
Abstract
Cyclooxygenase-1 (COX-1), a rate-limiting enzyme in the synthesis of prostanoids, is involved in selected vasodilatatory responses of the cerebral circulation. Cyclooxygenase-1–null mice were used to determine whether COX-1 influences cerebral ischemic damage. The middle cerebral artery was occluded in COX-1 −/− and +/+ mice (n = 9/group), and lesion volume was determined in thionin-stained sections 24 or 96 hours later. Middle cerebral artery occlusion produced larger infarcts in COX-1 −/− mice, both at 24 (35 ± 17%;P < 0.05) and 96 hours (41 ± 16%;P < 0.05) after ischemia. The enlargement was not due to increased susceptibility to glutamate excitotoxicity, because microinjection of N-methl- d -asparatate or kainate in the parietal cortex produced comparable lesions in COX-1 +/+ and −/− mice (P > 0.05; n = 8/group). To examine the contribution of hemodynamic factors to the enlargement of the infarct, cerebral blood flow was monitored by laser-Doppler flowmetry in the ischemic territory (n = 6/group). Although the reduction in cerebral blood flow was comparable in the ischemic core (P > 0.05), at the periphery of the ischemic territory the reduction was greater in COX-1 −/− mice (−58 ± 4%) than in COX-1 +/+ mice (−34 ± 5%;P < 0.05). It is concluded that mice lacking COX-1 are more susceptible to focal cerebral ischemia, an effect that can be attributed to a more severe cerebral blood flow reduction in vulnerable regions at the periphery of the ischemic territory. Thus, the vascular effects of COX-1 may contribute to maintain cerebral blood flow in the postischemic brain and, as such, play a protective role in ischemic brain injury.
Cyclooxygenase (COX) is a rate-limiting enzyme in the oxidation of arachidonic acid into prostaglandins and thromboxanes (see Vane et al., 1998 for a review). Two isoforms of COX have been described: COX-1 and COX-2. Cyclooxygenase-1 is ubiquitously expressed and is thought to produce prostanoids that play a role in normal cellular function (Vane et al., 1998). Cyclooxygenase-2 is present only in some organs, but its expression is markedly upregulated during inflammation, and COX-2 reaction products are thought to contribute to the cytotoxicity associated with inflammation (Dubois et al., 1998; Seibert et al., 1995).
Prostanoids have long been implicated in the mechanisms of cerebral ischemic injury (Chen et al., 1986; Moskowitz and Coughlin, 1981); however, most studies have led to contradictory results (Cole et al., 1993; Johshita et al., 1989; Petersen, 1989). A major problem has been the lack of tools needed to selectively study the role of COX-1 and COX-2. Mice who are COX-1 or COX-2 deficient have recently been developed (Langenbach et al., 1999). These mice provide the opportunity to study the role of the different COX isoforms in cerebrovascular regulation and cerebral ischemia. Cyclooxygenase-2–null mice have a reduction in the infarct produced by middle cerebral artery (MCA) occlusion, suggesting that this enzyme exerts a deleterious effect on the ischemic brain (Iadecola et al., 2001). However, COX-1–null mice have an attenuation in selected vasodilatatory responses of the cerebral circulation, including those evoked by the endothelium-dependent vasodilator bradykinin or by hypercapnia (Niwa et al., 2001). The role of COX-1 in the mechanisms of cerebral ischemia has not been investigated.
In this study, COX-1–null mice were used to examine this enzyme's role in ischemic brain injury. Cyclooxygenase-1–null mice had increased susceptibility to the brain damage produced by MCA occlusion, an effect that can be attributed to a more severe cerebral blood flow (CBF) reduction in regions at risk for infarction. Thus, COX-1 may promote collateral flow and protect marginally perfused regions of the ischemic territory through its hemodynamic actions. These findings, in concert with the observation that COX-2–null mice are relatively protected from ischemic brain injury (Iadecola et al., 2001), suggest that COX-1 and COX-2 exert opposite influences on ischemic brain damage.
MATERIALS AND METHODS
Techniques for MCA occlusion, microinjection of excitatory amino acids, measurement of injury volume, evaluation of neurologic deficits, and measurement of CBF in anesthetized ventilated mice have been described previously (Iadecola et al., 2001; Nagayama et al., 1999; Niwa et al., 2001) and will be summarized here.
