
Abstract
Serine proteases, such as thrombin and tissue-type plasminogen activator, play an important role in brain injury after intracerebral hemorrhage and other neurologic disorders. Plasminogen activator inhibitor-1 is one of the serine protease inhibitors, or serpins. The balance between serine proteases and serpins may affect the outcome of intracerebral hemorrhage. The purpose of this study was to determine whether plasminogen activator inhibitor-1 and tissue-type plasminogen activator are upregulated after intracerebral hemorrhage and the role that thrombin plays in that induction. Plasminogen activator inhibitor-1 protein levels were upregulated after intracerebral hemorrhage. Brain plasminogen activator inhibitor-1 content also increased after thrombin infusion in a dose-dependent manner. Hirudin, a specific thrombin inhibitor, blocked the upregulation of plasminogen activator inhibitor-1 after intracerebral hemorrhage. Time courses showed that plasminogen activator inhibitor-1 levels around the hematoma peaked at the first day. Plasminogen activator inhibitor-1–positive cells were detected in the perihematomal area and the ipsilateral basal ganglia after thrombin infusion, but not in the contralateral hemisphere. Plasminogen activator inhibitor-1 messenger RNA levels were increased at 24 hours after intracerebral hemorrhage and after thrombin infusion. However, tissue-type plasminogen activator protein levels were the same in the control, whole-blood, and thrombin-infusion groups. In conclusion, intracerebral hemorrhage and thrombin infusion stimulate plasminogen activator inhibitor-1 but not tissue-type plasminogen activator production in the brain. The upregulation of plasminogen activator inhibitor-1 may be neuroprotective by limiting thrombin or other serine protease-induced toxicity.
Keywords
Thrombin, a serine protease, plays an important role in brain injury after intracerebral hemorrhage (ICH) (Lee et al., 1996a, 1996b, 1997b; Xi et al., 1998b). Direct infusion of thrombin into brain parenchyma causes inflammation, edema, reactive gliosis, and scar formation (Lee et al., 1995, 1996b; Nishino et al., 1993). In addition, thrombin induces apoptosis in cultured neurons and astrocytes (Donovan et al., 1997).
The effects of thrombin are modulated by serine protease inhibitors (serpins) and other thrombin inhibitors, such as thrombomodulin. Serpins are a superfamily of proteins including antithrombin III, α1-protease inhibitor, plasminogen activator inhibitor-1 (PAI-1), protease nexin-1, neuroserpin, and colligin. However, only protease nexin-1, PAI-1 and neuroserpin are present in the normal brain. Plasminogen activator inhibitor-1 can inhibit thrombin activity in the presence of vitronectin (Seiffert et al., 1996). In addition, PAI-1 inhibits factor Xa (Urano et al., 1996), which may reduce thrombin formation. Plasminogen activator inhibitor-1 also can inhibit the activity of tissue-type plasminogen activator (tPA) effectively. Tissue-type plasminogen activator, a serine protease, mediates neuronal degeneration, activates microglia, potentiates thrombin-induced brain edema, and exacerbates hemoglobin-induced neurotoxicity (Figueroa et al., 1998; Rogove et al., 1999; Tsirka et al., 1995, 1996; Wang et al., 1999). Alterations in the concentrations of serpins affect brain injury; for example, infarct volume after cerebral ischemia is increased in PAI-1 knockout mice (Nagai et al., 1999).
Our previous studies have shown that previous intracerebral infusion of a low dose of thrombin (thrombin preconditioning) reduces brain injury caused by a subsequent intracerebral infusion of a high dose thrombin or cerebral ischemia (Masada et al., 2000; Xi et al., 1999). Although the mechanisms of thrombin-induced brain tolerance to hemorrhagic and ischemic stroke are not known, upregulation of serpins in the brain may be associated with the induced tolerance. Therefore, the present study examined whether PAI-1 is upregulated after intracerebral hemorrhage (ICH) or an intracerebral infusion of thrombin. We also examined whether tPA is upregulated after ICH or thrombin injection, another potential regulator of PAI-1.
MATERIALS AND METHODS
Animal preparation and intracerebral infusion
Animal study protocols were approved by the University of Michigan Committee on the Use and Care of Animals. A total of 143 male Sprague-Dawley rats (Charles River Laboratories, Portage, MI, U.S.A.) weighing 300 to 400 g were used in this study. Rats were anesthetized with intraperitoneal pentobarbital (40 mg/kg) and the right femoral artery was catheterized for blood pressure monitoring and blood sampling. Arterial blood was obtained for analysis of blood pH, blood gas, hematocrit, and blood glucose. The rectal temperature was maintained at 37.5°C using a feedback-controlled heating pad. Autologous whole blood, thrombin, or saline was infused stereotactically into the right caudate nucleus of rats at a rate of 10μL/minute through a 26-gauge needle (coordinates, 0.2 mm anterior, 5.5 mm ventral, and 3.5 mm lateral to the bregma) using a microinfusion pump (World Precision Instruments, Sarasota, FL, U.S.A.). The needle was removed and the skin incision was closed with suture after infusion.
