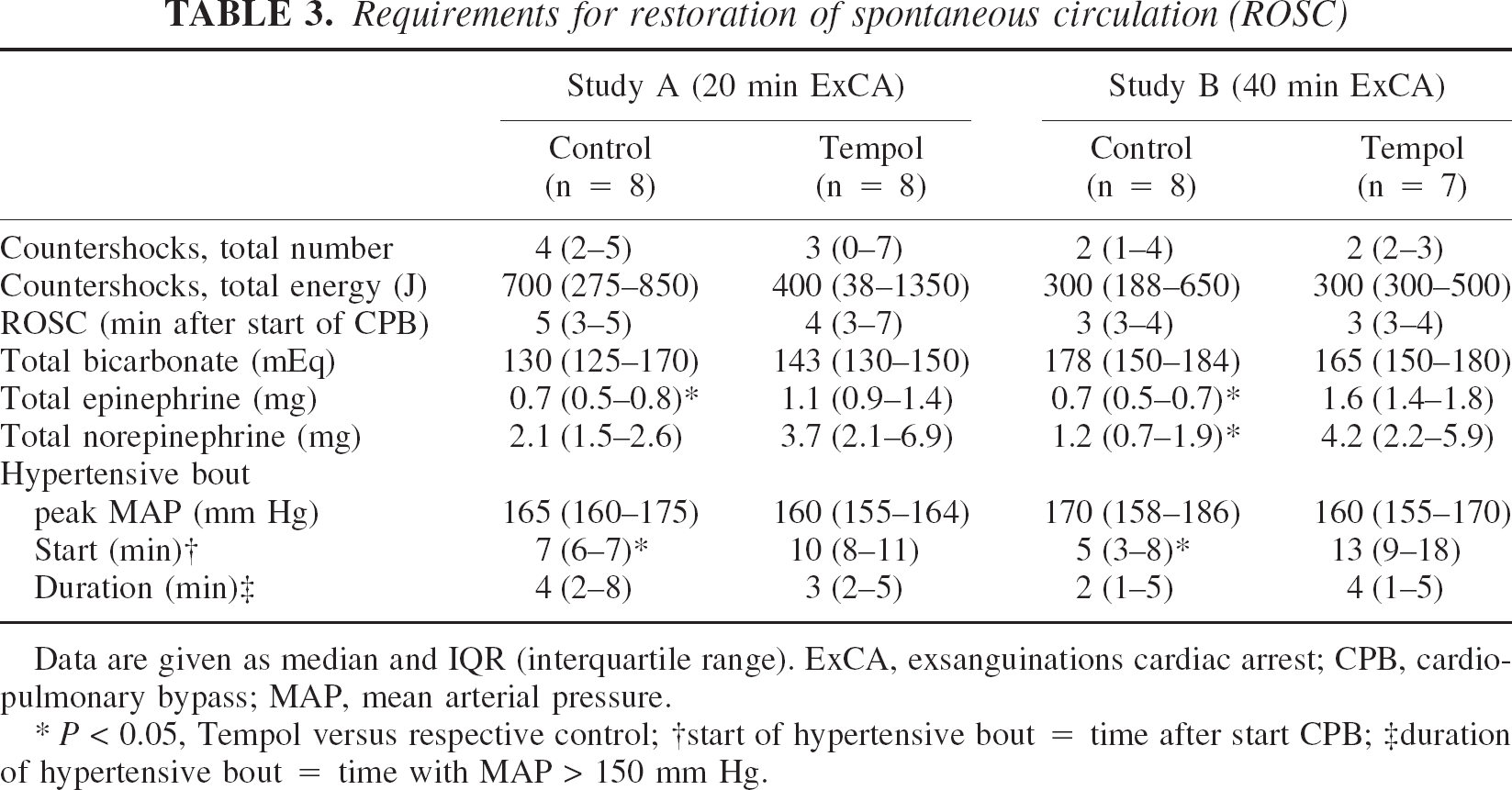
Data are given as median and IQR (interquartile range). ExCA, exsanguinations cardiac arrest; CPB, cardio-pulmonary bypass; MAP, mean arterial pressure.
P < 0.05, Tempol versus respective control
start of hypertensive bout = time after start CPB
duration of hypertensive bout = time with MAP > 150 mm Hg.
Temperatures
Tympanic membrane temperature just before exsanguination was 37.5 ± 0.1°C in all groups (Fig. 1). In study A (Fig. 1–I), the 25 mL/kg saline flush at 24°C decreased tympanic membrane temperature during CA within 2 minutes to a minimum of 35.4 ± 0.4°C in the control group versus 35.4 ± 0.3°C in the Tempol group (P = 0.9). Rectal temperature remained normothermic and did not differ from baseline values during CA. In study B (Fig. 1–II), the 50 mL/kg saline flush at 2°C decreased tympanic membrane temperature during CA within 5 minutes to a minimum of 27.5 ± 1.4°C in the control group versus 28.4 ± 0.6°C in the Tempol group (P = 0.1). Rectal temperature decreased from 37.8 ± 0.2°C to 37.2 ± 0.5°C (P = 0.03) in the control group and from 37.7 ± 0.2°C to 37.0 ± 0.7°C (P = 0.02) in the Tempol group during CA.
