
Abstract
Peroxynitrite is responsible for nitration in vivo, whereas myeloperoxidase can also catalyze protein nitration in the presence of high NO2− levels. Recent reports of myeloperoxidase-mediated enzyme inactivation or lipid peroxidation have suggested a role of myeloperoxidase in various pathological conditions. To clarify the role of myeloperoxidase in ischemic brain injury, the authors measured nitrotyrosine formation and infarct volume in myeloperoxidase-deficient or wild-type mice subjected to 2-hour focal cerebral ischemia-reperfusion. Twenty-four hours after reperfusion, infarct volume was significantly larger in myeloperoxidase-deficient mice than in wild-type mice (81 ± 20 mm3 vs. 52 ± 13 mm3, P < 0.01), and nitrotyrosine levels in the infarct region were higher in myeloperoxidase-deficient mice than in wild-type mice (13.4 ± 6.1 μg/mg vs. 9.8 ± 4.4 μg/mg, P = 0.13). Fourteen hours after reperfusion, the nitrotyrosine level was significantly higher in myeloperoxidase-deficient mice than in wild-type mice (3.3 ± 2.9 μg/mg vs. 1.4 ± 0.4 μg/mg, P < 0.05). The authors conclude that the absence of myeloperoxidase increases ischemic neuronal damage in vivo, and that the myeloperoxidase-mediated pathway is not responsible for the nitration reaction in cerebral ischemia-reperfusion.
Keywords
Protein nitration is implicated in the pathogenesis of ischemic neuronal damage (Beckman, 1991; Takizawa et al., 1999; Nakazawa et al., 2000; Choi et al., 2000), but its cellular mediators and reaction pathways have not been fully established. Among various nitrating pathways, peroxynitrite generated by the reaction of nitric oxide and superoxide is thought to be responsible for nitration in vivo (Beckman et al., 1990; Radi et al., 1991). However, myeloperoxidase in polymorphonuclear neutrophils and monocytes, which functions to synthesize hypochlorous acid from hydrogen peroxide and chloride as a defense against many types of microorganisms (Kettle and Winterbourn, 1997), was reported to catalyze protein nitration in the presence of high nitrite (NO2−) levels, and an injurious effect of myeloperoxidase in vitro through the nitration reaction was emphasized (van der Vliet et al., 1997; Eiserich et al., 1998). We have reported the formation of nitrotyrosine in brain (Fukuyama et al., 1998; Takizawa et al., 1999) or spinal cord (Sakurai et al., 1998) exposed to ischemia-reperfusion injury, and suggested that peroxynitrite was the mediator of the nitration. This study was designated to provide further support for our hypothesis by examining the role of myeloperoxidase in the nitration reaction in cerebral ischemia-reperfusion. We measured the formation of nitrotyrosine and infarct volume in myeloperoxidase-deficient or C57 Black/6 wild-type mice subjected to 2-hour focal ischemia-reperfusion. We found that myeloperoxidase-deficient mice exhibited a greater increase in nitrotyrosine formation and larger infarction than wild-type mice.
MATERIALS AND METHODS
All aspects of this study were approved by the Tokai University Animal Care and Use Committee. Myeloperoxidase-deficient mice were generated by using a gene-targeting technique described previously (Aratani et al., 1999). Briefly, the targeting vector was constructed by replacing a 0.7-kb Xba I-Eco RI fragment containing exon 6 through a part of intron 7 with the neo gene. In addition, a copy of the herpes simplex virus thymidine-kinase gene was placed at the 3′ end of the construct. Both of the selectable marker genes were inserted in the same transcriptional orientation as the myeloperoxidase gene. The myeloperoxidase gene mutation was transmitted through the germ line by mating male chimeric mice generated by correctly targeted embryonic stem cells with strain C57 Black/6 female mice. Homozygous myeloperoxidase −/− mice were produced by interbreeding heterozygous myeloperoxidase +/− mice and backcrossing onto C57 Black/6 mice for seven generations. The result of northern blot analysis of mRNA isolated from the bone marrow was negative for myeloperoxidase, and the peroxidase activity of polymorphonuclear neutrophils was below the detection limit (0.07 nmol·minute−1 ·10−6 cells).