Experimental groups
Part 1.
The study comprised six parts. In the first part, there were two sets of experiments. We measured PAI-1 messenger RNA (mRNA) using reverse transcription and polymerase chain reaction analysis. In the first set, 100 μL saline or autologous whole-blood infusion were administered to each of three rats. In the second set, four rats received an infusion of 50 μL saline, 1 U thrombin, or 5 U thrombin each. All rats were killed 24 hours later and the brains were sampled for PAI-1 mRNA measurement.
Part 2.
There were three sets of experiments in this part. In the first set, rats received either 100 μL autologous whole-blood (n = 5) or saline (n = 5). In the second set, rats received either 100 μL autologous whole blood (n = 5), blood plus hirudin (2.5 U, n = 6), or blood plus hirudin (5 U, n = 6). In the first two sets, rats were killed at 24 hours for PAI-1 quantitation. In the third set, rats received an infusion of 100 μL autologous whole blood and were killed at day 1 (n = 5), 3 (n = 5) or 7 (n = 5) for PAI-1 measurement. Plasminogen activator inhibitor-1 protein levels were quantitated by enzyme-linked immunosorbent assay (ELISA).
Part 3.
This part was divided into two sets. In the first set, rats received an intracerebral infusion of either 50 μL saline (n = 5), 1 U thrombin in 50 μL saline (n = 5), or 5 U thrombin in 50 μL saline (n = 5). The rats were killed at 24 hours for PAI-1 ELISA measurement. In the second set, rats received 1 U thrombin infusion and were killed at 4 hours (n = 5), 1 day (n = 5), 3 days (n = 5), or 7 days (n = 4) for PAI-1 quantitation.
Part 4.
Three rats in each group received an intracerebral infusion of either 50 μL saline, 100 μL saline, 1 U thrombin, 5 U thrombin, or 100 μL whole blood. Rats were killed at 24 hours for analysis of PAI-1 immunohistochemistry.
Part 5.
Four groups of five rats each received an intracerebral infusion of saline, 1 U thrombin, 5 U thrombin, or whole blood. Rats were killed 24 hours later, and tPA protein levels were measured by ELISA.
Part 6.
In this part, six groups of three rats each were used for Western blot analysis. Rats received an infusion of 1 U thrombin or 50 μL saline and were killed 1, 3, or 7 days later.
Reverse transcription and polymerase chain reaction
Animals were reanesthetized with intraperitoneal pentobarbital (60 mg/kg) and killed by decapitation. The brains were removed and a 3-mm thick coronal brain slice was cut with a blade approximately 4 mm from the frontal pole. The ipsilateral and the contralateral basal ganglia were sampled. Total RNA was extracted from the two areas as described previously with Trizol reagent (Gibco BRL Grand Island, NY, U.S.A.). One microgram RNA was digested with amplification-grade deoxyribonuclease I (Gibco BRL). Complimentary DNA was synthesized by reverse transcription, using the digested 1-μg RNA (11 μL) with 14 μL reaction buffer (Perkin Elmer, Foster City, CA, U.S.A.) containing dNTP (dATP, dCTP, dGTP, and dTTP), 25 mmol/L magnesium chloride, 10x polymerase chain reaction buffer II, Random Hexamer Primer, ribonuclease inhibitor, and murine leukemia virus reverse transcriptase. The reaction was performed at 42°C for 30 minutes and terminated at 99°C after 5 minutes. Diethylpyrocarbonate water (75 μL) was added to dilute the complimentary DNA to 100 μL and stored at −20°C for later use. Polymerase chain reaction was performed with 15 μL reverse transcriptase reaction mixture (Perkin Elmer) containing 25 mmol/L magnesium chloride, dNTP, 10x polymerase chain reaction buffer II, and AmpliTaq DNA polymerase in a final volume of 50 μL. The rat PAI-1 primers (NIH GenBank database) corresponded to nucleotides 438 to 455 (sense primer, 5′-GGTCATGGAACAAGAATG-3′) and 1203 to 1221 (antisense primer, 5′-GCTGAGACTAGAATGGCTG-3′). Rat GAPDH primers (5′-CTCAGTGTAGCCCAGGATGC-3′, 5′-ACCACCATGGAGAAGGCTGG-3′) were used to amplify GAPDH mRNA, a housekeeping gene used as a control. Amplification was performed in a DNA thermal cycler (MJ Research, Watertown, MA, U.S.A.). Samples were subjected to 40 cycles (94°C, 1 minute; 57°C, 1.5 minutes; and 72°C, 2 minutes). Polymerase chain reaction production was analyzed by electrophoresis on 1% agarose gel. Gels were observed with ethidium bromide staining and ultraviolet transillumination. Photographs were taken with black and white film (Polaroid Corp, Cambridge, MA, U.S.A.). The relative densities of the bands were analyzed with the NIH Image method (Version 1.61, Bethesda, MD, U.S.A.).
