
Abstract
Recent experimental work has shown that hypothermia with even small decreases in temperature is broadly neuroprotective, but the mechanism of this protection remains unclear. Although reduction of metabolism could explain protection by deep hypothermia, it does not explain the robust protection found with mild hypothermia. Several reports have suggested that ischemic apoptosis is reduced by hypothermia. The authors examined the effects of hypothermia on neuronal apoptosis using serum deprivation, a well-accepted model that induces neuronal apoptosis. Mild hypothermia (33°C) significantly reduced the number of morphologically apoptotic neurons to less than half the number seen in normothermic culture temperatures (37°C) after 48 hours. They examined the effect of hypothermia on several steps in the cascade. Caspase-3, −8, and −9 activity was significantly increased after 24 hours at 37°C, and was significantly lower in cultures deprived of serum at 33°C. Cytochrome c translocation was reduced by hypothermia. Western blot analysis failed to detect significant changes in Bax, bcl-2, or hsp-70 at early time points, whereas hypothermia significantly reduced cJun N-terminal kinase activation. The authors conclude that small decreases in temperature inhibit apoptosis very early, possibly at the level of the initiation of apoptosis, as suggested by reduced cJun N-terminal kinase activation and before the translocation of cytochrome c, with subsequent prevention of caspase activation.
Hypothermia is an established neuroprotectant, and recent work has shown that modest decreases in brain temperature are associated with robust protection both in vivo and in vitro (Ginsberg et al., 1992; Bruno et al., 1994; Barone et al., 1997; Maier et al., 1998, 2001). Although the protective effects of hypothermia have been attributed to reductions in the metabolic rate (Chopp et al., 1989), glutamate accumulation (Globus et al., 1988; Busto et al., 1989; Lo and Steinberg, 1992), and reductions in excitotoxicity (Bruno et al., 1994), these explanations cannot fully account for the degree of protection observed. Protection by deep hypothermia (temperature decreases ≦25°C) could be explained by the preservation of metabolic stores, but the mechanism or mechanisms of the robust protection with only a few degrees difference in brain temperature is less clear.
Programmed cell death, or apoptosis, occurs in a variety of pathological settings (e.g., stroke, trauma) that were once thought to be primarily necrotic. Histologic evidence of apoptosis after experimental stroke (Li et al., 1995a, 1995b) and trauma (Murakami et al., 1997; Conti et al., 1998; Kaya et al., 1999) is evident days after the initial insult. Caspase activation has also been noted in these models (Namura et al., 1998), with attenuation of injury after treatment with caspase inhibitors (Hara et al., 1997b; Namura et al., 1998) or in caspase-deficient animals (Friedlander et al., 1997; Hara et al., 1997a; Schielke et al., 1998). A few reports have shown that the density of neurons undergoing apoptosis in these settings is reduced with modest reductions in brain temperature (Edwards et al., 1995; Maier et al., 1998; Xu et al., 1998b). Further evidence favoring the idea that apoptosis is induced during ischemic injury includes observations that antiapoptotic bcl-2 family members are downregulated whereas proapoptotic members are upregulated in some instances (Gillardon et al., 1996), and cytochrome c is translocated (Fujimura et al., 1998) after experimental stroke. However, these studies have focused on complex whole-animal models where necrotic death is also an important component of injury and other types of programmed cell death may also occur.
Several studies suggest that the occurrence of apoptosis in ischemic brain injury is reduced by hypothermia (Edwards et al., 1995; Maier et al., 1998; Xu et al., 1998 b), but this effect has not been well quantitated and the mechanism of this reduction has not been elucidated. The use of in vitro paradigms of primarily apoptotic neuronal injury offers a unique opportunity to investigate the detailed mechanisms of mild hypothermic neuroprotection. One study using hypoxia-induced apoptosis of neurons showed reduced apoptosis at 32°C, but 23% of the cells showed morphologic features of apoptosis (Bossenmeyer-Pourie et al., 2000). We test the effect of mild hypothermia, in the range that protects from cerebral ischemia, on neuronal apoptosis induced by serum deprivation, which induces morphologic features of apoptosis in more than 60% of the neurons. This in vitro model allows easy quantification of cell death and evaluation of biochemical changes. We demonstrate that mild hypothermia markedly reduces neuronal apoptosis, and test whether the reduction in apoptosis is associated with reductions in caspase activation, cytochrome c translocation, or changes in apoptosis regulatory proteins.
MATERIALS AND METHODS
Cortical neuronal cultures
After approval by the Stanford University Administrative Panel on Laboratory Animal Care, pure neuronal-cell cultures were prepared from cortices of fetal mice at 14 or 15 days of gestation as previously described with minor modifications (Dugan et al., 1995). The plating medium contained 5% fetal bovine serum and 5% equine serum (HyClone, Logan, Utah). Twenty-four hours after plating the single-cell suspension, half of the culture medium was replaced with a glial-conditioned medium containing 5% fetal bovine serum, and B-27 (2%, Gibco BRL, Rockville, MD, U.S.A.) was added. Cytosine arabinoside (3 μmol/L) was added to inhibit glial proliferation. Cultures were kept at 37°C in a humidified incubator in a 5% carbon dioxide atmosphere without further medium changes until used for experiments after 4 or 5 days in vitro. These conditions result in cultures containing less than 1% astrocytes (glial fibrillary acidic protein immunopositive cells).