Transient focal ischemia was achieved in male myeloperoxidase-deficient (n = 29; body weight, 26.6 ± 1.2 g) and male C57 Black/6 wild-type (n = 28; body weight, 25.7 ± 1.2 g) mice through intraluminal filament occlusion of the middle cerebral artery using previously described techniques (Hara et al., 1996; Kitagawa et al., 1998). Briefly, mice were anesthetized with halothane (induction 3%, maintenance 1% in 30% oxygen and 70% nitrous oxide), and the left common and external carotid arteries were isolated and ligated. A 6–0 nylon filament was introduced into the internal carotid artery and advanced to a position 10 mm distal from the carotid bifurcation for occlusion of the middle cerebral artery. Two hours after occlusion, animals were briefly reanesthetized with halothane and the filament was withdrawn through the internal carotid artery. Cortical microperfusion was measured by laser-Doppler flowmetry 5 minutes before and after occlusion and 20 minutes after reperfusion. Neurologic deficit was evaluated by neurologic scores 30 minutes after the start of occlusion (Bederson et al., 1986).
For the estimation of infarct volume, animals (myeloperoxidase-deficient mice, n = 11; wild-type mice, n = 8) were killed with an overdose of pentobarbital 24 hours after the start of occlusion. The brain was removed and cut into 2-mm thick coronal sections. The brain slices were quickly incubated in TTC (2% 2,3,5-triphenyltetrazolium chloride) in phosphate-buffered saline for 30 minutes at room temperature and stored in 4% paraformaldehyde. Both surfaces of the 2-mm-thick TTC-stained sections were photographed using color slide film. Infarct volume (mm3) was measured using an image analysis system (MCID, Imaging Research, Canada). Corrected infarct volume adjusted for brain swelling was calculated from the difference between the volume of the contralateral cortex and the volume of the TTC-stained portion (nonischemic) of the ipsilateral cortex of each mouse (Lin et al., 1993). This indirect measure of infarct volume, based on the assumption that the volumes of the ipsilateral and contralateral cortices were the same before ischemia, corrects the total infarct volume for the edema component (Swanson et al., 1990).
For the measurement of nitrotyrosine, animals (myeloperoxidase-deficient mice, n = 18; wild-type mice, n = 20) were killed with an overdose of pentobarbital 14 or 24 hours after the start of occlusion. The brain was excised and cerebral hemispheres were separated. Nitrotyrosine residue in proteins was quantified by a competitive enzyme-linked immunosorbent assay (Khan et al., 1998) with some modifications. Briefly, the assay was performed in 96-well plates coated with 10 μg/mL nitrobovine serum albumin (immobilized antigen) and blocked with 1% casein in phosphate-buffered saline to prevent nonspecific binding. Homogenized brain (50 μL) was added to each well and incubated with immunoaffinity-purified polyclonal antinitrotyrosine rabbit immunoglobulin G (1:500, 100 μL) for 2 hours at 37°C. The plate was washed with phosphate-buffered saline containing 0.05% Tween-20 and sequentially incubated with donkey antirabbit immunoglobulin G horseradish peroxidase complex. After further washing, color development was initiated by the addition of an o-phenylenediamine-hydrogen peroxide mixture, and was allowed to proceed for up to 5 minutes at room temperature. The development was terminated by the addition of 2.5 mol/L sulfuric acid. Antibody binding was determined by measuring the absorbency at 490 nm using a microplate reader (Spectra Max 250, Molecular Devices, Sunnyvale, CA, U.S.A.). The concentrations of nitrated proteins were estimated from a standard curve obtained by the serial dilution of nitrated bovine serum, and results were expressed as nitrated bovine serum albumin equivalents.
The statistical significance of differences in infarct volume or 3-nitro-L-tyrosine in each region among groups and in physiologic parameters among groups was analyzed by one-way analysis of variance, followed by Fisher protected least-significant difference. Values are expressed as mean ± SD.
RESULTS
In the 14-hour reperfusion model, only one of nine myeloperoxidase-deficient mice and zero of nine wild-type mice died. However, in the 24-hour reperfusion model, 6 of 20 myeloperoxidase-deficient mice and 3 of 19 wild-type mice died of convulsion or apnea, with no significant difference in mortality rate. All brains of dead mice macroscopically exhibited severe edema involving a large infarct. The rectal body temperature in both groups was 37.0 ± 0.6°C, but this difference was nonsignificant. Neurologic deficit scores during the occlusion of the middle cerebral artery did not differ between the myeloperoxidase-deficient and wild-type mice (2.4 ± 0.6 vs. 2.3 ± 0.6). Cortical microperfusion 5 minutes after the occlusion of the middle cerebral artery (10.3 ± 10.0% vs. 9.4 ± 7.7% of preocclusion flow) and 20 minutes after reperfusion (63.0 ± 24.5% vs. 61.8 ± 22.7% of preocclusion flow) did not differ between myeloperoxidase-deficient and wild-type mice.