Enzyme-linked immunosorbent assay
Rats were reanesthetized with intraperitoneal pentobarbital (60 mg/kg) and killed at different times. The brain was removed and sampled as described previously. Weighed tissue was immediately snap-frozen in liquid nitrogen and transferred to a −80°C freezer for later use. Specimens were subsequently thawed and sonicated with 37.5 mmol/L Tris-hydrochloride, 0.75 mmol/L ethylenediaminetetraacetic acid, 75 mmol/L sodium chloride, and 15 mmol/L lysin (pH 7.4). The weight/volume was 40%. The homogenate was centrifuged at 4°C for 20 minutes at 7,000 g and the supernatants were used for ELISA. Quantification of antigen was immunologically measured with an ELISA kit (Biopool, Umea, Sweden). Data were expressed as nanogram per gram brain tissue.
Immunohistochemistry
Immunohistochemistry was performed as previously described (Xi et al., 1999). Briefly, rats were reanesthetized with intraperitoneal pentobarbital (60 mg/kg) and underwent intracardiac perfusion with 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (pH 7.4). The brains were removed and kept in 4% paraformaldehyde for 6 hours, then were immersed in 25% sucrose for 3 to 4 days at 4°C. Specimens were then placed in embedding compound and sectioned (18-μm slices) on a cryostat. Sections were incubated using avidin-biotin complex technique. Polyclonal rabbit antirat PAI-1 antibody (1:400 dilution; American Diagnostica, Greenwich, CT, U.S.A.) was used as the primary antibody. Normal rabbit immunoglobulin G was used as a negative control.
Western blot analysis
Rats were anesthetized and killed at different times for Western blot analysis (Xi et al., 1999). Rats underwent intracardiac perfusion with saline, and brain tissues were sampled as described previously. Brain samples were sonicated with Western blot lysis buffer. Protein concentration was determined with a Bio-Rad protein assay kit. Protein (50 μg) from each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a hybond-C–pure nitrocellulose membrane (Amersham). Membranes were probed with a 1:2500 dilution of the primary antibody and a 1:2000 dilution of the secondary antibody (peroxidase-conjugated goat antirabbit antibody). Rat PAI-1 was used as a positive control. The antigen-antibody complexes were observed with the ECL chemiluminescence system (Amersham) and exposed to Kodak (Rochester, NY, U.S.A.) X-OMAT film. The relative densities of bands were analyzed with the NIH Image program (Version 1.61).
Statistical analysis
All data are presented as mean ± SD. Data were analyzed with Student t-test or analysis of variance with a Scheffé F test. Statistical significance was set at P < 0.05.
RESULTS
All physiologic variables were measured before intracerebral infusions. Mean arterial blood pressure, pH, arterial oxygen and carbon dioxide tensions, hematocrit, and blood glucose were controlled within normal range.
Brain plasminogen activator inhibitor-1 messenger RNA
Reverse transcription and polymerase chain reaction analysis followed by scanning densitometry of bands revealed that brain PAI-1 mRNA levels were upregulated in ICH and after thrombin injection. The PAI-1 mRNA levels were increased in the perihematomal area 24 hours after ICH (1274 ± 250 vs. 457 ± 436 pixels in saline control, P < 0.05, Fig. 1), and PAI-1 mRNA expression in the ipsilateral basal ganglia increased 24 hours after intracerebral infusion of 1 U thrombin (Fig. 2). A large dose of thrombin (5 U) also induced PAI-1 expression in the ipsilateral basal ganglia 24 hours after infusion (2541 ± 1391 vs. 360 ± 310 pixels in saline control, P < 0.05). In all contralateral samples, PAI-1 mRNA expression was negative.
Perihematomal plasminogen activator inhibitor-1 messenger RNA levels 24 hours after intracerebral infusion of 100 μL saline (lanes 1–3) or autologous whole blood (lanes 4–6).

