
Abstract
Carboxypeptidase E, an exoprotease involved in the processing of bioactive peptides released by a regulated secretory pathway, was identified in a subtractive complementary DNA library derived from an ischemic rat brain by differential screening. In situ hybridization and immunocytochemical analysis showed the presence of carboxypeptidase E messenger RNA and protein in the cerebral cortex, thalamus, striatum, and hippocampus of a healthy rat brain. After 15 minutes of transient global ischemia followed by 8 hours of reperfusion, increased levels of carboxypeptidase E messenger RNA and protein were observed in the hippocampal CA1 and CA3 regions and in the cortex, as detected by Northern and Western blot analyses and in situ hybridization. After extended reperfusion (24 to 72 hours), both carboxypeptidase E messenger RNA and protein levels were decreased. The ischemia-induced changes in carboxypeptidase E expression suggest that this enzyme may play a role in modulating the brain's response to ischemia.
Current understanding of delayed cell death in the CA1 sector of hippocampus after transient global ischemia implicates cell death-regulatory genes, several of which have been identified (Choi, 1996; MacManus and Linnik, 1997). These regulatory genes include nerve growth factor, brain-derived neurotrophic factor, and other neuropeptides that protect against neuronal death after transient global cerebral ischemia and focal cerebral ischemia with reperfusion in vivo (Akins at al., 1996; Henrich-Noack et al., 1996; Saito et al., 1996; Sakata et al., 1998). The withdrawal of these factors results in apoptotic neuronal death in in vitro cell culture models (Barnes, 1988; Deckwerth and Johnson, 1993; Deckwerth et al., 1998; Raff et al., 1993), providing additional evidence that neuropeptides play a role in programmed neuronal cell death after ischemic injury.
Most known peptide growth factors and hormones are synthesized initially as larger inactive precursors, which are processed posttranslationally by endopeptidases in a restricted subcellular compartment, the secretory pathway. For many peptide precursors in the secretory pathway, such cleavage takes place at the carboxyl terminus of monobasic, dibasic, or multiple-basic amino acids, of which dibasic amino acids is the most abundant. After endoproteolytic cleavage, the remaining carboxyl basic residues are removed by the action of a carboxypeptidase (for reviews, see Fricker LD and Leiter EH, 1999; Rouille et al., 1995; Steiner DF, 1998; Seidah et al., 1999; Zhou et al., 1999). In parallel with posttranslational processing, the secretory peptides are sorted to either the constitutive secretory pathway or the regulated secretory pathway. In the nervous system and endocrine cells, neuropeptide neurotransmitters and peptide hormones are sorted into and released by the calcium-dependent, regulated secretory pathway.
Carboxypeptidase E (CPE; EC 3.4.17.10), also designated carboxypeptidase H or enkephalin convertase (Fricker and Snyder, 1983; Fricker et al., 1986; Fricker and Herbert, 1988; Smyth et al., 1989; Fricker et al., 1990), is a carboxypeptidase B-like enzyme expressed in neuroendocrine tissues. Carboxypeptidase E mediates the biosynthetic processing of many neuropeptide precursors, and may play a role in the sorting of neuropeptides (Cool et al., 1997). Therefore, CPE is used as a marker for peptide biosynthesis in the central nervous system (Fricker and Herbert, 1988; MacCumber et al., 1990), where its messenger RNA (mRNA) and protein distribution is well characterized (Lynch et al., 1990; MacCumber et al., 1990; Schafer et al., 1993). However the extent to which altered expression of CPE may be associated with acute cerebral dysfunction is unclear.
In the present study, we isolated a rat CPE gene from a subtractive ischemic complementary DNA (cDNA) library using differential screening. The CPE mRNA and protein were detected in a healthy brain. After ischemia, both mRNA and protein levels were upregulated in the selectively vulnerable CA1 and CA3 hippocampal neurons, and in the cytosol and apical dendrites of pyramidal cells in the cortex. These findings suggest that CPE may help to regulate the vulnerability of certain neuronal populations to ischemia.