Induction of apoptosis
Neuronal cultures were deprived of the glial-conditioned medium and serum by triple exchange of the medium with balanced salt solution (BSS5.5) as previously described (Dugan et al., 1995). The balanced salt solution consisted of 5.5 mmol/L glucose, 1.8 mmol/L calcium chloride, 0.8 mmol/L magnesium sulfate, 5.4 mmol/L potassium chloride, 1 mmol/L sodium hydrogen phosphate, 10 mmol/L HEPES [N-(2-hydroxyethyl) piperazine-N′-ethanesulfonic acid], and 26 mmol/L sodium bicarbonate at pH 7.4. The cells were kept at 37°C (normothermic) or 33°C (hypothermic) in a humidified incubator in an atmosphere of 5% carbon dioxide for 2 days.
Injury assessment
Injury was assessed at the indicated times by nuclear morphology and quantitated by cell counting after staining with propidium iodide and Hoechst dye 33258. In addition, cell lysis was quantified by measuring lactate dehydrogenase (LDH) efflux to the medium (Koh and Choi, 1987). The maximum amount of LDH release from each culture was determined by freeze-thaw disruption at the end of the experiment. All experiments were carried out at least three times using cells from different dissections. Statistical significance was determined using analysis of variance and the Student-Neuman-Keuls test, t-test, or paired t-test (Sigmastat; Jandel Scientific, San Rafael, CA, U.S.A.).
Nuclear morphology
Nuclear morphology was assessed using a Nikon Diaphot microscope equipped for epifluorescence with ultraviolet filterblock after a 10-to 15-minute preincubation of live cultures at 37°C with Hoechst dye 33258 (Xu et al., 1998a) (final concentration, 5μg/mL) followed by the addition of propidium iodide (Sigma, St. Louis, MO, U.S.A.) (final concentration, 5μg/mL). Nuclei exhibiting bright fluorescence and the fragmented nuclear morphology typical of apoptotic bodies were counted as apoptotic. Diffusely fluorescent nuclei, which stained with Hoechst dye, were scored as alive and viable, whereas diffusely fluorescent nuclei stained with propidium iodide were scored as necrotic (Fig. 1).
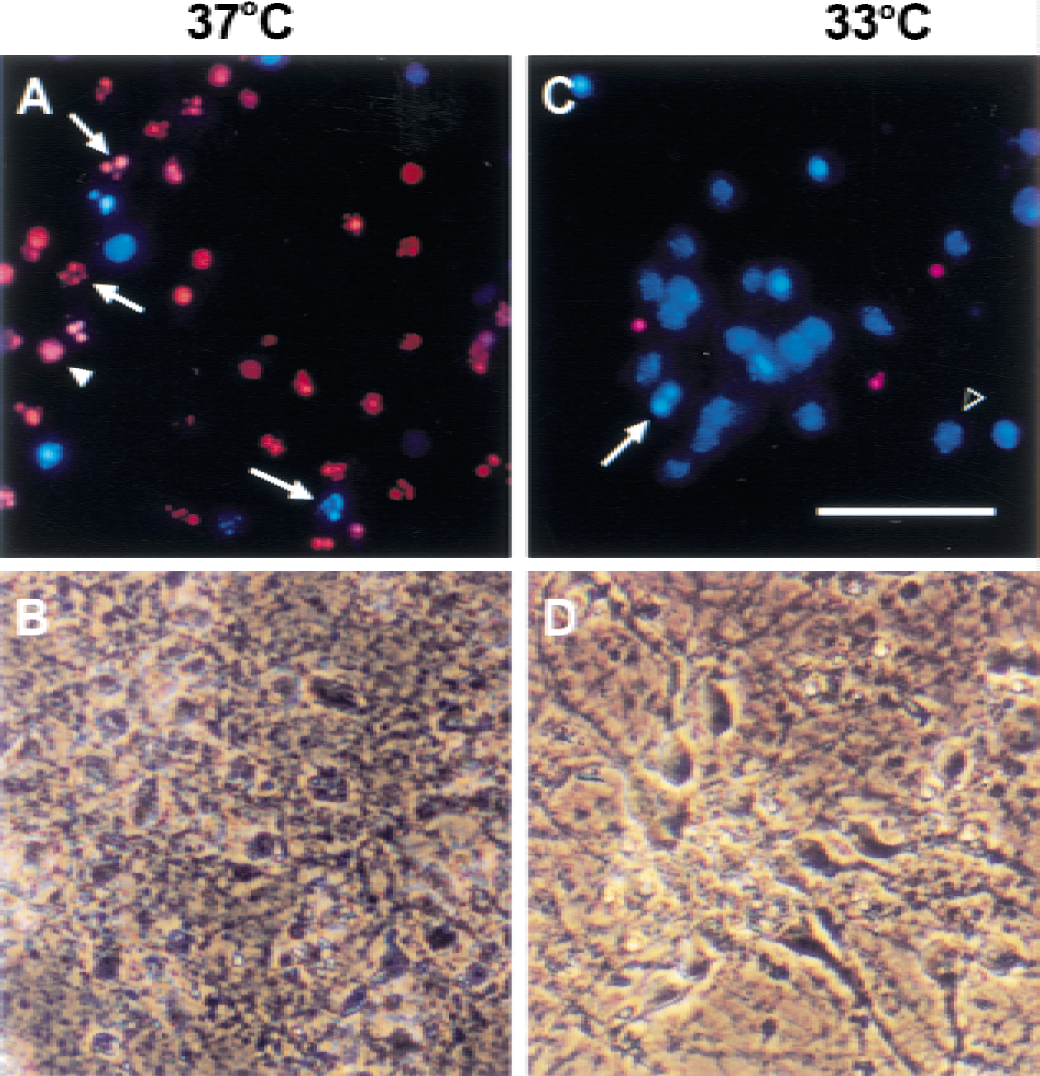
Light morphology of neurons deprived of serum at 37°C and 33°C. After 5 days in culture, pure neuronal cultures were deprived of glial-conditioned medium and serum for 48 hours, and the same field was photographed using fluorescence
Caspase-3, −8, and −9 activity assays
The fluorogenic substrates for caspase-3 (Ac-DEVD-AFC), caspase-8 (Ac-IETD-AFC), and caspase-9 (Ac-LEHD-AFC) were obtained from Enzyme Systems Products (CaspAFC Assay System; Livermore, CA). When excited by ultraviolet light, the 7-amino-4-trifluoromethyl coumarin (AFC)-labeled substrates produce blue fluorescence at 440 nm. The fluorescence emission shifts to 530nm when AFC is cleaved from the substrate, and this shift is proportional to the amount of caspase activity in the sample. The cells were collected by scraping with a rubber policeman, washed once with phosphate-buffered saline, and gently suspended in lysis buffer (100 mmol/L HEPES, 1% Triton-X100, 1 mmol/L edetic acid, and 10% sucrose). Samples were collected after 8, 16, 24, and 48 hours of serum deprivation. Fluorescence was measured with a Cytofluor II fluorescence plate reader (Applied Biosystems, Foster City, CA, U.S.A.). Fluorescence was recorded in arbitrary units immediately after and 30 minutes after incubation at 37°C. Increases in fluorescence were normalized to the protein in each sample.
Immunoblotting