Plasminogen activator inhibitor-1 messenger RNA in the ipsilateral basal ganglia 24 hours after intracerebral infusion of 50 μL saline (lanes 1–4) or 1 U thrombin (50 μL, lanes 5–8).
Enzyme-linked immunosorbent assay analysis
Brain PAI-1 protein levels were upregulated in the perihematomal zone 24 hours after ICH (173 ± 60 vs. 82 ± 44 ng/g in saline control, P < 0.05, Fig. 3A), but PAI-1 was almost undetectable in the contralateral basal ganglia of ICH and saline-control samples (Fig. 3A). Hirudin, a specific thrombin inhibitor, blocked the perihematomal upregulation of PAI-1 (Fig. 3B). The time-course study showed that PAI-1 concentrations reached high levels 24 hours after ICH and returned to normal by 7 days (Fig. 3C).
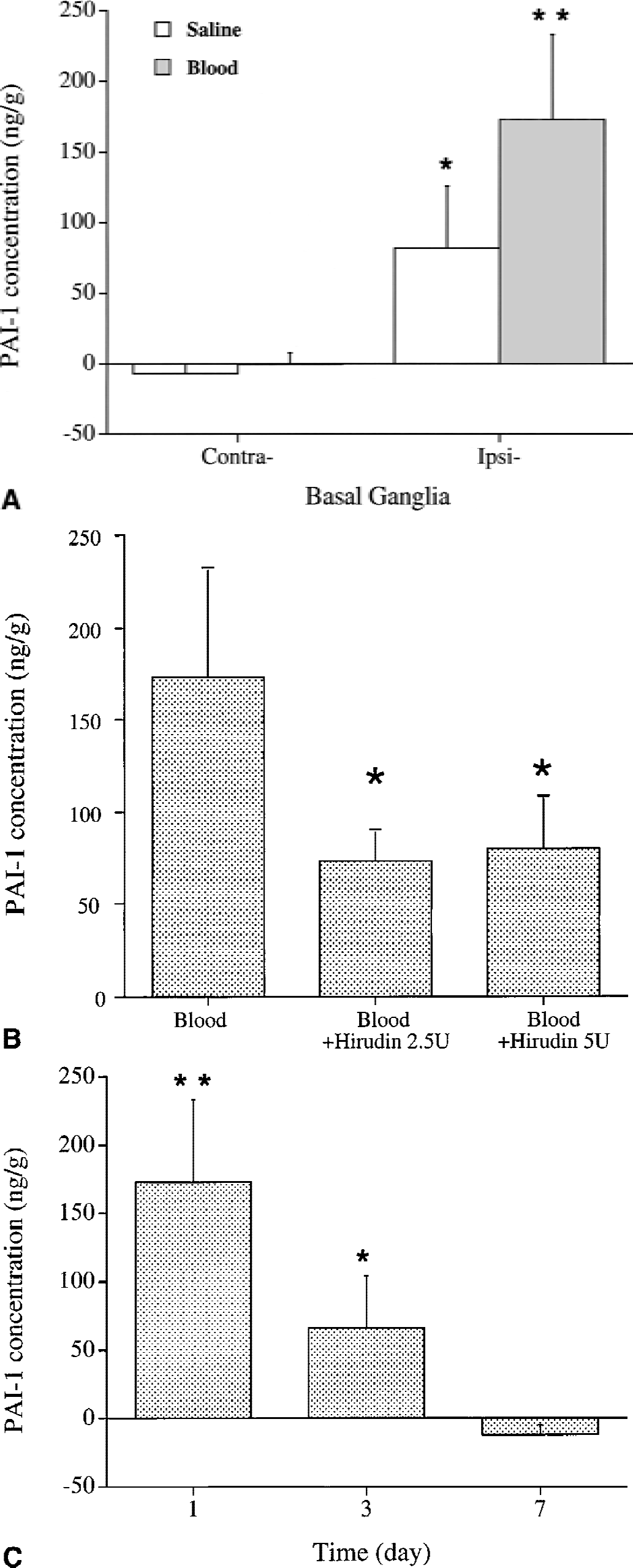
Ipsilateral brain PAI-1 content also increased after thrombin infusion. The upregulation of PAI-1 by thrombin seemed to be dose dependent (Fig. 4). The time-course study showed that PAI-1 levels peaked 3 days after a 1-U thrombin infusion and returned to baseline at 7 days (Fig. 5). However, brain tPA protein level in the ipsilateral basal ganglia did not change 24 hours after ICH or thrombin infusion (Table 1).