Animals
Cyclooxygenase-1–null mice [homozygous (−/−) mice and their wild-type (+/+) littermates] (Langenbach et al., 1995), were obtained from breeding pairs provided by Jackson laboratories (Niwa et al., 2001). Mice were backcrossed to C57BL/6J mice seven to nine times, and were studied at age 2 or 3 months. The genotype of all COX-1 mice was determined by polymerase chain reaction analysis (Langenbach et al., 1995).
Induction of focal cerebral ischemia
Focal cerebral ischemia was produced by MCA occlusion (Nagayama et al., 1999). Mice were anesthetized with 2% halothane in 100% oxygen. Body temperature was maintained at 37 ± 0.5°C by a thermostatically controlled infrared lamp. A 2-mm hole was drilled in the inferior portion of the temporal bone to expose the left MCA. The MCA was elevated and cauterized distal to the origin of the lenticulostriate branches. Mice in whom the MCA was exposed but not occluded served as sham-operated controls. Wounds were sutured and mice were allowed to recover before they were returned to their cages. Rectal temperature was controlled until mice regained full consciousness. Thereafter, rectal temperature was measured daily until the time of death. There were no significant differences in rectal temperature among COX-1 +/+, and −/− mice before and after MCA occlusion (P > 0.05). For example, 24 hours after MCA occlusion, rectal temperature was 36.2 ± 01°C in COX-1 +/+ mice and 36.0 ± 0.2°C in COX-1 −/− mice (P > 0.05, t-test).
N-methl- d -asparatate microinjection in neocortex
In halothane-anesthetized mice, the dura overlying the parietal cortex was exposed and N-methl- d -asparatate (NMDA) (20 nmol in 200 nL sterile 0.1 mol/L PBS, pH 7.4) or kainate (1 nmol in 200 nL) was injected using a glass micropipette (tip, 40–50 μm) connected to a microinjection device (Iadecola et al., 2001). The concentration of NMDA and kainate were chosen in preliminary experiments to produce maximal damage. The micropipette was inserted into the parietal cortex at a site 1.5 mm caudal to bregma, 4.0 mm from the midline, and 0.8 mm below the dural surface. The micropipette was left in place for 10 minutes to minimize back flux, and was then removed. Mice were returned to their cages and allowed to survive for 24 hours before lesion volume was determined.
Determination of lesion volume
Mice were killed 1 and 4 days after MCA occlusion, or 1 day after NMDA or kainate injection. Their brains were removed and frozen in cooled isopentane (−30°C). Coronal forebrain sections (thickness, 30 μm) were serially cut in a cryostat, collected at 90-μm intervals, and stained with thionin for determination of lesion volume by an image analyzer (MCID, Imaging Research, St. Catherine, Ontario, Canada;Iadecola et al., 2001; Nagayama et al., 1999). In studies of cerebral ischemia, infarct volume in cerebral cortex was corrected for swelling to factor out the contribution of ischemic edema to the total volume of the lesion (Iadecola et al., 2001; Lin et al., 1993; Nagayama et al., 1999). The correction method is based on the determination of ischemic swelling by comparing the volume of ischemic and nonischemic hemispheres (Lin et al., 1993).
Neurologic evaluation
Neurologic deficits were assessed using a modification of the postural reflex test, as previously described (Bederson et al., 1986; Nagayama et al., 1999). The examiner was not aware of the identity of the mice. Because the internal capsule and the striatum were relatively spared by the infarct, neurologic deficits ranged between 0.5 (flexion of contralateral forelimb) and 1 (flexion of torso and contralateral forelimb when the mouse was lifted by the tail). Mice were evaluated before MCA occlusion and at 24-hour intervals up to 96 hours after MCA occlusion.