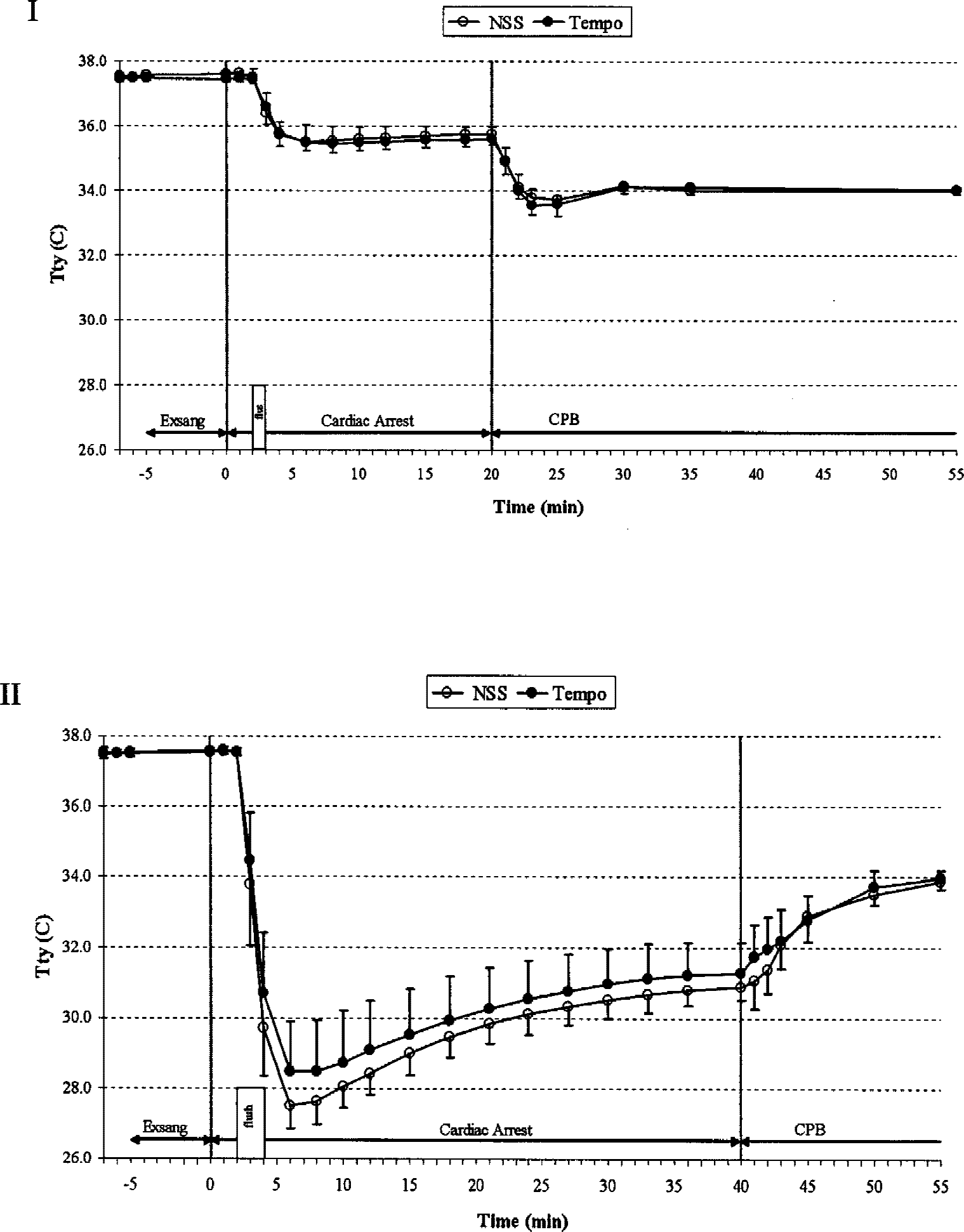
Tympanic membrane temperatures during exsanguination cardiac arrest of 20 (study A, I) and 40 minutes no-flow (study B, II). Resuscitation with cardiopulmonary bypass. Aortic flush with balloon catheter was into the aortic arch in study A (saline, 25 mL/kg at 24°C) and into the abdominal aorta in study B (saline, 50 mL/kg at 4°C). Temperature areas under the curve during arrest were not different (P = 0.5 in I, and P = 0.2 in II).
Methemoglobinemia
In the first dogs administered Tempol, we observed that the blood in the bypass tubing remained dark colored after passing through the oxygenator. Dogs receiving 150 mg/kg Tempol showed methemoglobinemia peak levels of 2.5% to 4.3% at 30 minutes of reperfusion, whereas those receiving 300 mg/kg Tempol showed 4.5% to 13.4% levels at 1 to 2 hours of reperfusion. Twelve hours after Tempol administration, methemoglobin levels were below 1%.
General outcome evaluation
In both studies, OPC at 72 hours were significantly improved in the Tempol groups compared with the control groups (Table 4). In study A, OPC 1 was achieved in three of four dogs receiving 150 mg/kg Tempol and in two of four dogs receiving 300 mg/kg Tempol. In study B, dogs receiving Tempol listed as OPC 2 showed normal cerebral function, and were rated as OPC 2 because of severe weakness in all extremities resulting in the inability to sit, walk, or stand. Histologic evaluation of the spinal cord in four of these dogs revealed no damage under light microscopy, except in one dog with a small focal area of spongiform changes in the dorsal horn of the gray matter in the thoracic segment.
Outcome in terms of final overall performance categories (OPC 1–5) at 72 hours after exsanguinations cardiac arrest (ExCA)

Each dot represents one dog.
In both studies, final NDS at 72 hours were significantly better in the Tempol groups than in the control groups (Fig. 2). In study A, six of eight dogs in the Tempol group had an NDS below 10% (i.e., were normal). In study B, all seven dogs in the Tempol group had NDS ranging between 24% and 31% (reflecting the weakness in all extremities described previously, with seemingly normal cerebral function and behavior) compared with NDS ranging between 22% and 57% in the eight control dogs.