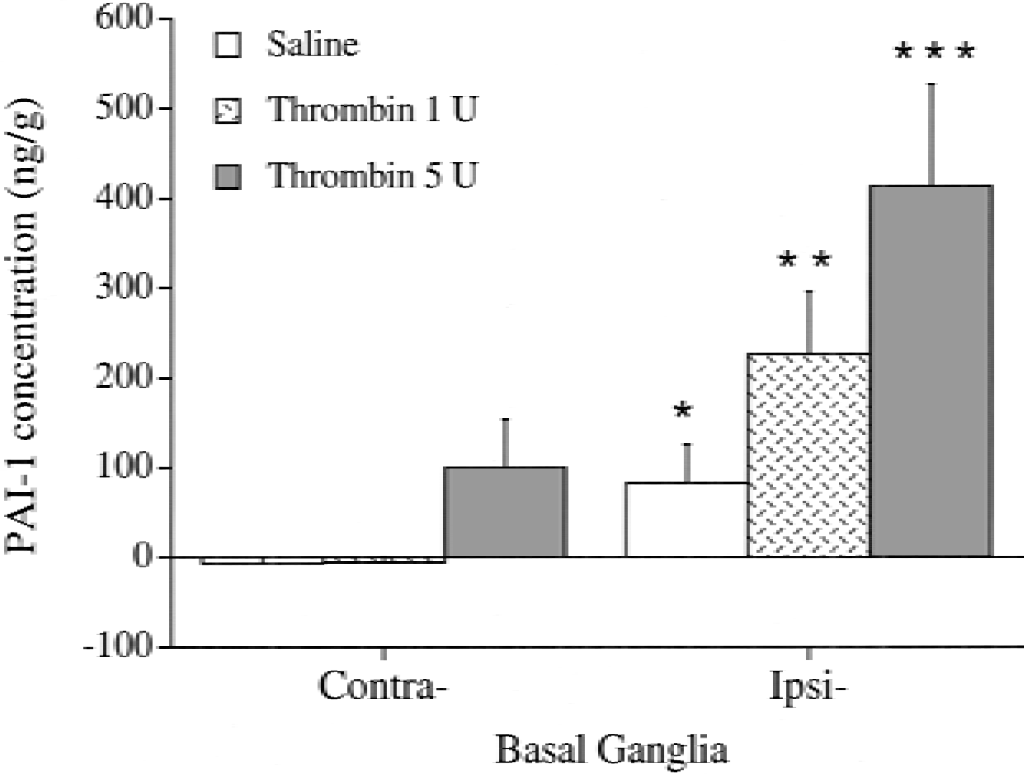
Plasminogen activator inhibitor-1 concentration (ng/g) in the basal ganglia 24 hours after infusion of saline, 1 U thrombin, or 5 U thrombin. Values are mean ± SD (n = 5). * P < 0.05 versus contralateral basal ganglia. ** P < 0.05 versus saline ipsilateral basal ganglia, and P < 0.01 versus contralateral basal ganglia. *** P < 0.01 versus contralateral basal ganglia and saline ipsilateral basal ganglia, and P < 0.05 versus thrombin (1 U).
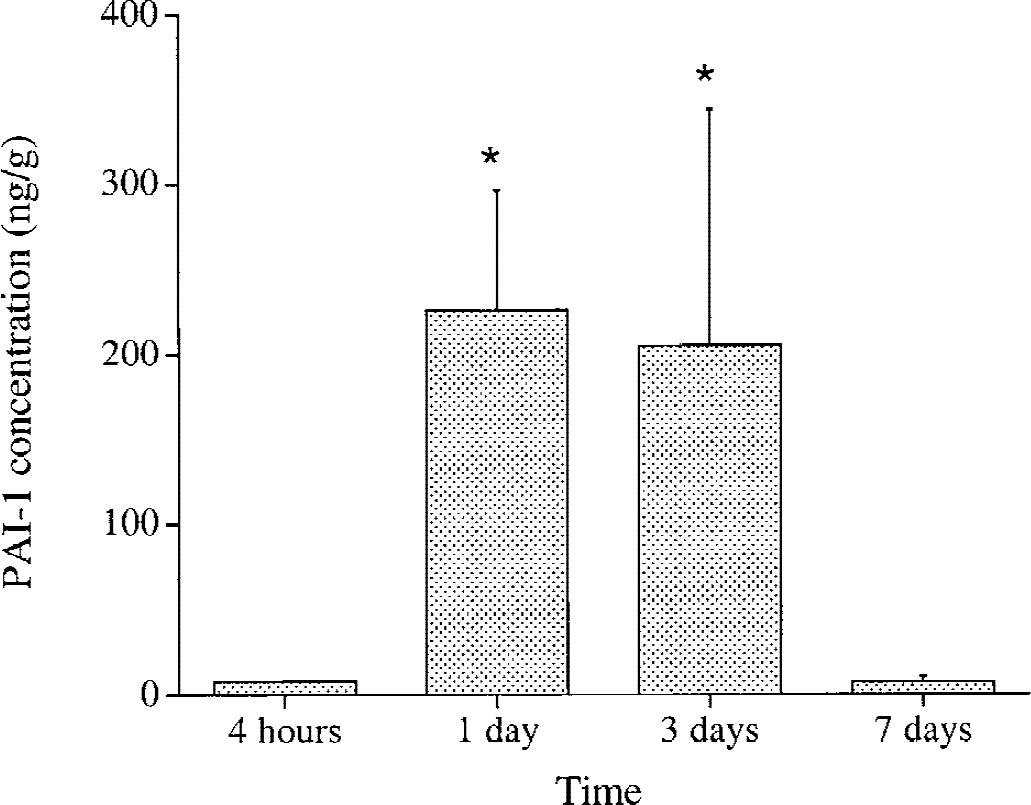
Time course of plasminogen activator inhibitor-1 upregulation in the brain after intracerebral infusion of 1 U thrombin. Values are mean ± SD (n = 5). * P < 0.05 versus 4-hour and 7-day groups.