Procaspase-3 and its cleaved forms were detected by Western blot analysis using a rabbit polyclonal antibody to caspase-3 (CPP32) at a 1:2,000 dilution, which reacts with both full-length procaspase-3 (P32) and the cleaved forms of 20 and 10 kDa (Idun Pharmaceuticals, San Diego, CA, U.S.A.). Activated mitogen-activated protein kinase was detected by immunoblot analysis using the antiphospho-p44/42 mitogen-activated protein kinase monoclonal antibody (1:1,500; Cell Signaling Technology, Beverly, MA, U.S.A.) specific for activated extracellular signal regulated kinase (ERK) 1 (p44) and 2 (p42); activated stress-activated protein kinase/cJun N-terminal kinase (SAPK-JNK) (P54 and P46) were detected with antiphospho-SAPK-JNK (1:1,500, Cell Signaling Technology). The rabbit polyclonal antibody to Bax was used at a 1:1,000 dilution (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), monoclonal antibody to hsp 70 was used at a 1:1,000 dilution (Stressgen Biotechnology, Victoria, BC, Canada), mouse monoclonal antibody to rat cytochrome c was used at 1:1000 dilution (Pharmingen, San Diego, CA, U.S.A.), and rabbit polyclonal antibody to bcl-2 was used at a 1:1,000 dilution (Santa Cruz Biotechnology). Protein concentration was determined by the bicinchoninic acid method (Pierce, Rockford, IL, U.S.A.). Equal amounts of protein for each condition were separated on 12.5% or 15% polyacrylamide gel and electrotransferred onto Immobilon PVDF membrane (Millipore, Bedford, MA) using a mini Trans-Blot cell (BioRad Laboratories, Hercules, CA, U.S.A.). After transfer, the membranes were blocked with 5% nonfat dry milk and incubated in primary antibody at the previously noted dilutions for 2 hours. The membranes were then incubated in secondary antibody conjugated to horseradish peroxidase (Amersham, Piscataway, NJ, U.S.A.). Immunoreactive bands were observed with the enhanced chemiluminescence detection system (ECL, Amersham) using Kodak X-AR film. Membranes were stained with 0.1% ponceau S in 5% acetic acid for 5 minutes; after transfer, gels were stained with Coomassie brilliant blue to verify equal loading and transfer. Densitometric quantification analysis of the chemiluminescent bands was performed using a video camera equipped with densitometer software (Labworks, UVP Laboratory Products).
Cytochrome c translocation
Cell fractionation and western blot analyses were performed as described previously with modifications (Li et al., 1997; Fujimura et al., 1998). Cells were collected at the indicated times after serum deprivation and gently homogenized with a glass tissue homogenizer (Wheaton, Millville, NJ, U.S.A.) in ice-cold suspension buffer containing 20 mmol/L HEPES-potassium hydroxide (pH 7.5), 250 mmol/L sucrose, 10 mmol/L potassium chloride, 1.5 mmol/L magnesium chloride, 1 mmol/L edetic acid, 1 mmol/L ethyleneglycoltetracetic acid, 1 mmol/L dithiothreitol, 0.1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 2 μg/mL aprotinin, 10 μg/mL leupeptin, 5 μg/mL pepstatin. and 12.5 μg/mL N-acetyl- l eu- l eu-norleucinal. The homogenates were centrifuged at 4°C at 750 g for 10 minutes, and the resulting supernatants were centrifuged at 8,000 g for 20 minutes. The pellet from the second centrifugation represented the mitochondrial fraction and was saved. The supernatants were further centrifuged at 100,000 g for 60 minutes, and the resulting supernatants were analyzed as the cytosolic fraction. Protein samples from both the cytosolic and mitochondrial fractions were examined. Purified rat cytochrome c was used as a positive control.