Infarct volume
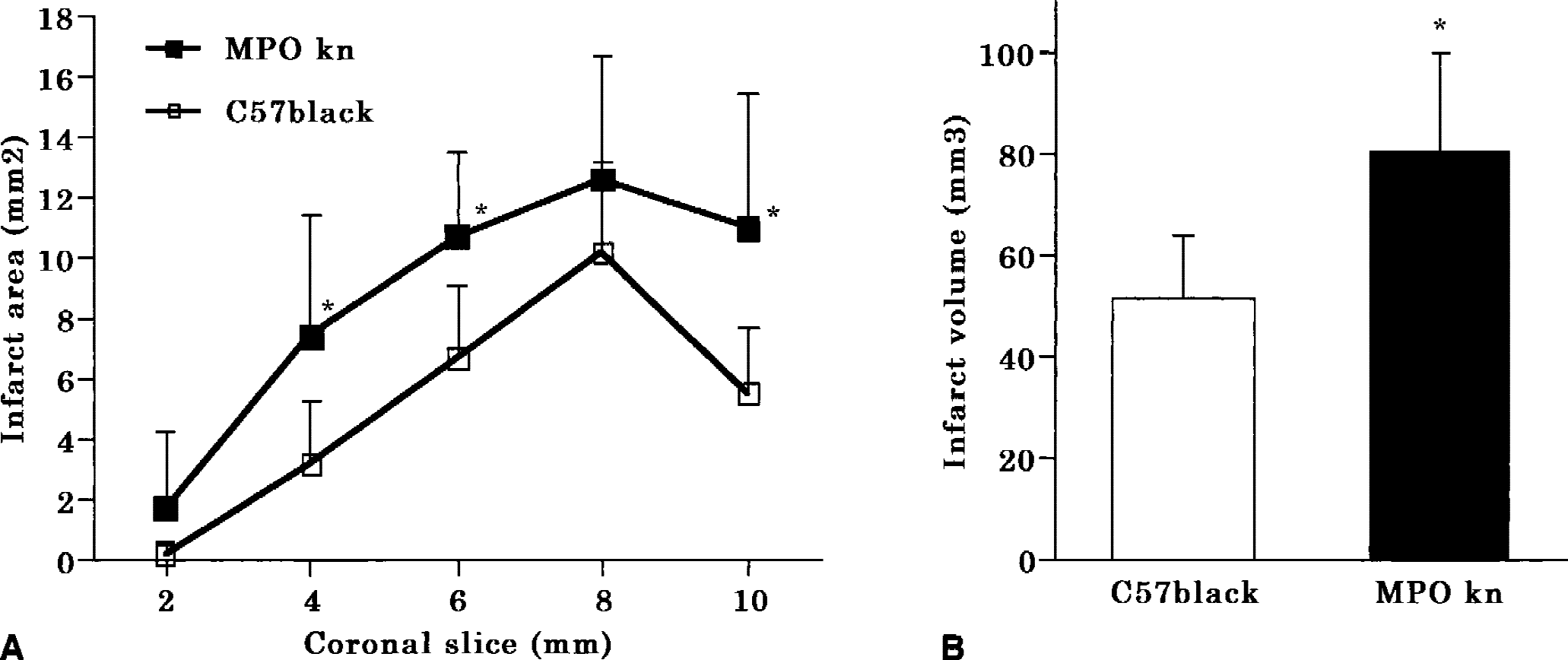
Twenty-four hours after reperfusion, infarct areas with TTC-stained coronal slices 4, 6, and 10 mm from the frontal tip were significantly larger in myeloperoxidase-deficient mice than in wild-type mice (P < 0.05;Fig. 1A). Infarct volume corrected for brain swelling was significantly larger in myeloperoxidase-deficient mice than in wild-type mice (81 ± 20 mm3 vs. 52 ± 13 mm3, P < 0.01;Fig. 1B). The difference in directly measured infarct volume between myeloperoxidase-deficient and wild-type mice was also significant (93 ± 24 mm3 vs. 66 ± 21 mm3, P < 0.05).