tPA protein levels in the ipsilateral basal ganglia 24 hours after intracerebral infusion of saline, blood, or thrombin

No significant difference among all groups (n = 5). Mean ± SD.
tPA, tissue-type plasminogen.
Immunohistochemistry
Only a few PAI-1–positive cells were detected in the ipsilateral and contralateral basal ganglia in saline-control animals (Figs. 6A and 6B). The PAI-1 immunoreactivities were detected in the perihematomal area 24 hours after ICH, but not in the contralateral basal ganglia (Figs. 6C and 6D). Twenty-four hours after 1-U thrombin infusion, PAI-1–positive cells were also found in the ipsilateral basal ganglia (Fig. 6F). Most PAI-1–positive cells found in the basal ganglia seemed to be neurons and astrocytes, whereas most PAI-1–positive cells found in the white matter were astrocytes. With larger doses of thrombin (5 U), PAI-1–positive cells were found in the ipsilateral basal ganglia and the ipsilateral cortex (data not shown). Plasminogen activator inhibitor-1–positive cells were not found in the brain sections of negative controls.
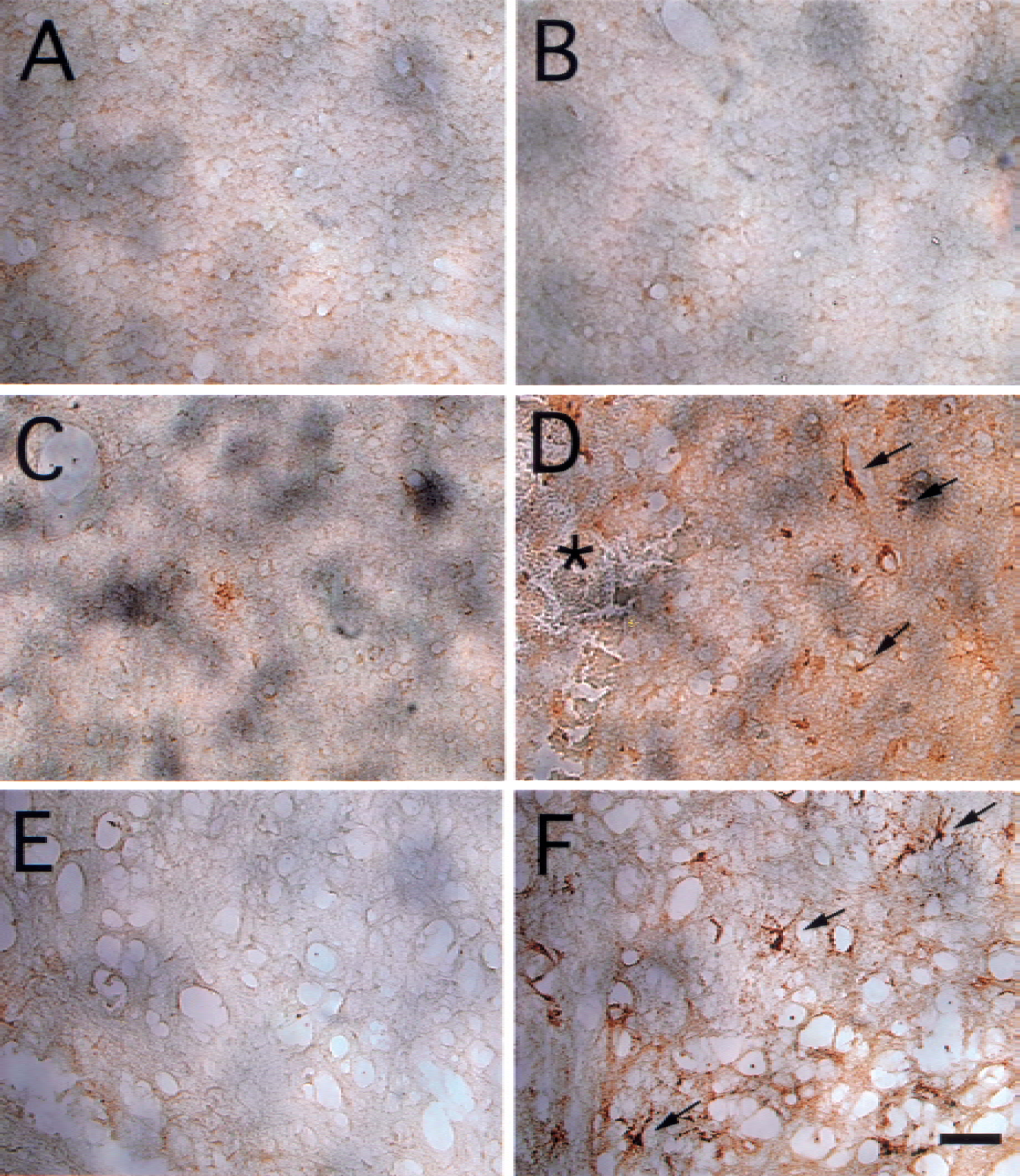
Immunohistochemistry of plasminogen activator inhibitor-1 (PAI-1) in the contralateral
Western blot analysis
Plasminogen activator inhibitor-1 protein in the normal brain was not detectable by Western blot analysis. However, PAI-1 was detected in the brain after the intracerebral infusion of thrombin. The time-course study found that PAI-1 in the ipsilateral basal ganglia peaked at the third day after 1-U thrombin infusion (Fig. 7).
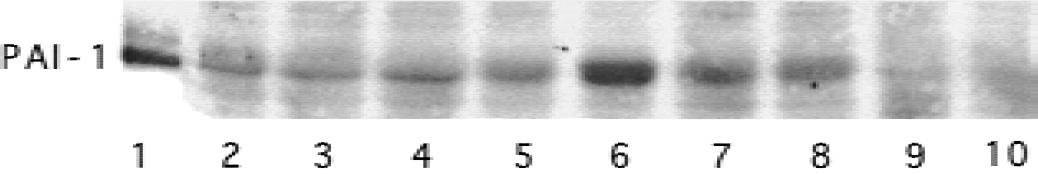