Monitoring of cerebral blood flow
Mice were anesthetized with halothane (maintenance 1%) and the femoral artery and trachea were cannulated. Mice were artificially ventilated with an oxygen-nitrogen mixture with a mechanical ventilator (SAR-830; CWI, Ardmore, PA, U.S.A.). The oxygen concentration in the mixture was adjusted to maintain an arterial oxygen tension of 120 to 140 mm Hg (Table 1). End-tidal carbon dioxide was continuously monitored using a carbon dioxide analyzer (Capstar-100, CWI) (Iadecola et al., 2001; Niwa et al., 2001). Cerebral blood flow was monitored with two laser-Doppler flow probes (Vasamedic, St. Paul, MN, U.S.A.) placed through burr holes drilled in the center (3.5 mm lateral to the midline and 1 mm caudal to bregma) and the periphery (1.5 mm lateral to the midline and 1.7 mm rostral to lambda) of the ischemic territory (Chan et al., 1993; Iadecola et al., 2001). The location of the probe was chosen in preliminary experiments to correspond to the brain region that is recruited into infarction in COX-1–deficient mice (see Zhang and Iadecola, 1993 for the general location of the penumbral probe). After placement of the probes, the MCA was occluded and CBF was monitored for 90 minutes. Cerebral blood flow data are expressed as percent of the preocclusion value. Blood gases did not differ between