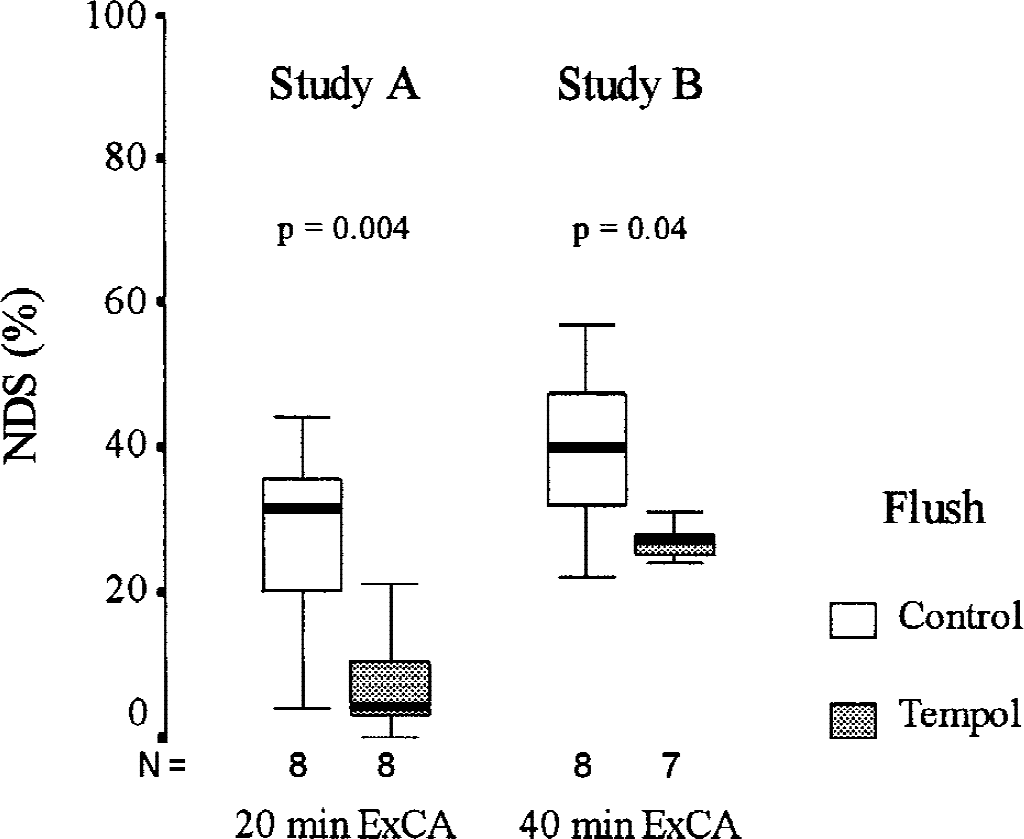
Neurologic deficit scores 72 hours after exsanguination of cardiac arrest of 20 (study A) and 40 minutes (study B). Boxes represent interquartile ranges. The line across each box indicates the median, and the whiskers (⊥T) are the highest and lowest values.
Brain histopathology
Light microscopic scores.
In both studies, there were no differences in total brain HDS between Tempol and control groups at 72 hours (Fig. 3). None of the dogs in either study had a normal brain on light microscopic examination. All HDS were scattered ischemic neuronal scores; scores for macroinfarcts and edema were zero. No microinfarcts, hemorrhages, edema, or lesions were detected in white matter at 72 hours. In study A, regional brain HDS in the temporal cortex, hippocampus, and amygdala were significantly higher in the Tempol group, and HDS in the dentate gyrus were significantly lower in this group (Fig. 4–I). In study B, regional brain HDS in the dentate gyrus was significantly lower in the Tempol group (Fig. 4–II).
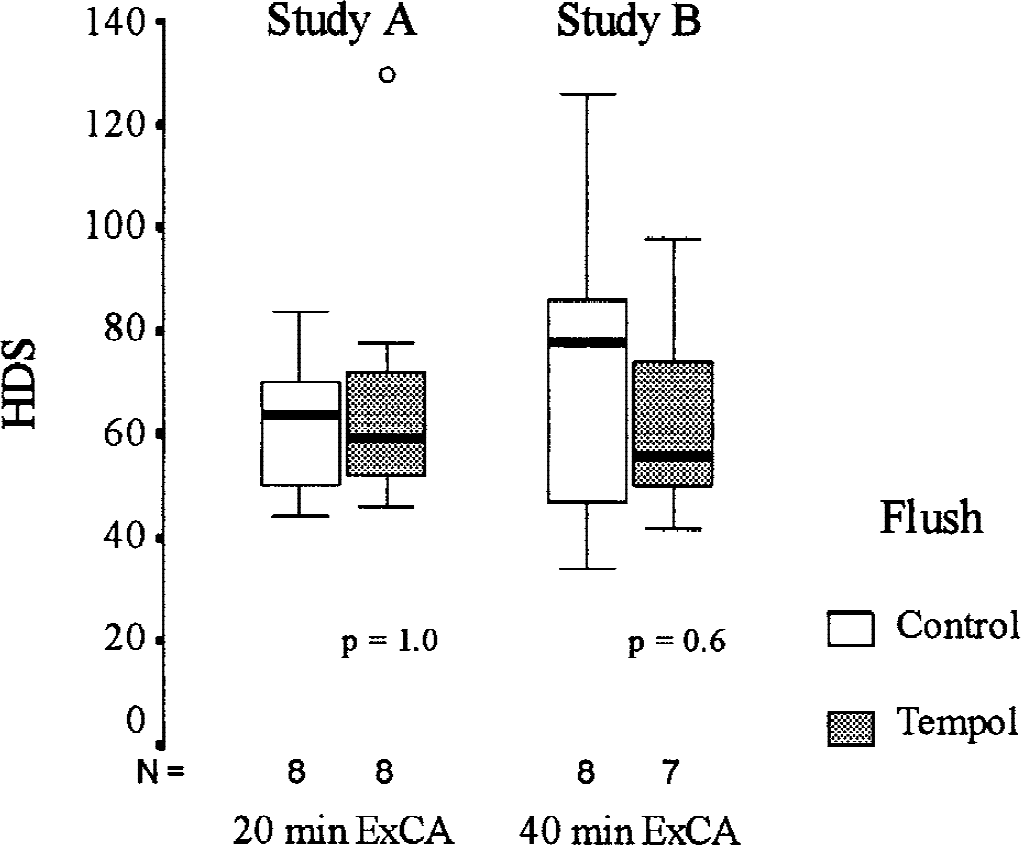
Total brain histologic damage scores with light microscopy 72 hours after exsanguination cardiac arrest of 20 (study A) and 40 minutes (study B). Boxes represent interquartile ranges. The line across each box indicates the median, and the whiskers (⊥T) are the highest and lowest values. The o indicates outliers (values between 1.5–3 box-lengths from the upper or lower edge of the box).