Western blot analysis of plasminogen activator inhibitor-1 (PAI-1) protein levels in the ipsilateral basal ganglia 1 (lanes 2–4), 3 (lanes 5–7) and 7 days (lanes 8–10) after 1-U thrombin infusion. Lane 1 is the PAI-1–positive control.
DISCUSSION
The present study shows that PAI-1 mRNA expression is increased after ICH or an intracerebral infusion of thrombin. The PAI-1 protein levels are also increased in the rat brain after an ICH or thrombin infusion, whereas brain tPA levels remain unchanged. Plasminogen activator inhibitor-1–positive cells were found in the brain around the hematoma and in the ipsilateral basal ganglia after thrombin infusion. These results indicate that PAI-1, a serine proteases inhibitor, modulates thrombin activity in the brain after ICH and, thus, after injury.
Plasminogen activator inhibitor-1 is an inhibitor of tPA and urokinase, but it can also inhibit thrombin effectively in the presence of vitronectin (Keijer et al., 1991; Seiffert et al., 1996). Plasminogen activator inhibitor-1 may also inhibit thrombin production by inhibiting the cleavage of prothrombin to thrombin by activated factor X (Urano et al., 1996); factor X expression is found in rat brain (Shikamoto and Morita, 1999). Ahn et al. (1999) found that PAI-1 mRNA is increased after cerebral ischemia. In addition, PAI-1 may mediate neuroprotective activity against N-methyl- d -aspartate–induced neurotoxicity (Buisson et al., 1998), and ischemic infarct volume is increased in PAI-1 knockout mice (Nagai et al., 1999). In combination with our finding that PAI-1 mRNA and protein are increased in the brain after ICH or thrombin infusion, these results suggest that PAI-1 may be involved in modulating a variety of brain injury forms.