COX-1 +/+ and −/− mice 10 minutes after MCA occlusion (arterial carbon dioxide tension, 33.8 ± 1.0 vs. 33.1 ± 1.0mm Hg; arterial oxygen tension, 140 ± 8 vs. 128 ± 9 mm Hg; pH 7.32 vs. 7.34, respectively).
Data analysis
Data in text and figures are expressed as means ± SD. Two-group comparisons were analyzed by the two-tailed Student t-test for independent samples. Neurologic deficits were statistically evaluated by the Mann-Whitney nonparametric test. For all procedures, probability values of less than 0.05 were considered statistically significant.
RESULTS
Effect of MCA occlusion on infarct volume in COX-1–null mice
In mice killed 24 hours after MCA occlusion, the volume of the lesion was larger in COX-1 −/− mice than in COX-1 +/+ mice (Fig. 1;P < 0.05, t-test). The enlargement of the lesion persisted after volumetric data were corrected for swelling (Fig. 1). Cyclooxygenase-1 −/− mice had larger infarcts also in the striatum. To determine whether the enlargement of the infarct persisted, separate mice were killed 96 hours after MCA occlusion, and infarct volume was larger in COX-1 −/− mice (Fig. 1). In agreement with the infarct volume data, the motor deficits produced by MCA occlusion were worse in COX-1 −/− mice than in COX-1 +/+ mice (Fig. 2).
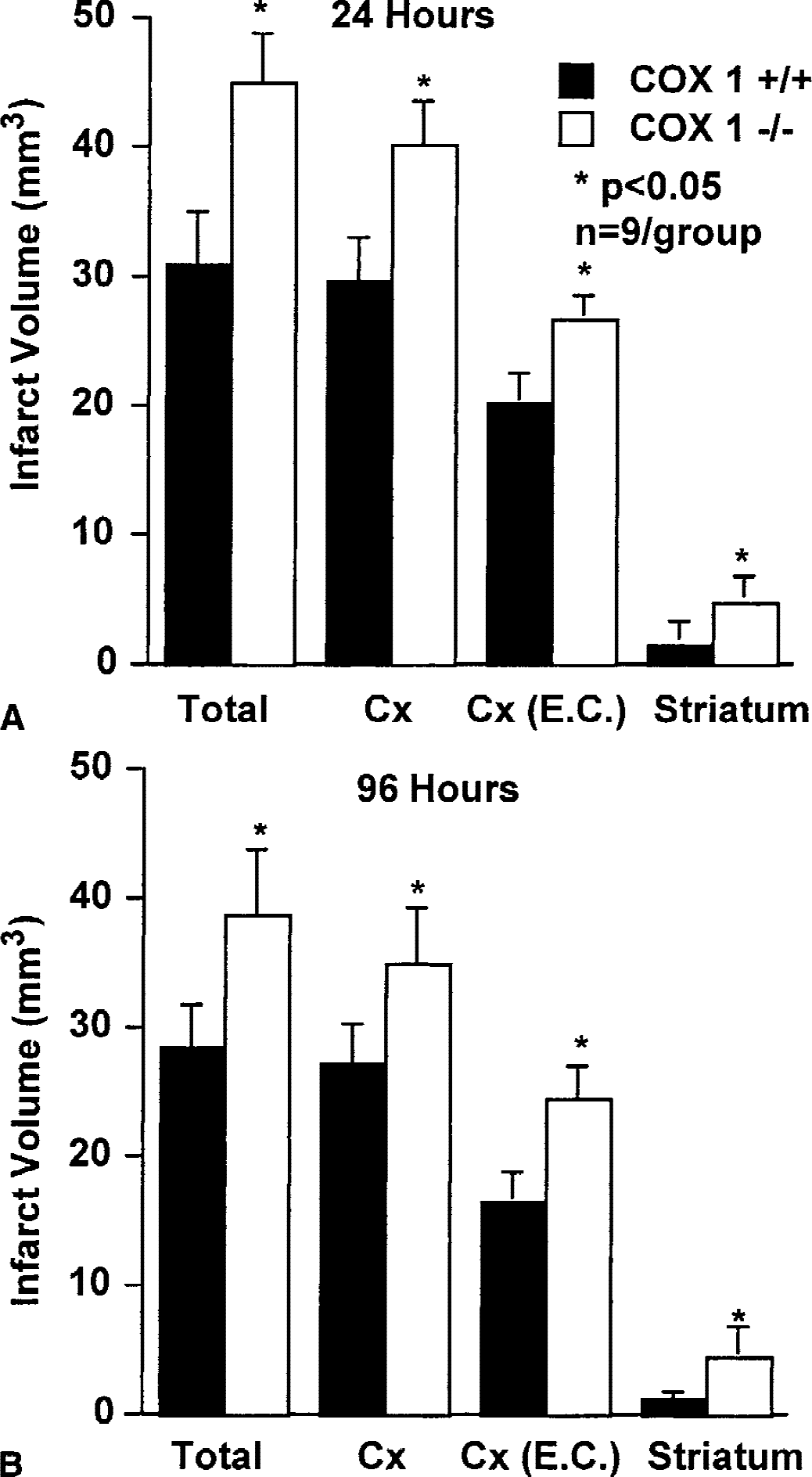