EXPERIMENTAL PROCEDURES
Ischemia model
Animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize animal suffering and reduce the number of animals used. Global ischemia was induced using the four-vessel occlusion method (Pulsinelli et al., 1982) with modifications described previously (Jin et al., 1999a; Simon et al., 1991). Male Sprague-Dawley rats (300–330 g) were ventilated with 1.5% isoflurane in a mixture of 25% oxygen and 73.5% nitrous oxide. The left femoral artery was cannulated for blood pressure monitoring and blood gas sampling. Rectal temperature was continuously monitored and kept at 37 to 37.5°C with a heating pad. Brain temperature was monitored with a 29-Ga thermocouple implanted in the left striatum and allowed to decrease to 36°C during ischemia, but was otherwise maintained at 37°C. Animals were placed in a Kopf stereotaxic frame. Both vertebral arteries were coagulated and transected at a level between the first and the second cervical vertebrae. After the external carotid arteries were ligated, global ischemia was induced by common carotid artery occlusion with microvascular clips for 15 minutes and confirmed electroencephalographically (isoelectric within 5 seconds). This duration of ischemia causes injury to selectively vulnerable neurons of the hippocampus and cortex (Simon et al., 1991). After sham surgery (nonischemic control) or at 4, 8, 16, 24, or 72 hours of reperfusion after ischemia (n = 4 per time point), animals were killed and their brains were perfused with 0.9% saline followed by 4% paraformaldehyde in phosphate-buffered saline (pH 7.4).
Construction of subtractive cDNA library and differential screening
Rat mRNA was pooled from animals subjected to 15 minutes of global ischemia produced by the four-vessel occlusion method with death at 0, 4, 8, 16, 24, and 72 hours of reperfusion after ischemia (n = 4 per time point). A subtractive cDNA library from the ischemic rat brain was constructed and differentially screened as described previously (Jin et al, 1999b).
Northern blot analysis
Total RNA (20 mg) from control and ischemic (15 minutes) rat hippocampi (n = 4 per experimental condition) was denatured, separated by electrophoresis through a 1.2% agarose-formaldehyde gel, and blotted onto a Hybond-N+ membrane (Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.). Northern blotting was performed using 32P-dATP–labeled CPE cDNA as a probe, as previously described (Jin et al., 1999b). The integrity and amount of RNA loaded onto gels were assessed by hybridization with an 18S rRNA oligonucleotide probe (5′-ACGGTATCTGATCGTCTTCGAACC-3′) designed from the rat DNA sequence.
Sequence analysis
The cDNA clones were sequenced on both strands using a Sequenase II kit (United States Biochemical, Cleveland, OH, U.S.A.) according to the manufacturer's instructions. BLAST searching of the GenBank database was performed using the National Center for Biotechnology Information on-line service. Sequence analysis was performed using MacVector software (International Biotechnologies, New Haven, CT, U.S.A.).
In situ hybridization
In situ hybridization was performed according to Jin et al. (1999b) using 20-mm frozen sections from control and ischemic (15 min) rat brains (n = 4 per condition). The hybridization probes of specific 35S-labeled RNA transcripts complementary to the mRNA were obtained after in vitro transcription of the cDNA insert from the linearized CPE plasmid, using SP6 RNA polymerase for sense probes or T7 RNA polymerase for antisense probes. The transcription was performed using a transcription kit (Stratagene, La Jolla, CA, U.S.A.). The transcription mixture, containing 0.5 mM each of CTP, GTP, and ATP, 12 mM 35S-UTP (NEN Life Science Products, Boston, MA, U.S.A.), 40 U RNase inhibitor, 2 mmol/L spermidine, 10 mmol/L dithiothreitol, and 10 U T7-or SP6-RNA polymerase, was incubated for 1 hour at 37°C, followed by DNase digestion of the template (RQI DNase, Promega, Madison, WI, U.S.A.), phenol/chloroform extraction, and ethanol precipitation. In situ hybridization was performed as previously described (Jin et al., 1999b).
Western blot analysis
Proteins were extracted from control and ischemic hippocampi (n = 4 per experimental condition) and protein concentration was determined using a Bio-Rad protein assay (Bio-Rad, Hercules, CA, U.S.A.). Western blot analyses were performed as described previously (Jin et al., 1999b); 100 mg protein was added per lane, and filters were stripped and reprobed for β-actin to control for possible lane-to-lane differences in loading. The primary antibody for CPE was a mouse monoclonal anti-CPE antibody (Transduction Laboratory, Bluegrass-Lexington, KY, U.S.A.) used at a 1:500 dilution. The secondary antibody was a horseradish peroxidase-conjugated sheep antimouse immunoglobulin G (Amersham Pharmacia Biotech). Protein-bound antibody was detected by chemiluminescence methods (NEN Life Science Products).