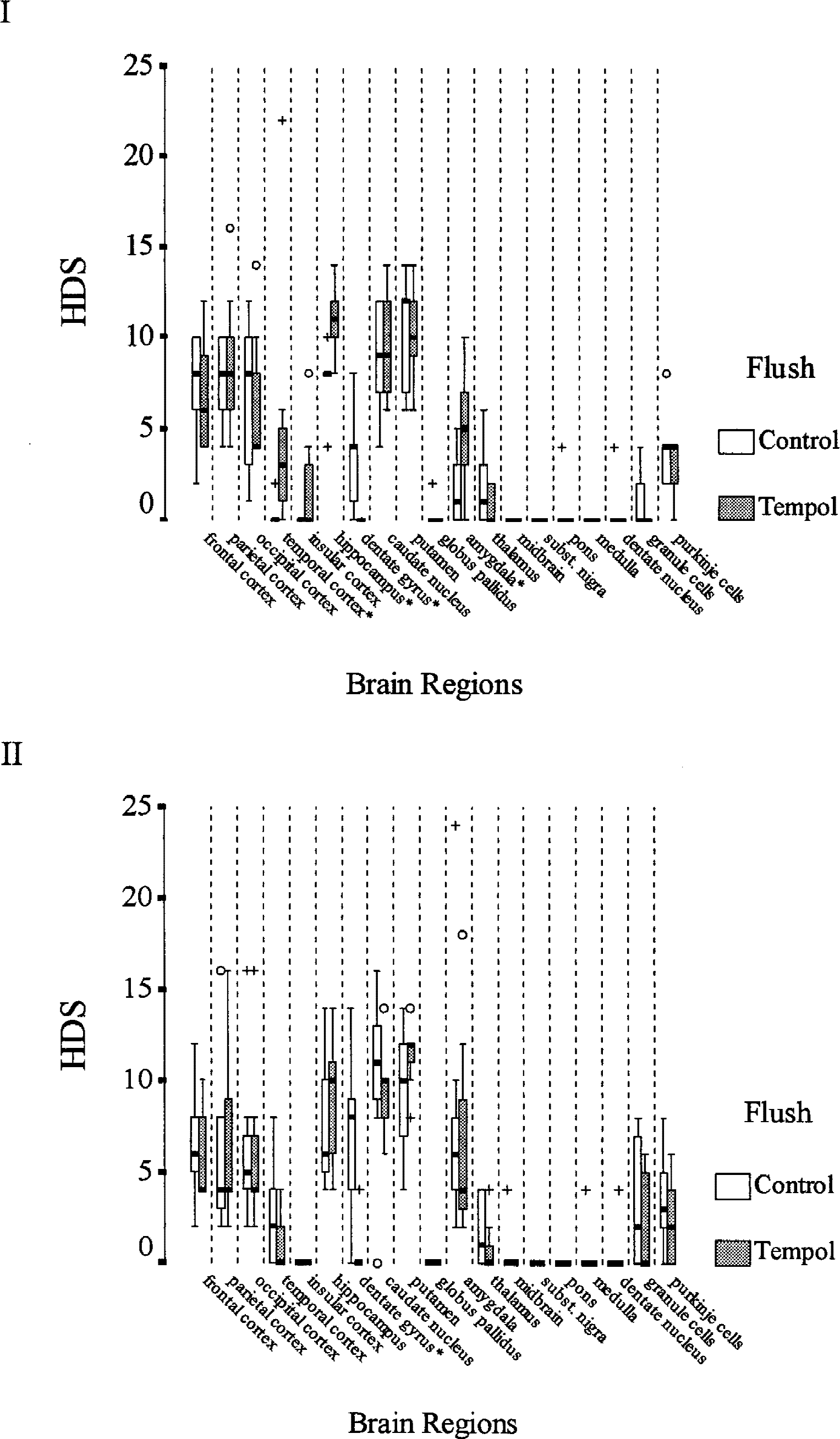
Regional brain histologic damage scores 72 hours after exsanguination of cardiac arrest of 20 (study A, I) and 40 minutes (study B, II). Boxes represent interquartile ranges. The line across each box indicates the median, and the whiskers (⊥T) are the highest and lowest values. The o indicates outliers (values between 1.5–3 box-lengths from the upper or lower edge of the box). The + indicates extremes (values > 3 box-lengths from the upper or lower edge of the box). * P < 0.05.
Nitrotyrosine immunohistochemistry (study B).
Nitrotyrosine brain damage scores at 72 hours were not different between groups [median 3 (IQR, 3–4) in the control group vs, 3 (IQR, 2–4) in the Tempol group, P = 0.2]. Increased nitrotyrosine immunoreactivity in both groups was detected in cells with the morphologic appearance of neurons, glia, and endothelium; in some sections, prominent dendritic labeling was seen, and immunoreactive cells with the morphologic appearance of oligodendrocytes were observed in white matter regions of all slides examined. No staining was observed in slides incubated without primary antibody.
DNA damage (study B).
The DNA damage scores by TUNEL staining showed no difference between groups [median 2 (IQR, 1–4) in the control group vs. 4 (IQR, 2–5) in the Tempol group, P = 0.2]. The PANT stain for single-strand DNA damage was not detected in either group.
Biochemical mechanisms
Ascorbate radicals and Tempol.