Effect of middle cerebral artery occlusion in COX-1 +/+ and −/− mice, 24
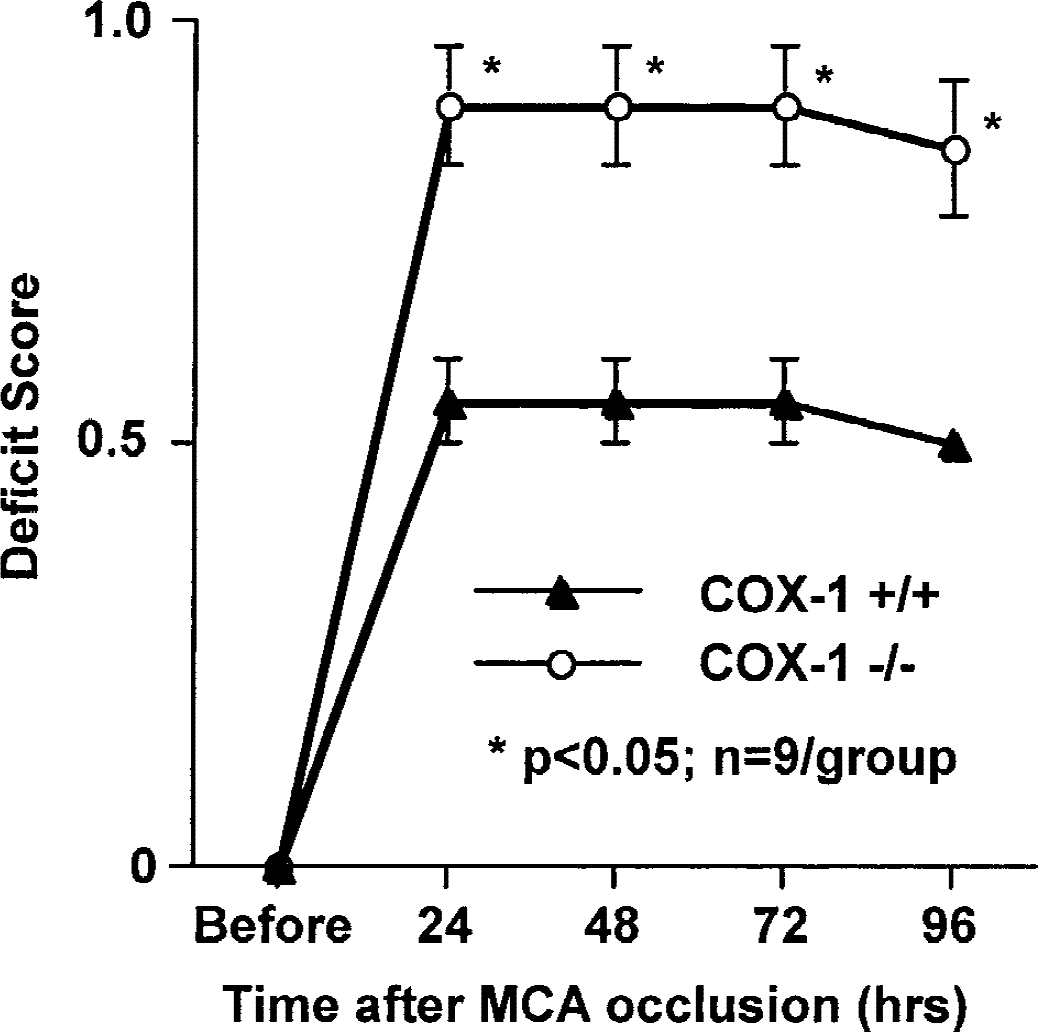
Neurologic deficits produced by middle cerebral artery occlusion in COX-1 +/+ and −/− mice. * P < 0.05 Mann-Whitney test.
Effect of excitotoxic injury in COX-1–null mice
Glutamate receptors play a critical role in the initiation of ischemic brain injury (Lee et al., 2000). Therefore, we sought to determine whether the increase in ischemic damage in COX-1–null mice could be attributed to increased susceptibility to glutamate receptor-mediated damage. The glutamate receptor agonists NMDA or kainate were microinjected directly in the cerebral cortex and injury volume was determined in thionin-stained brain sections 24 hours later. The damage produced by NMDA or kainate did not differ between COX-1 +/+ and −/− mice (P > 0.05)(Fig. 3).
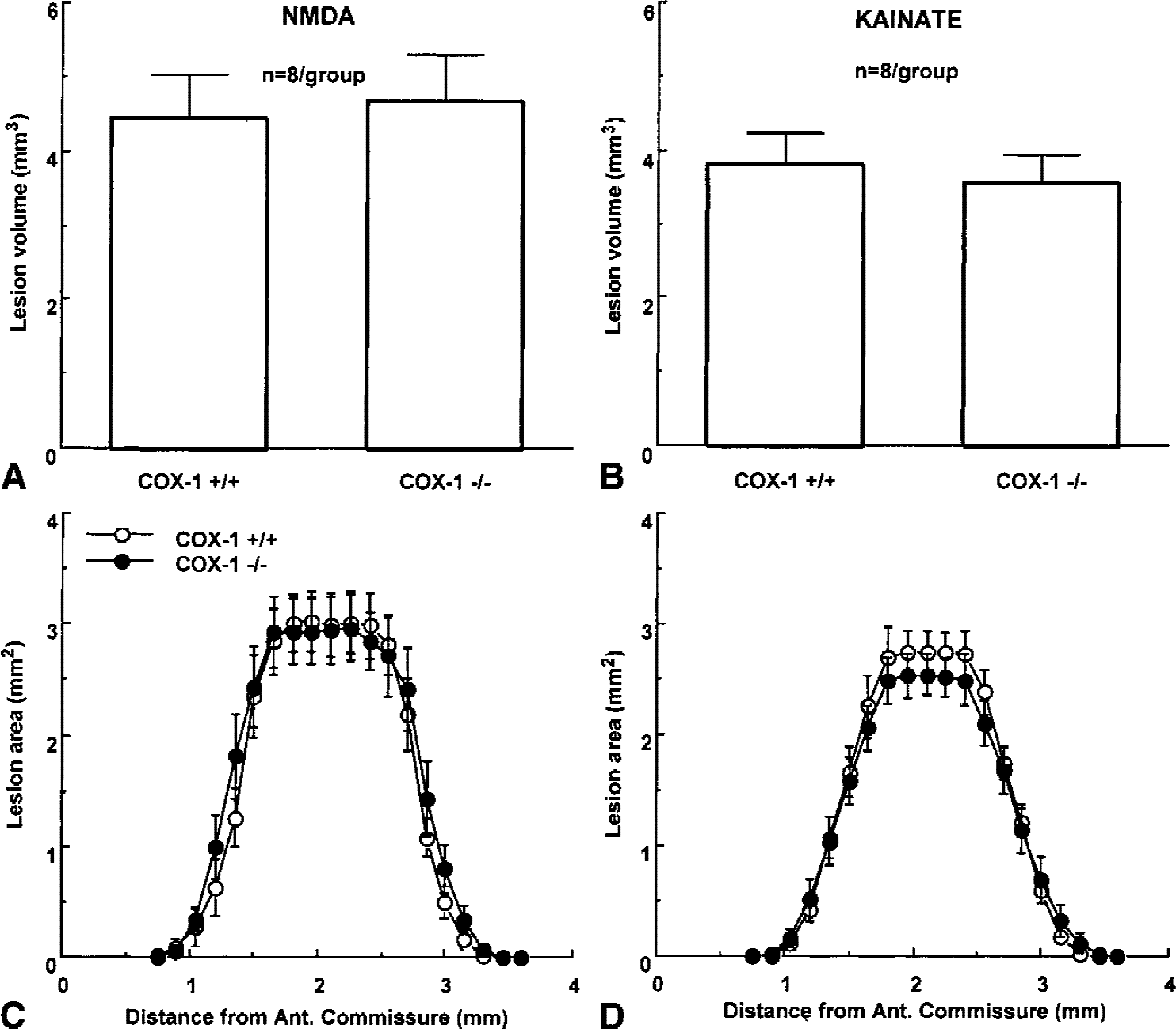
Effect of microinjection of NMDA
Effect of MCA occlusion on CBF in COX-1–null mice
The intensity of the ischemic insult has a profound impact on the ensuing brain damage (see Hossmann, 1994 for a review). Therefore, the effect of MCA occlusion on CBF was studied in COX-1 +/+ and −/− mice. Cerebral blood flow was monitored both in the center of the ischemic territory and in the peripheral region that is recruited into infarction in COX-1 −/− mice. As illustrated