Nitrotyrosine level
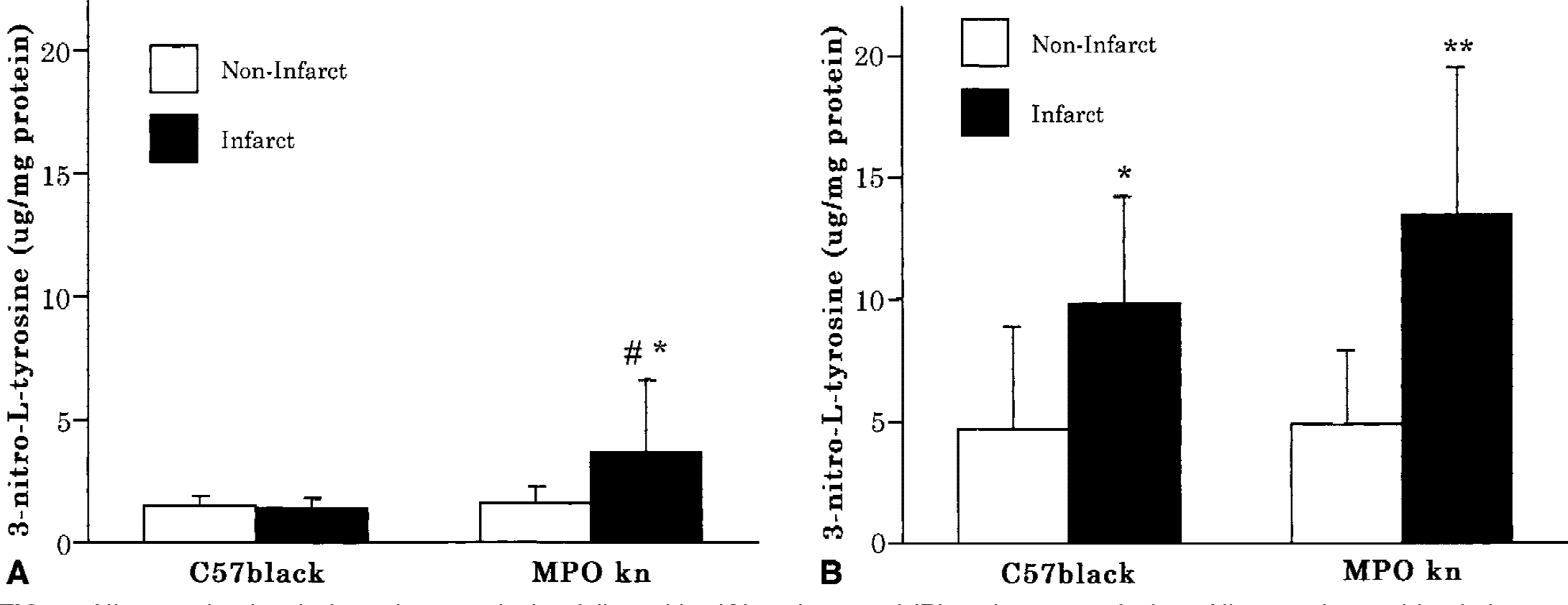
Nitrotyrosine levels of the infarct region were not markedly increased 14 hours after reperfusion compared with those in the noninfarct region in either group. However, the nitrotyrosine level of the infarct region in myeloperoxidase-deficient mice was significantly increased compared with that in wild-type mice (3.3 ± 2.9 μg/mg vs. 1.4 ± 0.4 μg/mg;P < 0.05;Fig. 2A). Twenty-four hours after reperfusion, nitrotyrosine levels in the infarct region were markedly increased in both groups and again were higher in myeloperoxidase-deficient mice than wild-type mice (13.4 ± 6.1 μg/mg vs. 9.8 ± 4.4 μg/mg;P = 0.13;Fig. 2B). Although the nitrotyrosine increase was significant at 14 hours of reperfusion, the nonsignificant nitrotyrosine increase in myeloperoxidase-deficient mice at 24 hours after reperfusion may be because 6 of 20 myeloperoxidase-deficient mice and 3 of 19 wild-type mice died before the 24-hour period and were not included in the data analysis.