In brain tissue homogenates at 72 hours, EPR signals of ascorbate radicals (Fig. 5–I) were detected in all dogs. Because Tempol can be reduced by ascorbate to the EPR-silent Tempol-hydoxylamine (Tempol-OH), we used the oxidant potassium-ferricyanide to convert Tempol-OH to its EPR-detectable radical form Tempol (Fig. 5–II). The Tempol signal was detected in study A in one of three available brains of dogs treated with 150 mg/kg Tempol, in all three available brains of dogs treated with 300 mg/kg Tempol, and in study B in five of seven brains treated with 300 mg/kg Tempol. The addition of potassium-ferricyanide to brain homogenates from dogs that did not receive Tempol produced no EPR signal of Tempol. In study A, the amplitude of the Tempol signal in plasma was highest at 1 minute of reperfusion and was detectable until 8 hours of reperfusion in dogs treated with 150 mg/kg Tempol, and until 36 hours in dogs treated with 300 mg/kg Tempol. The addition of potassium-ferricyanide did not change the amplitude of the signal in measurements at 1 or 30 minutes and 1 hour. The ascorbate radical signal of control dogs showed a threefold increase above baseline at 1 minute of reperfusion, was not detectable at 30 minutes and 1 hour, and slowly returned to baseline values at 72 hours of reperfusion. In study B, EPR signals were measured only at 1 minute and at 3 and 72 hours of reperfusion; the Tempol signal was detectable at 1 minute and at 3 hours, but not at 72 hours of reperfusion, The addition of potassium-ferricyanide increased the amplitude of the Tempol signal 10-fold at 1 minute and 100-fold at 3 hours.
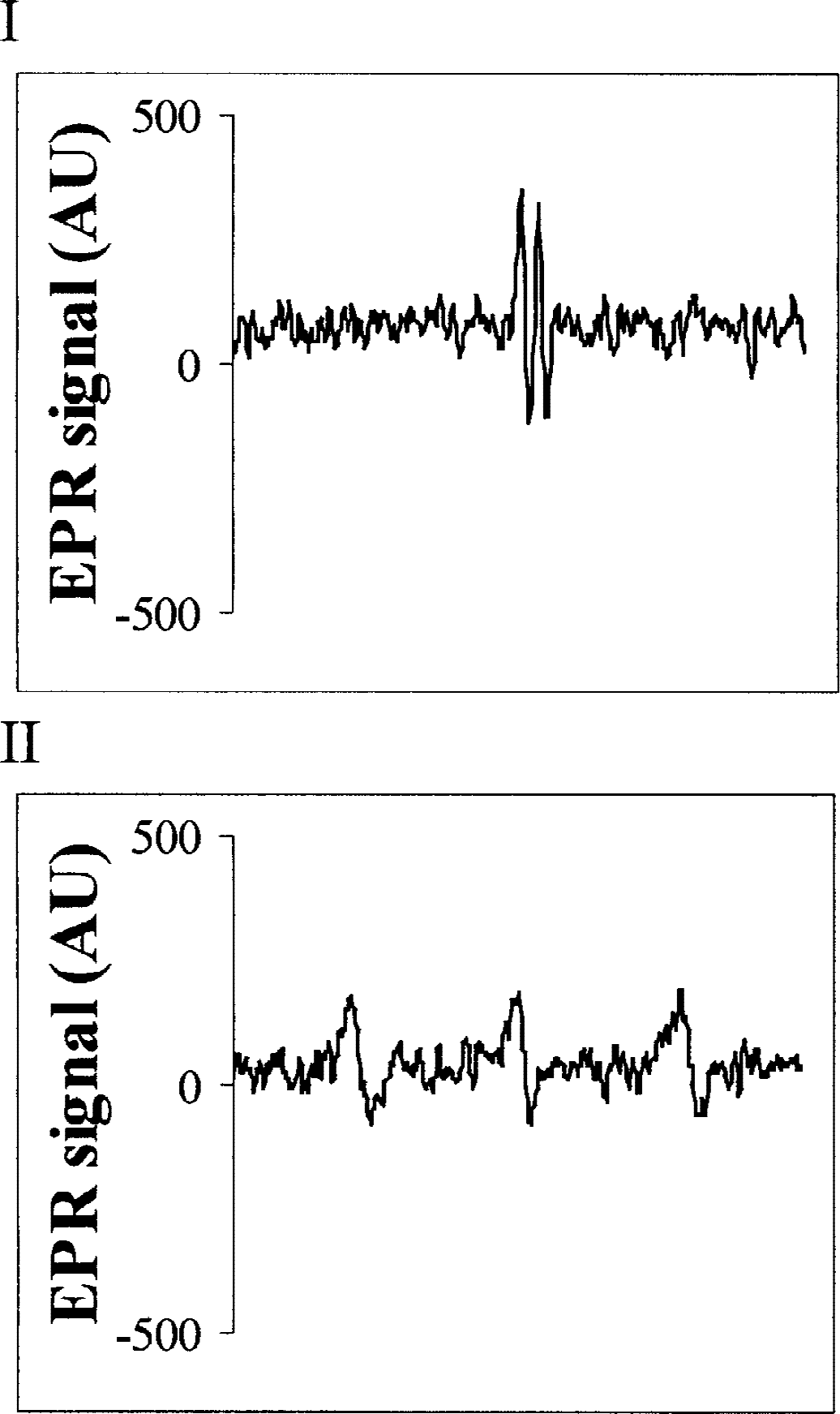
Electron paramagnetic resonance spectroscopy of brain frontal cortex 72 hours after exsanguination cardiac arrest in one dog in study B. I indicates representative typical doublet signal of the ascorbate radical; II, representative typical triplet signal of Tempol after adding the oxidant potassium-ferricyanide to the sample.
Total antioxidant reserve.