in Fig. 4, the reduction in CBF in the center of the ischemic territory did not differ between COX-1 +/+ and −/− mice (P > 0.05). However, the CBF reduction at the periphery of the ischemic area was more marked in COX-1 −/− mice than in COX-1 +/+ mice (Fig. 4;P < 0.05). These data show that the reduction in CBF in areas that are recruited into infarction in COX-1 −/− mice is more severe than that of COX-1 +/+ mice.
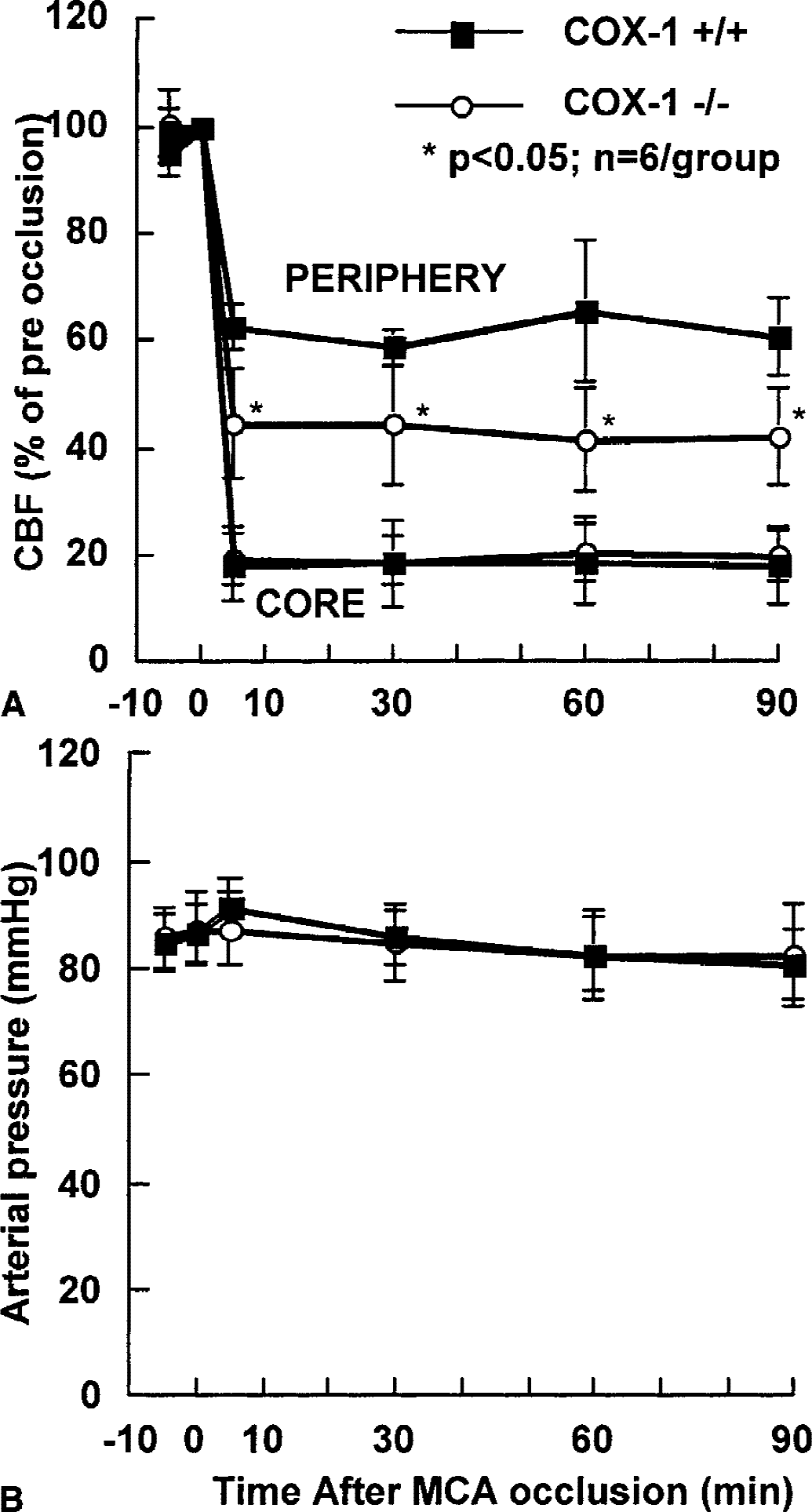
Effect of middle cerebral artery occlusion on cerebral blood flow
DISCUSSION
It has been demonstrated that COX-1–null mice subjected to MCA occlusion have larger infarcts than their wild-type littermates. The enlargement is observed both 1 and 4 days after ischemia, and is unrelated to the brain swelling that accompanies ischemic injury. The increase in infarct volume was associated with a worsening of the neurologic deficits, indicating that the increased neuropathological damage resulted in a poorer functional outcome. To provide insight into the mechanisms of the effect of COX-1 deletion, the role of COX-1 in the injury produced by activation of glutamate receptors, a major factor in the initiation of ischemic brain injury (Lee et al., 2000), was investigated. It was found that the damage produced by direct microinjection of NMDA or kainate into the cerebral cortex did not differ between COX-1 +/+ and −/− mice. This observation indicates that the increase in cerebral ischemic damage observed in COX-1–null mice is not caused by enhanced susceptibility to glutamate receptor-mediated neurotoxicity.
The role of hemodynamic factors in the increased brain damage produced by MCA occlusion was then investigated. Cyclooxygenase-1 is involved in selected vasodilatatory responses of the cerebral circulation (Niwa et al., 2001), which could play a role in the maintenance of CBF at the borderzone between the ischemic and normally perfused brain. In regions at the periphery of the ischemic territory, CBF reduction produced by MCA occlusion was more marked in COX-1–null mice than in wild-type littermates. This finding indicates that the collateral flow that arises from patent arteries adjacent to the occluded MCA is relatively impaired in COX-1–null mice. These observations collectively suggest that COX-1 reaction products facilitate collateral flow to the brain tissue supplied by the occluded MCA and, through this mechanism, protect marginally perfused regions at risk for infarction at the periphery of the ischemic territory. It must be stressed that the present results pertain to permanent cerebral ischemia and that that role of COX-1 in transient ischemia remains to be defined.