RESULTS
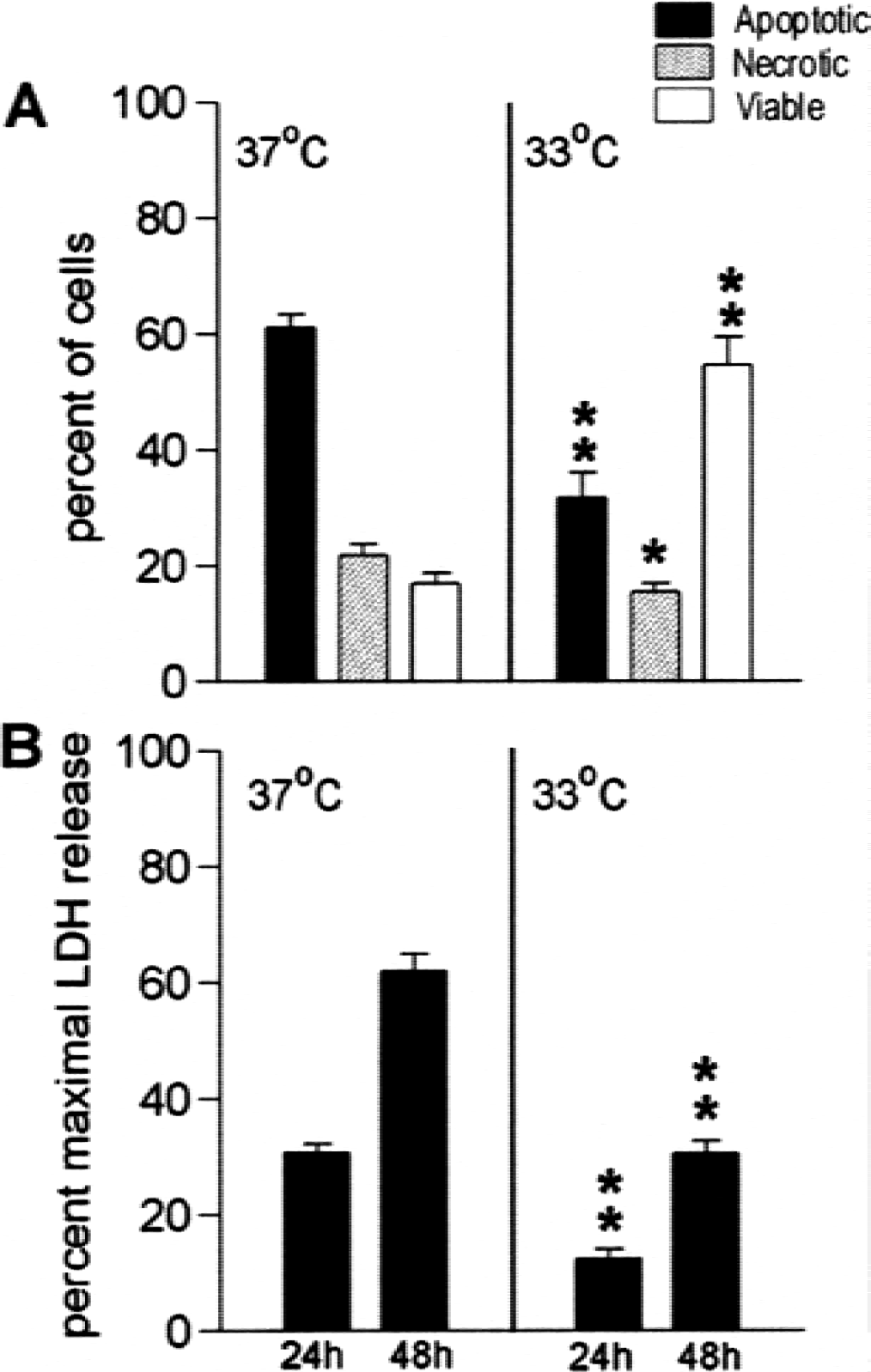
Hypothermia protects neurons from serum deprivation-induced apoptosis: Neurons deprived of serum at 33°C suffered less injury than those deprived at 37°C (Figs. 1, 2). Neuronal processes showed early damage with disruption of cell bodies evident at 48 hours at 37°C (Figs. 1A and 1B). Cell morphology was relatively preserved at 33°C (Figs. 1C and 1D) and fewer apoptotic cells are evident. Nuclear morphology was assessed using Hoechst dye and propidium iodide staining 48 hours after serum deprivation for quantitation. Apoptotic nuclei and necrotic nuclei were counted as a percentage of the total cells in several high-power (400X) fields (Fig. 2A). Cultures deprived of serum at 37°C showed 71.4 ± 3.3% cell death (all propidium iodide-positive cells by cell count) compared with 34.1 ± 2.9% in cultures deprived of serum at 33°C (P < 0.001) for 48 hours. This finding was similar to the extent of cell lysis assessed by quantifying the amount of LDH released into the media; LDH release at 33°C was less than half of that seen at 37°C (Fig. 2B) after 24 and 48 hours (Fig. 2B). To identify the areas where hypothermia might have an effect, we analyzed several of the steps in the apoptosis cascade.