The total antioxidant reserve in brain frontal cortex at 72 hours was not different between groups in both studies (Fig. 6–I) and was only numerically lower than in sham experiments without CA.
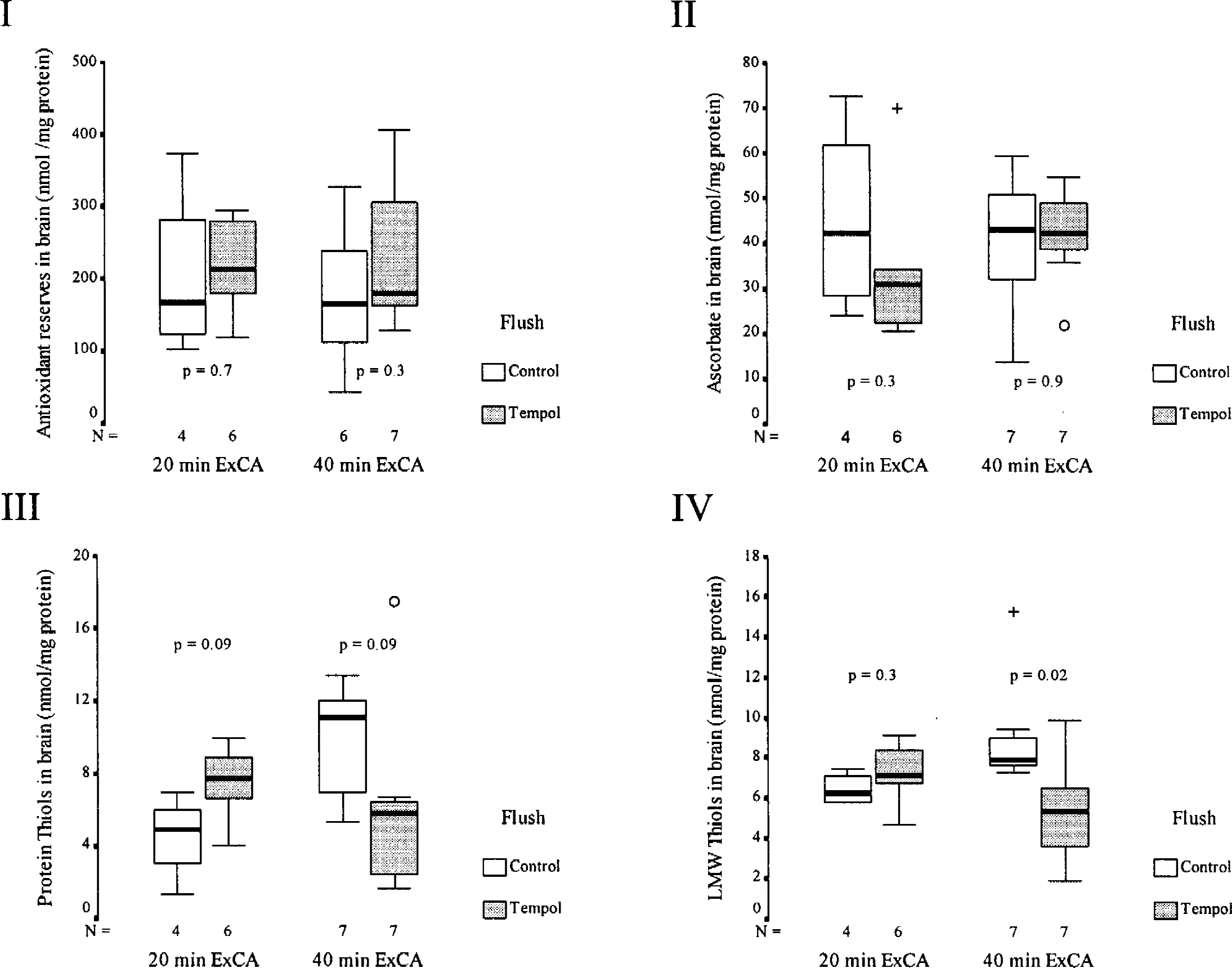
Total antioxidant reserve (I), ascorbate (II), protein thiols (III), and low molecular weight thiols (IV), in brain frontal cortex 72 hours after exsanguination cardiac arrest of 20 (study A) and 40 minutes (study B). Boxes represent interquartile ranges. The line across each box indicates the median, and the whiskers (⊥T) are the highest and lowest values. The o indicates outliers (values between 1.5–3 box-lengths from the upper or lower edge of the box). For reference, in shams (n = 4) without cardiac arrest or Tempol, brain values were 202 ± 60 nmol/mg protein (n4) for antioxidant reserve, 45 ± 11 for ascorbate, 4.5 ± 1.2 for protein thiols, and 6.4 ± 0.4 for low molecular weight thiols.
Ascorbic acid.
Ascorbate levels in brain frontal cortex at 72 hours were not different between groups in both studies (Fig. 6–II) and were similar to that in dogs without CA (Fig. 6).
Thiols.
Protein sulfhydryl content at 72 hours in brain frontal cortex in study A showed a trend toward slightly higher concentrations of protein thiols in the Tempol group, and toward decreased concentration of protein thiols in the Tempol group in study B (Fig. 6–III). Low molecular weight thiols in brain frontal cortex were not different in study A, but were significantly lower in the study B Tempol group (Fig. 6–IV). Values were similar to those in dogs without CA.
DISCUSSION
In this study, Tempol in an aortic flush at the start of prolonged CA improved NDS and OPC after CA compared with saline flush, augmenting the preservation effect of mild hypothermia (tympanic membrane temperature, 35.5°C) for a 20-minute CA and of moderate hypothermia (tympanic membrane temperature, 28°C) for a 40-minute CA. This is the first documentation of mitigation of functional deficit by an antioxidant in a clinically relevant CA outcome model in dogs. However, Tempol did not prevent overall histologic brain damage.