The differences in infarct volume between COX-1 +/+ and −/− mice cannot be attributed to heterogeneity of the genetic background of the mice (Choi, 1997), because COX-1–null mice were back crossed multiple times into the C57BL/6 strain. Furthermore, COX-1 +/+ littermates were used as controls. Therefore, the effects of genetic background on the outcome of cerebral ischemia were minimized (Choi, 1997). Similarly, the differences in postischemic CBF between COX-1 +/+ and −/− mice cannot be attributed to differences in arterial pressure or blood gases because these variables were carefully controlled and did not differ between groups. Furthermore, the cerebrovascular anatomy is not altered in COX-1–null mice (Niwa et al., 2001), a finding that rules out the participation of morphologic abnormalities of cerebral vessels in the greater CBF reduction observed in COX-1 +/+ mice after MCA occlusion. It is also unlikely that abnormalities in platelet aggregation contributed to the enlargement of infarct size in COX-1 −/− mice because COX-1–null mice have normal platelet aggregation (Langenbach et al., 1995). Therefore, the enlarged infarct in COX-1–null mice cannot be explained on the basis of differences in genetic background, cerebrovascular anatomy, and physiologic parameters or platelet aggregation.
Previous investigations on the role of COX in ischemic brain injury have pharmacologic inhibitors that do not discriminate between COX-1 and COX-2. Consequently, these studies have led to contradictory findings. Some investigators found that COX inhibition ameliorates ischemic injury, whereas others reported no effect or worsening of the damage (Cole et al., 1993; Johshita et al., 1989; Petersen, 1989). The results of the present study, in concert with recent data in COX-2–null mice (Iadecola et al., 2001), help reconcile these conflicting observations. Although COX-1 protects the brain from the consequences of focal cerebral ischemia, COX-2 contributes to ischemic damage. Thus, COX-2 inhibition with NS-398 or COX-2 genetic deletion reduces infarct volume (Iadecola et al., 2001; Nagayama et al., 1999). It is therefore likely that the results of studies using nonselective COX inhibitors differed depending on the degree of COX-1 or COX-2 inhibition achieved in the specific experimental preparation used.
Cyclooxygenase-1 has emerged as an important factor in the regulation of the cerebral circulation. Although resting CBF is reduced by COX-1 inhibition or genetic deletion, COX-1 reaction products are involved in the increases in CBF produced by hypercapnia and by the endothelium-dependent vasodilators bradykinin and A23196 (Niwa et al., 2001). Resting CBF is reduced by 15% to 20% in the cerebral cortex of COX-1–null mice, as assessed by the iodoantipyrine technique with quantitative autoradiography (Niwa et al., 2001). This finding indicates that following MCA occlusion in COX-1 −/− mice, absolute CBF is even lower than anticipated on the basis of the greater relative reduction detected by laser-Doppler flowmetry. However, measurement of CBF by quantitative techniques is needed to more firmly establish this finding. The exact cellular localization of COX-1 and the COX-1 reaction products responsible for the effects on postischemic CBF have not been established. However, anatomical and functional studies suggest that COX-1 is localized to cerebral blood vessels (Breder et al., 1992; Niwa et al., 2001).
Acetylsalicylic acid, an agent that inhibits COX-1, has been reported to reduce cerebral ischemic injury both in vivo and in vitro (Grilli et al., 1996; Khayyam et al., 1999). However, the effects of acetylsalicylic acid have been attributed to the finding that this agent, in addition to inhibiting platelet aggregation, blocks the action of transcription factors that contribute to ischemic brain injury (Grilli et al., 1996; Moro et al., 2000). Therefore, it would seem that in studies using acetylsalicylic acid, the deleterious hemodynamic effects of COX-1 inhibition were offset by the beneficial actions of inhibition of platelet aggregation and blockade of expression of deleterious genes.
In conclusion, the data show that COX-1–null mice have larger infarcts following MCA occlusion. This effect can be attributed to more severe ischemia in brain regions at the periphery of the ischemic territory. The present findings, in concert with data from COX-2–null mice (Iadecola et al., 2001), suggest that COX-1 and COX-2 play opposing roles in the mechanisms of focal cerebral ischemia, and help reconcile the conflicting results obtained by previous studies using nonselective COX inhibitors.
Footnotes
Acknowledgments:
The authors thank Ms. Joan Tetrault for maintaining the mouse colony and Ms. Andrea Hyde for editorial assistance.