Caspase activation
Caspase-3 is one of the final caspases responsible for carrying out apoptosis. Procaspase-3 is activated by cleavage by caspase-9, which is proteolytically activated by a complex often referred to as the apoptosome, which includes the cytochrome c that has been released from mitochondria. We began the analysis with caspase-3 and worked up the activation pathway. Caspase-3 protein was examined by Western blot analysis. After serum deprivation, full-length procaspase-3 (P32) decreased, whereas the P20 and P10 cleavage products increased from a just-detectable level in control cultures without injury to easily viewed bands after 24 hours of serum deprivation (Fig. 3A), a time at which less than 30% cell lysis has occurred. Cultures subjected to serum deprivation at 33°C showed less P20 and more P32 after 24 hours of serum deprivation compared with normothermic cultures (37°C). The time course of increase in caspase-3 and −9 activity is shown in Figs. 3B and C. Both activities were significantly increased by 16 hours of serum deprivation and showed peak activity at 24 hours. Both caspases showed approximately twice the activity after 16 and 24 hours of serum deprivation at 37°C compared with 33°C (P < 0.05). By 48 hours at 37°C, a point at which significant cell lysis had already occurred, the activity of both caspases decreased dramatically.
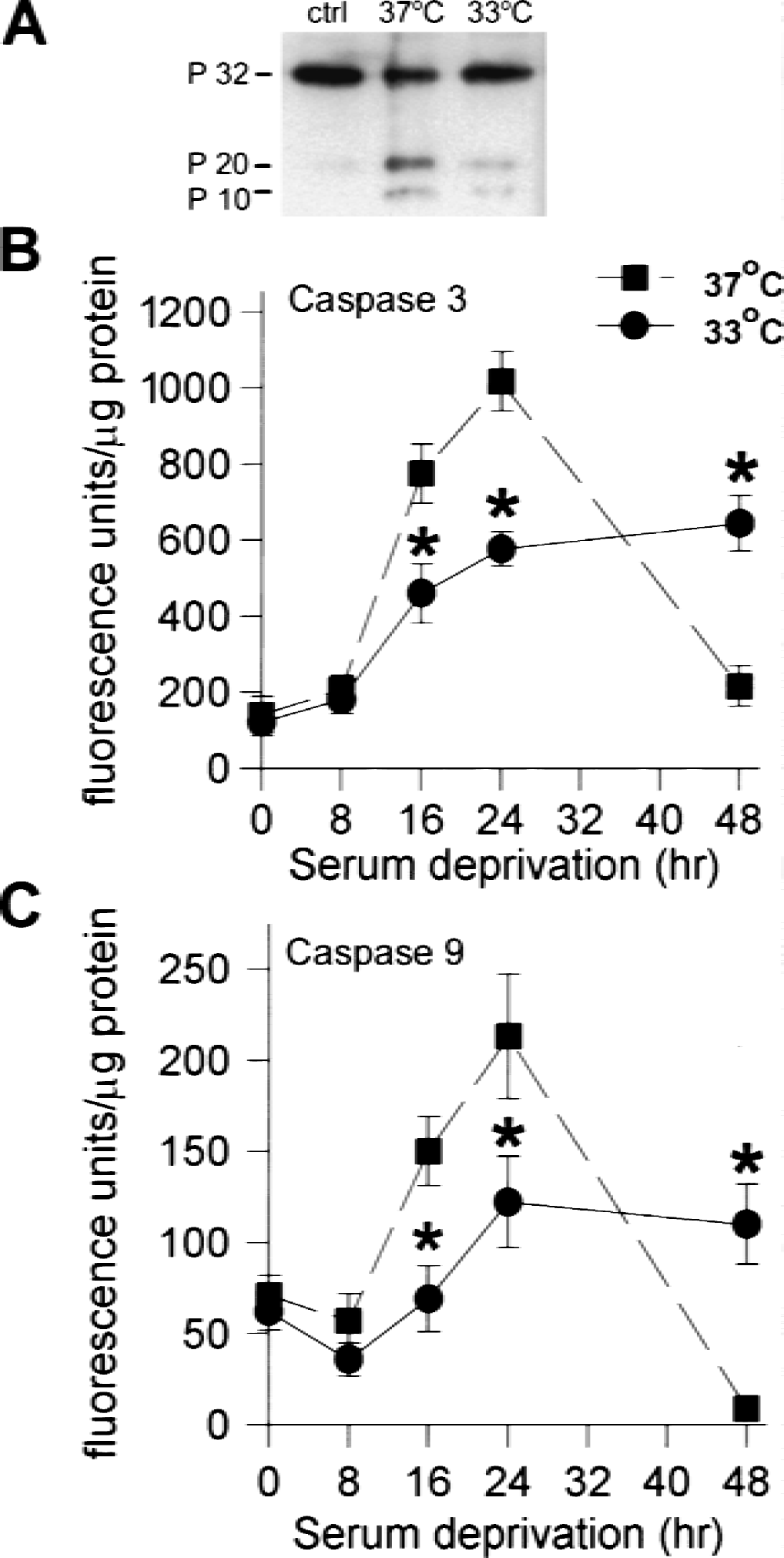
We also measured caspase-8 activity to see if this alternate pathway was induced in this model of neuronal apoptosis, and whether it too was affected by temperature. We observed that caspase-8 was activated, but later than caspases-3 and −9, and this activation was also significantly reduced by hypothermia (Fig. 4). As noted for the other caspases, there was a sharp decrease after 48 hours of serum deprivation at 37°C in association with cell lysis.