The discrepancy between the beneficial effects of Tempol on functional deficits but not on histologic damage is puzzling. In our previous CA dog studies to 72- to 96-hour outcomes, a normothermic VF-CA no-flow time of 10 minutes followed by normothermic resuscitation resulted in the same total HDS found in this study [i.e., 75 ± 15 (Sterz et al., 1991) or 106 ± 44 (Vaagenes et al., 1984)], but functional outcome (NDS) was invariably poor. One of several possible explanations might be found in a selective preservation of synaptic structure and function by Tempol in neurons not considered doomed in this study (not shrunken and eosinophilic with pyknotic nuclei). Tempol could reduce reactive oxygen species (ROS), which can impair N-methyl- d -aspartate antagonist receptors and various mechanisms necessary for signal transduction between surviving neurons. Recovery of integrative function despite cellular damage is possible (Bothe et al., 1986). Because Tempol reduced histologic damage in both studies in the dentate gyrus, the least vulnerable region of the hippocampal formation, it may be more likely to detect a preservative effect of a treatment in this region. The possibility that longer life support could have resulted in further recovery of control dogs is unlikely, because life support for more than 4 to 7 days did not show improvement beyond the NDS at 48 hours. The final outcome evaluation at 72 hours, though it could not be blinded, was agreed upon by at least two team members to diminish potential observer bias. The detailed components of OPC and NDS called for objective recording.
In this study, we again demonstrated the feasibility of inducing preservative hypothermia during CA within a few minutes, using a cold saline flush through a balloon catheter into the aorta. Our study also shows that Tempol, a water-soluble, inexpensive, commercially available compound that improves cerebral function after CA, deserves additional investigation because it penetrates the blood-brain-barrier (Mitchell et al., 1990). That aspect is important because temporary complete, normothermic global cerebral ischemia up to 30 minutes does not grossly disrupt the blood-brain-barrier (Schleien et al., 1990), as does focal ischemia or brain trauma. Another novel finding is that the beneficial effect of moderate or mild hypothermia, which itself mitigates free-radical reactions (Lei et al., 1994), can still be augmented using a pharmacologic antioxidant. Ideally, cerebral preservation should be induced before anoxic depolarization (e.g., within 3 minutes of CA). From another dog study with 30-minute exsanguination CA (Behringer, 2001, unpublished data) we learned that cold aortic flush with 100 mL/kg saline at 2°C at 2 minutes of CA (as in this study) lead to normal outcome and minimal histologic brain damage. The same flush delayed to 5 minutes of CA lead to normal outcome with moderate histologic brain damage, and delay to 8 minutes lead to poor outcome with severe histologic brain damage. Delayed administration of Tempol may reduce its efficacy as well.
The objectives of suspended animation for temporarily unresuscitable conditions have been discussed elsewhere (Tisherman et al., 1991; Capone et al., 1996; Bellamy et al., 1996; Behringer et al., 2000b) and include (1) helping to save military or civilian trauma victims without severe brain trauma with presently unresuscitable exsanguination CA, (2) helping to save persons experiencing seemingly unresuscitable, nontraumatic sudden cardiac death, and (3) enabling selected elective surgical procedures to be performed that are feasible only during a prolonged state of no blood flow.
Reactive oxygen species seem to be important pathophysiologic mediators of postischemic anoxic encephalopathy. Glutamate release and increases in intracellular calcium activate several free-radical pathways during brain ischemia, which markedly increase during reperfusion, leading to neuronal death. Based on the various sources of free radicals, there exist different mechanisms to inhibit ROS-induced brain damage (Hall, 1997). Indirect-acting antioxidants that inhibit formation of eicosanoids include ibuprofen and indomethacin; xanthine oxidase inhibitors include allopurinol. Enzymatic agents include superoxide dismutase and catalase. Spin-trapping compounds like PBN (n-tert-butyl-α-phenyl-nitrone) scavenge hydroxyl radical; NOS inhibitors, or a peroxy nitrite scavenger reduce the generation or effects of peroxy nitrite. αTocopherol and 21-aminosteroid tirilazad mesylate inhibit lipid peroxidation, and deferoxamine chelates iron. However, intravenous administration of these compounds does not assure adequate delivery to brain tissue because of the blood-brain barrier.
Tempol, a stable nitroxide radical, readily penetrates the blood-brain barrier and enters cells (Mitchell et al., 1990), which should permit the molecule to scavenge both intracellular and extracellular ROS. Nitroxides are universal antioxidants, combining some of the previously mentioned mechanisms, and have direct radical scavenging effects. They act as superoxide dismutase mimetics by removal of O2− · in a catalytic process. Nitroxides are first reduced by O2− · to a hydroxylamine intermediate that can be oxidized by another O2− · to the initial nitroxide, allowing the molecule to act as a self-replenishing antioxidant (Samuni et al., 1990). Nitroxides oxidize Fe2+ to preempt the Fenton reaction (Mitchell et al., 1990).
The mechanisms of Tempol effects are not thoroughly explored. In study B, immunostaining for nitrotyrosine was used to determine if Tempol administration prevented peroxynitrite-mediated nitrosylation of proteins (Cuzzocrea et al., 2000). We did not find a difference in immunostaining for nitrotyrosine between controls and Tempol-treated dogs. In our model, Tempol may not influence the peroxynitrite pathway. We used TUNEL and PANT staining to elucidate the effect of Tempol on downstream aspects of the cascade of ischemic cell death. Nitroxides were shown to prevent free-radical–induced DNA oxidative damage in vitro (Damiani et al., 2000). In our study, TUNEL staining revealed double-strand DNA damage in both groups at 72 hours, without group differences. The lack of protection against double-strand DNA damage by Tempol was unanticipated because free-radical damage to the mitochondria is known to be a major initiator of apoptotic pathways (Chan, 2001). One possible explanation is that enzymatic rather than oxidative cleavage is the major source of DNA damage in our model. TUNEL staining, however, is not specific for apoptosis. It is consistent with, but not confirmative of, apoptosis. We did not detect any PANT staining at 72 hours in controls or in the Tempol group. Most likely, 72 hours after the insult is too late to detect single-strand DNA damage. At this point, cells have recovered from the insult, or damage to the cell structure is too severe to be detected by PANT staining.