Nitrotyrosine level after 2-hour occlusion followed by
DISCUSSION
The present study showed exacerbation of infarction in myeloperoxidase-deficient mice, and formation of nitrotyrosine in the brain of myeloperoxidase-deficient and wild-type mice, with focal ischemia-reperfusion injury. These findings indicate that peroxynitrite-mediated reaction, not myeloperoxidase-mediated reaction, plays the key role in this nitration reaction in vivo. We do not dispute that myeloperoxidase catalyzes the nitration reaction in the presence of NO2− because there are several convincing biochemical studies (Sampson, et al., 1996; Sampson, et al., 1998; Hazen et al., 1999) showing that nitrotyrosine formation was augmented by myeloperoxidase, and we have obtained similar results in a solution experiment. However, in the in-vivo condition, the peroxynitrite-mediated pathway appears more likely to be relevant because the concentration of NO2− may not reach the levels used in these previous experiments (2–50 μM), unless marked induction of inducible nitric oxide synthase occurs. The peroxynitrite-mediated pathway can function under conditions where the generation of superoxide is increased, but inducible nitric oxide synthase is not yet induced. This fact is relevant in the ischemic brain, in which a low concentration of nitric oxide is produced by neuronal nitric oxide synthase in neuronal cells in response to massive glutamate release (Choi, 1991; Eliasson et al, 1999; Hirabayashi et al., 2001). Because the rate constant of the reaction of nitric oxide with superoxide is far greater than that of the degradation of nitric oxide to NO2− or the reaction of NO2− and myeloperoxidase (Goldstein and Czapski, 1995; Furtmuller et al., 2000), most of the nitric oxide would react with superoxide to produce peroxynitrite. Even though a high flux of nitric oxide is generated after the induction of inducible nitric oxide synthase, nitration via myeloperoxidase may be limited to the vicinity of vessels, because inducible nitric oxide synthase immunoreactivity is observed predominantly in vascular cells in cerebral ischemia followed by 12-hour to 24-hour reperfusion (Iadecola et al., 1996; Coeroli et al., 1998; Hirabayashi et al., 2000). The present finding of increased formation of nitrotyrosine in myeloperoxidase-deficient mice is consistent with that of our previous study (Fukuyama et al., 1996), which showed that the nitration reaction in human polymorphonuclear neutrophils was increased in the presence of a myeloperoxidase inhibitor. Further, a recent finding that atherosclerosis was increased in myeloperoxidase-deficient mice (Brennan et al., 2001) also supports the findings of the present study.
The possible underlying mechanisms by which infarct volume was increased in myeloperoxidase-deficient mice include the increased nitration of tyrosine residues of functionally crucial proteins, such as ribonucleotide reductase (Guittet et al., 2000), cytochrome c (Cassina et al., 2000), and c-SRC tyrosine kinase (MacMillan-Crow et al., 2000). The cytotoxic effect of nitrotyrosine may have contributed to the injury, based on the recent findings that nitrotyrosination of α-tubulin (Eiserich et al., 1999) and DNA fragmentation (Mihm et al., 2000) are induced by nitrotyrosine supplementation. Alternatively, myeloperoxidase-dependent attenuation of hydroxyl radical production may have played a neuroprotective role because myeloperoxidase reacts with hydrogen peroxide, a hydroxyl-radical precursor, and purified myeloperoxidase may inhibit the hydroxyl radical production of activated neutrophils in a concentration-dependent manner (Winterbourn, 1986; Kettle and Winterbourn, 1997).
In this study, we used the enzyme-linked immunosorbent assay to evaluate nitrotyrosine in tissue. Although other methods have been used to quantify nitrotyrosine in tissues, such as high-performance liquid chromatography with an optical absorbance detector (Fukuyama et al., 1998; Sakurai et al., 1998; Takizawa et al., 1999) and a dual-channel electrochemical detector combined with tissue hydrolysis (Hirabayashi et al., 2000), an unknown peak superimposed on the nitrotyrosine peak may interfere with quantification (Kaur et al., 1998). In fact, we confirmed that results obtained by high-performance liquid chromatography correlated well with those obtained by the assay (data not shown).
In summary, we conclude that the myeloperoxidase-mediated pathway does not play a major role in the nitration reaction in cerebral ischemia-reperfusion, and the absence of myeloperoxidase increases neuronal damage in vivo. This finding suggests that the optimum therapeutic strategy to inhibit nitration in the brain is blocking the peroxynitrite-mediated nitration pathway.
Footnotes
Acknowledgments:
The authors thank Dr. K. Kitagawa (Osaka University) for advice concerning the mouse-occlusion model. The skillful technical assistance of C. Kawaguchi, S. Kohara, and Y. Takahari (Tokai University) is gratefully acknowledged.