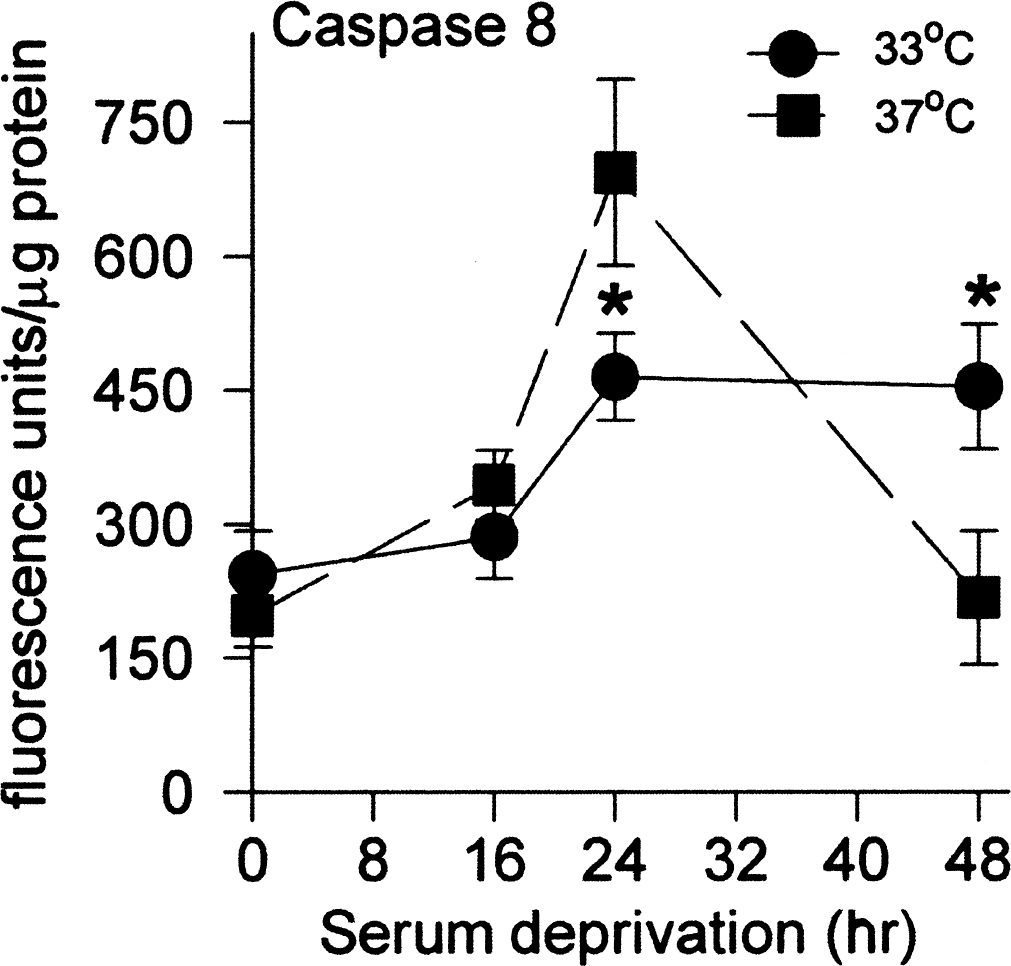
Caspase-8 activity increases after serum deprivation at 37°C are reduced at 33°C. Activity was measured using the fluorescent substrate; fluorescence is measured in arbitrary units and normalized to the amount of protein. The values shown at 0 hours represent control cultures. Data are mean ± SE for n = 12 cultures; *difference from the same point at 37°C is P < 0.05.
Cytochrome c translocation
We then went one step further upstream and looked for translocation of cytochrome c from the mitochondria to the cytosol by cell fractionation after detection by Western blot analysis. The averages for three experiments are shown in Fig. 5. There is a significant increase in cytochrome c detected in the cytosol after 14 and 20 hours of serum deprivation at 37°C compared with uninjured cells, whereas cells deprived of serum at 33°C did not differ significantly from controls. Conversely, there is less cytochrome c in mitochondria of cells subjected to serum deprivation at 37°C for 20 hours compared with control, a change that is less evident in cells after 20 hours of serum deprivation at 33°C.