The EPR Tempol signal in the brain was detected at 72 hours in almost all dogs treated with 300 mg/kg Tempol. This finding is consistent with a sustained effect of the drug, even when given only with the flush at the start of CA. The EPR Tempol signal in plasma was highest at the start of reperfusion and was detectable for up to 36 hours. This finding shows that Tempol was delivered and present in the brain. After 40-minute but not 20-minute CA, the addition of the oxidant potassium-ferricyanide to plasma samples early at reperfusion increased the Tempol signal, probably because reducing equivalents (in part O2− ·), which reduce Tempol to the EPR-silent Tempol-OH, accumulate more during 40-minute CA. The addition of potassium-ferricyanide converts Tempol-OH back to its EPR-detectable radical form Tempol.
We assume that ischemia-reperfusion causes a free-radical attack in the brain that consumes the antioxidant reserve. Only total antioxidant reserve in brains of four dogs without CA were slightly higher, whereas thiols and ascorbate levels were similar. Ascorbate is the primary water-soluble antioxidant in the brain, where the EPR detectable ascorbate radical intermediate reflects its interactions with oxidative stress-inducing free-radical species. We would anticipate detecting a greater antioxidant reserve in dog brains treated with Tempol. When ROS are generated in the brain, these radicals will be likely scavenged by Tempol and Tempol-OH, thereby protecting other targets from oxidative damage. Total antioxidant reserve in the brain frontal cortex 72 hours after CA was not different between groups (Fig. 6–I). As a recycling antioxidant, Tempol is reduced to Tempol-OH at the expense of other intracellular reductants, such as ascorbate and thiols. One can easily envision a cascade in which the radical form of Tempol is reduced to Tempol-OH at the expense of ascorbate oxidation. Thiols, in turn, can recycle ascorbate from its oxidation products at the expense of their own oxidation. This idea is in line with our observation of unchanged ascorbate levels (Fig. 6–II) and decreased levels of thiols (Figs. 6–III and 6–IV) in study B dogs treated with Tempol. Consumption of thiols yields increased amounts of Tempol-OH, which is a strong electron-donating antioxidant. The finding that the total antioxidant reserve in brain at 72 hours did not differ between groups suggests that Tempol preserves other antioxidants, and is finally recycled at the expense of thiols. Obviously, thiols are not the major components of the total antioxidant reserve in the brain because the decrease in thiols was not reflected in a decrease in total antioxidant reserve. Measurements of biochemical markers of the oxidative pathways only at 72 hours after CA limit the conclusion. Measurements at different times would be desirable, but were not feasible in this outcome-oriented study.
Antioxidants are expected to be beneficial when administered during reperfusion. In two pilot experiments with the same model, we added 100 and 150 mg/kg Tempol to the flush at start of CA and 50 and 150 mg/kg added during reperfusion in the two dogs, respectively. With these preservative doses, which are lower than those in the present study, both dogs remained as severely brain damaged, as did the controls. This finding suggests that reoxygenation injury triggers occur during ischemia. Indeed, there is an increase in free-radical formation during ischemia (Nelson et al., 1992). In rats with hemorrhagic shock, Tempol given early but not late improved outcome (Kentner et al., 2000). Although mild or moderate hypothermia in itself is beneficial through synergism of multiple mechanisms, adding Tempol for an antioxidant effect apparently adds to that of hypothermia. Further improvement in preservation potency might necessitate broader combination treatments.
The introduction of Tempol to clinical resuscitation medicine might be problematic. Tempol (but not Tempol-OH) can oxidize the iron moiety of hemoglobin, which can lead to the methemoglobinemia seen in this study. Clinically, the maximal values of 4.5% to 13.4% methemoglobin seen at 1 to 2 hours of reperfusion (after 300 mg/kg Tempol) causes only minor symptoms (Wright et al., 1999). At 12 hours, methemoglobin levels were below 1% (presumably harmless). An alternative approach would be to use Tempol-OH instead of Tempol because our results suggest that the reducing environment during and after CA favors accumulation of Tempol-OH. Recovery of Tempol from Tempol-OH is achieved after adding the oxidant potassium-ferricyanide.
In a swine model, Tempol produced dose-related hypotension accompanied by reflex tachycardia, agitation, somnolence, and seizures (Hahn et al., 1999). In our model, the mild hypotensive effect of Tempol was easily manageable with higher doses of epinephrine to restore spontaneous circulation, and norepinephrine after restoration of spontaneous circulation to maintain normotension (Table 2). We did not observe agitation, somnolence, or seizure activity in the dogs treated with Tempol after anesthesia was discontinued after 24 to 72 hours.
We conclude that in dogs, the antioxidant Tempol enhances the beneficial effect of mild or moderate cerebral hypothermia on functional outcome when induced at the start of prolonged CA by aortic flush. Tempol in the doses used seems to produce no major complications. The mechanism of the beneficial effect on neurologic dysfunction without the mitigation of histologic damage is complex and needs further clarification. Future research exploring optimal preservative and resuscitative strategies for CA should include investigation of Tempol and other brain-penetrating nitroxides.
Footnotes
Acknowledgments:
The authors thank Drs. Lyn Yaffe, Carleton Hsia, and Larry W. Jenkins for their valuable suggestions, Keri Janesko and Paula Nathaniel for preparing tissue slides for histologic evaluation, Sherman Culver, Nikolas Dedousis, Yuichi Sakai, William Stezoski, and Jason Stezoski for helping with intensive care unit life support, and Patricia Boyle for helping with the editing. The Cardeon Corporation provided the flush catheter.