Current evidence suggests that thrombin plays a key role in brain injury after cerebral hemorrhage and ischemia (Gingrich and Traynelis, 2000). A direct infusion of thrombin into rat parenchyma causes brain edema and seizures, and thrombin is linked to brain edema formation after ICH (Lee et al., 1995, 1996a, 1996b, 1997a, 1997b; Xi et al., 1998a, 1998b). In addition, high concentrations of thrombin kill neurons and astrocytes in vitro (Striggow et al., 2000; Vaughan et al., 1995). Weinstein et al. (1998), examining Ad12 HER 10 cells, found that thrombin concentration as low as 1.2 × 10−3 U/mL cause cell death in hypoglycemic (0.1mmol/L glucose) but not normoglycemic conditions, suggesting that ischemic cells may be particularly susceptible to thrombin-induced injury. An intraventricular infusion of hirudin, a specific inhibitor of thrombin, increases the survival of hippocampal CA1 neurons after global cerebral ischemia in gerbils, indicating that thrombin may also contribute to ischemic brain injury (Striggow et al., 2000).
Such evidence implies that endogenous inhibitors of thrombin may limit brain injury, a hypothesis supported by our finding that PAI-1 is upregulated after ICH. Thrombin is produced immediately as blood clots after ICH, but it may also result from blood-brain barrier breakdown, which occurs in many kinds of brain injury, and entry of prothrombin from blood. In addition, the brain and blood may be sites of thrombin production. In vitro studies have shown that prothrombin mRNA is expressed in the cells of the nervous system (Dihanich et al., 1991) and prothrombin mRNA is upregulated after spinal cord injury in vivo (Citron et al., 2000). In our experiments, PAI-1 protein upregulation took several hours. Thus, such upregulation would not be expected to protect the brain from the adverse effects of thrombin generated in the ICH as clotting proceeds. It should be noted, however, that some of the thrombin produced during coagulation may remain within a hematoma associated with fibrin (Hsieh, 1997), and be released slowly into the surrounding brain. Such thrombin, and that produced after blood-brain barrier breakdown or by brain parenchymal cells, may be inhibited by the upregulated PAI-I.
The mechanisms of PAI-1 upregulation after ICH are still unknown. Our results suggest that thrombin plays an important role because an intracerebral infusion of thrombin also upregulated PAI-1, whereas the addition of hirudin to the ICH model blunted PAI-1 upregulation. Other studies also found that thrombin can mediate PAI-1 production in vitro. Thrombin treatment results in upregulation of PAI-1 in endothelial cells, which is blocked by argatroban, a specific thrombin inhibitor (Ueshima et al., 2000). The effect of thrombin on PAI-1 may be mediated by tumor necrosis factor-α. Tumor necrosis factor-α increased endothelial PAI-1 production both in vitro and in vivo, and its levels were increased after ICH (van Hinsbergh et al., 1988; Xi et al., 2001). Furthermore, PAI-1 expression was increased in focal cerebral ischemia, and thrombin may be involved in ischemic brain injury (Ahn et al., 1999; Striggow et al., 2000).
Plasminogen activator inhibitor-1 is also a tPA inhibitor. Tissue-type plasminogen activator mediates neuronal degeneration, activates microglia, potentiates thrombin-induced brain edema, and exacerbates hemoglobin-induced neurotoxicity (Figueroa et al., 1998; Rogove et al., 1999; Tsirka et al., 1995, 1996; Wang et al., 1999). Therefore, we also examined whether an increase in tPA activity might be a signal for PAI-1 upregulation after ICH or thrombin injection, but tPA activity was not upregulated in either case.
The decrease in PAI-1 levels 3 days after ICH and their return to normal by day 7 may be important factor in regulating t-PA levels during clot lysis. In the rat, the hematoma starts to lyse after approximately 3 days (Xi et al., 1998a) and lysis would presumably be impeded if the high levels of PAI-1 found at day 1 were maintained.
Although large doses of thrombin cause brain injury and cell death, an intracerebral infusion of low doses of thrombin induces tolerance in a variety brain injuries. Thus, pretreatment with a low dose of thrombin attenuates the brain edema induced by thrombin or ICH, and reduces the infarct size in a rat middle cerebral artery occlusion model (Masada et al., 2000; Xi et al., 1999). Our present results show that PAI-1 contents were upregulated after thrombin treatment, suggesting that PAI-1 may contribute to thrombin-induced brain tolerance.
In conclusion, both PAI-1 mRNA and protein are increased after ICH. Increases of PAI-1 contents in the brain may result from thrombin production and may be neuroprotective.