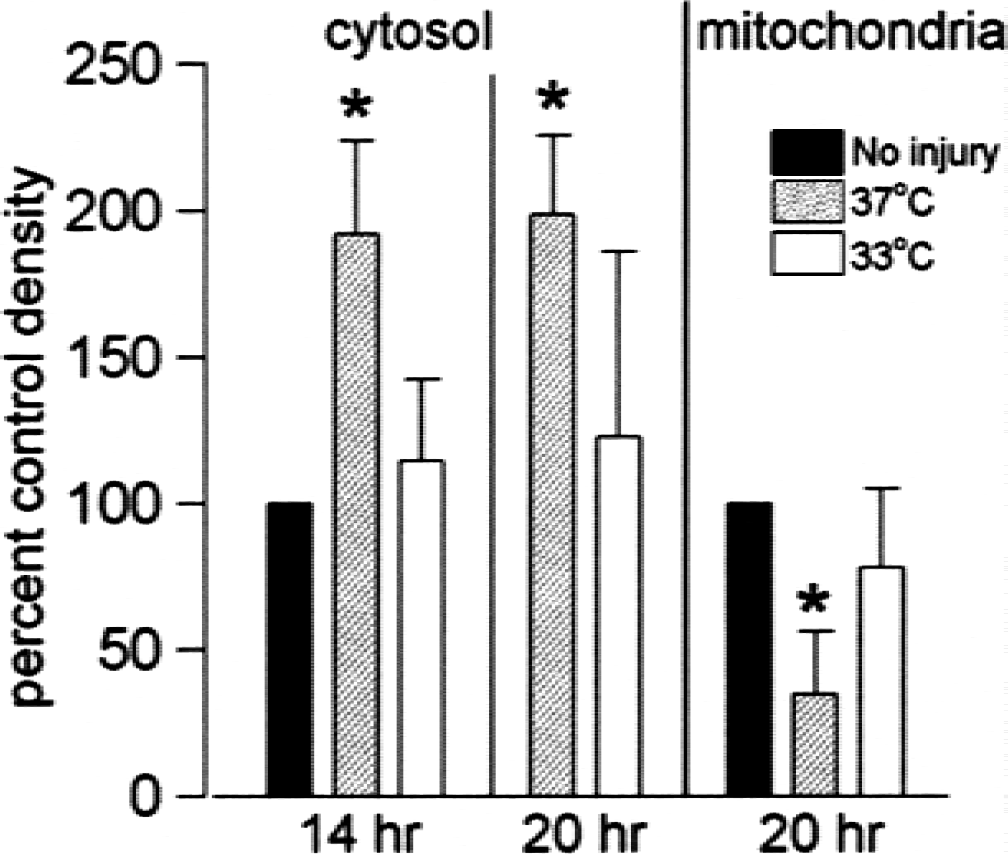
Cytochrome c translocation induced by serum deprivation is reduced at 33°C. Cell fractionation followed by Western blot analysis showed an increase in cytosolic cytochrome c at 37°C after 14 and 20 hours of serum deprivation that was not seen at 33°C, and a decrease in mitochondrial cytochrome c compared with the no-injury control. Values are mean ± SE of scanned-band density normalized to uninjured control-band density in the same experiment, n = 3–4; *difference from control is P < 0.05.
Apoptosis regulatory proteins
Several additional apoptosis regulatory proteins were studied by immunoblot analysis. Bax was easily detected in controls and in injured cells, with no difference in levels after 5 or 8 hours of serum deprivation at either temperature. Hsp-70 and bcl-2 levels were low or barely detectable in these neuronal cultures, and did not increase significantly in the first 24 hours. Activated ERK was not detected after serum deprivation. Activated SAPK/JNK, both phospho-p46 and phospho-p54, were increased by serum deprivation at 37°C. The increase in p46 was much greater (Fig 6A). The increase in activated JNK was affected by temperature, and significantly lower levels were observed at 33°C compared with 37°C (Fig. 6B). These results are consistent with the well-documented involvement of this kinase in the transcriptional control of nerve growth factor deprivation-induced apoptosis of sympathetic neurons (Ham et al., 2000), suggesting that JNK activation is also a key step in initiating serum deprivation-induced apoptosis of cortical neurons.
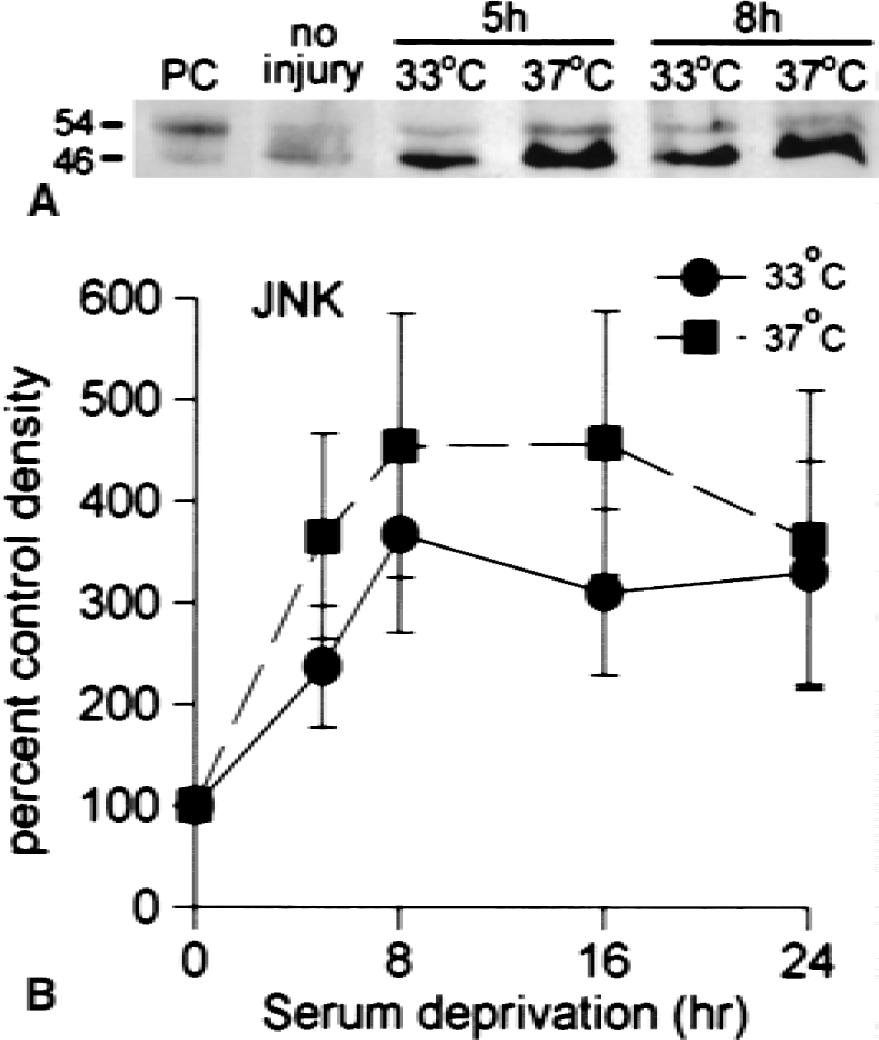
cJun N-terminal kinase activation induced by serum deprivation is greater at 37°C than at 33°C.
DISCUSSION
Our findings show that mild hypothermia is associated with decreased injury in a model of neuronal apoptosis induced by serum deprivation. This apoptosis is associated with JNK activation, cytochrome c translocation, and activation of caspases-3, −8, and −9, all of which are reduced by mild hypothermia. We are the first to demonstrate that mild hypothermia attenuates neuronal injury in an in vitro model resulting in primarily apoptotic death, with reduced activation of regulatory and apoptosis executing proteins throughout the apoptosis cascade. These are the first data linking JNK activation to serum deprivation-induced apoptosis of cortical neurons. Although previous work in in vivo ischemia models suggests that hypothermia can inhibit apoptosis, these models combine apoptosis, necrosis, and inflammatory response, depending on the time of analysis. To clarify the effects of hypothermia on several steps in the process of apoptosis, we used relatively pure neuronal cultures in which nearly all the cell death induced by serum deprivation was apoptotic.
Earlier work has shown that mild hypothermia (temperature decreases to 32–34°C) is associated with decreases in excitatory amino acid accumulation (Lo and Steinberg, 1992; Baker et al., 1995; Huang et al., 1998) and reactive oxygen species (Kil et al., 1996; Zhao et al., 1996). However, some studies have shown that mild hypothermia is protective even when brain cooling is applied 1 to 2 hours after the insult (Baker et al., 1992; Colbourne and Corbett, 1994; Maier et al., 2001). Given that metabolic stores are depleted within 1 hour (Hata et al., 2000) and that glutamate release occurs within 60 to 90 minutes after middle cerebral artery occlusion (Busto et al., 1989; Baker et al., 1995; Huang et al., 1998), the protective effect of hypothermia is not due only to reduction of these early events. It is becoming increasingly clear that ischemic injury evolves over hours to days, and that apoptosis plays some role.
As noted previously, evidence from many laboratories suggests that apoptosis or a closely related type of cell death contributes to ischemic injury; several different antiapoptotic strategies have been shown to reduce ischemic brain injury, including the overexpression of antiapoptotic proteins (Martinou et al., 1994; Linnik et al., 1995; Lawrence et al., 1996, 1997) and caspase inhibition (Hara et al., 1997a, b ; Schielke et al., 1998). The effect of hypothermia on changes in apoptosis regulatory proteins has been assessed in a few examples of global and focal cerebral ischemia models. In a global ischemia model, mild hypothermia increased bcl-2 expression (Zhang et al., in press). After focal ischemia, hypothermic protection was accompanied by increased bcl-2 expression and decreased Bax expression and cytochrome c release (Prakasa Babu et al., 2000). Mild hypothermia has also been shown to decrease cytochrome c translocation 5 hours after transient focal ischemia, while not altering levels of caspases-3 or −9 (Hoehn et al., in press). Improved recovery of potassium homeostasis (Sick et al., 1999) is another mechanism of protection documented with hypothermia.
Bossenmeyer-Pourie et al. (2000) have demonstrated that imposing hypothermia during hypoxia in vitro reduces both necrotic and apoptotic neuronal death, and reduces early hypoxia-induced changes in protein synthesis. These authors did not assess caspase activation or cytochrome c translocation in their system. We demonstrate here that the neuronal apoptotic cascade is significantly reduced by hypothermia as early as the point of JNK activation. The association of JNK activation with this injury paradigm suggests a parallel to the better-studied growth factor deprivation-induced apoptosis of sympathetic neurons by the removal of nerve growth factor. Our results suggest that apoptosis is sensitive to temperature at perhaps the earliest step in initiation and before translocation of cytochrome c from the mitochondria to the cytosol. This finding is consistent with the presumed early effect on induction of apoptosis observed by Bosenmeyer-Pourie et al. (2000).
Although JNK activation is itself the result of previous kinase activation, we have now traced the pathway of apoptotic signaling induced by serum deprivation back to nearly its origin, suggesting that hypothermia reduces the initiation of apoptosis at one of the earliest steps. It is difficult to determine whether the delay in activation of the apoptotic cascade observed here represents complete inhibition of apoptosis or simply the slowing of induction, but we have observed that the readdition of glial-conditioned medium and serum after 48 hours allows for neuronal survival for at least an additional 24 hours (data not shown). This finding suggests that hypothermia extends the period during which rescue of neurons can occur. These results support the current optimism about the clinical use of hypothermia for brain protection against ischemic and traumatic injury.
Footnotes
Acknowledgments:
The authors thank Beth Houle for expert assistance with the figures and IDUN Pharmaceuticals for providing the anticaspase-3 antibody.