Immunocytochemistry
Immunocytochemical analysis of CPE on paraffin-embedded brain sections was performed as previously described (Jin et al., 1999b). Briefly, sections were deparaffinized with xylene and ethanol and were rehydrated. Endogenous peroxidase activity was blocked by incubating the sections with 1% H2 O2 for 15 minutes. Incubation with primary antibody was perform at 4°C overnight in a phosphate-buffered saline buffer system containing 2% horse serum, 0.2% Triton X-100, and 0.1% bovine serum albumin. The subsequent incubation with fluorescein isothiocyanate-conjugated goat antimouse immunoglobulin G (Vectastain Elite ABC, Vector Co., Burlingame, CA, U.S.A.; 1:200) was performed at room temperature for 1 hour in the same buffer system. After immunostaining, sections were examined using a fluorescence microscope. Alternate sections used as controls were incubated with buffer and secondary antibody without the primary antibody.
For triple-fluorescence labeling, deparaffinized sections were first treated with unmasking solution (Vector Co.), as recommended by the manufacturer. Sections were then incubated with a mixture of two appropriate primary antibodies raised in different species (mouse anti-CPE with rabbit anti-NF-68 (Chemicon International, Temecula, CA, U.S.A.; 1:100 or rabbit antiglial fibrillary acidic protein (Sigma Chemical Co., St. Louis, MO, U.S.A.; 1:150), respectively, at 4°C overnight. The following day, after extensive washing steps, sections were incubated with two different secondary antibodies: fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin G, and rhodamine-conjugated rat-absorbed donkey antirabbit immunoglobulin G (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, U.S.A.; 1:200). Hoechst 33258 (2′-[4-hydroxyphenyl]-5-[4-methyl-1-piperazinyl]-2,5′-bi-1H-benzimidazole, Sigma Chemical) was added to the secondary antibody mixture (20 μg/mL) to counterstain nuclei. The three fluorescence signals were detected with a Nikon E800 epifluorescence microscope (Nikon Inc., Melville, NY, U.S.A.) with excitation/emission wavelengths of 535/565 nm for rhodamine (red), 470/505 nm for fluorescein isothiocyanate (green), and 360/400 nm for Hoechst 33258 (blue). Results were recorded with a Magnifire digital color camera (Optronics Corporation, Goleta, CA, U.S.A.).
Means for quantifying mRNA and protein expression
Differences in mRNA or protein expression observed with in situ hybridization and Northern or Western blotting analyses were quantified by densitometric methods using the MCID Image Analysis System (Image Research, St. Catherine's, Ontario, Canada). Statistical analyses for differences between groups were performed by analysis of variance and post hoc t-tests. A value of P < 0.05 was considered significant.
RESULTS
Isolation of carboxypeptidase E cDNA
To identify enhanced expression of cell death-regulatory genes after transient global ischemia, a subtractive cDNA library was constructed using hippocampal mRNA from ischemic rat brains (Jin et al., 1999b). One gene isolated by differential screening was 1.8 kb long and encoded a complete open-reading frame predicting 476 amino acids. A BLAST search of the GenBank online database showed that its sequence was identical to rat CPE at both the nucleotide and amino acid levels (GenBank accession number: NM013128) (Fricker et al., 1989; Manser et al., 1990).
Expression of carboxypeptidase E mRNA
Identification of CPE cDNA in the previously described subtractive cDNA library study clearly indicated an upregulation of CPE expression in the rat brain after ischemia. To determine the temporal pattern of changes in CPE expression, Northern blot analysis was performed using a CPE cDNA probe. Total RNA was isolated from the hippocampi of brains taken after different intervals of reperfusion (n = 4 per experimental condition) after 15 minutes of global ischemia. As shown in Fig. 1, a single transcript was detected in the sham-operated hippocampi at approximately 2 kb, which is close to the predicted size of the CPE transcript. The CPE mRNA level was increased to the control levels at 8 hours of reperfusion after ischemia. At 24 hours, however, the CPE mRNA level was greatly decreased. Filters were stripped and rehybridized with an 18S rRNA oligonucleotide probe to verify that identical amounts of RNA were loaded per lane.
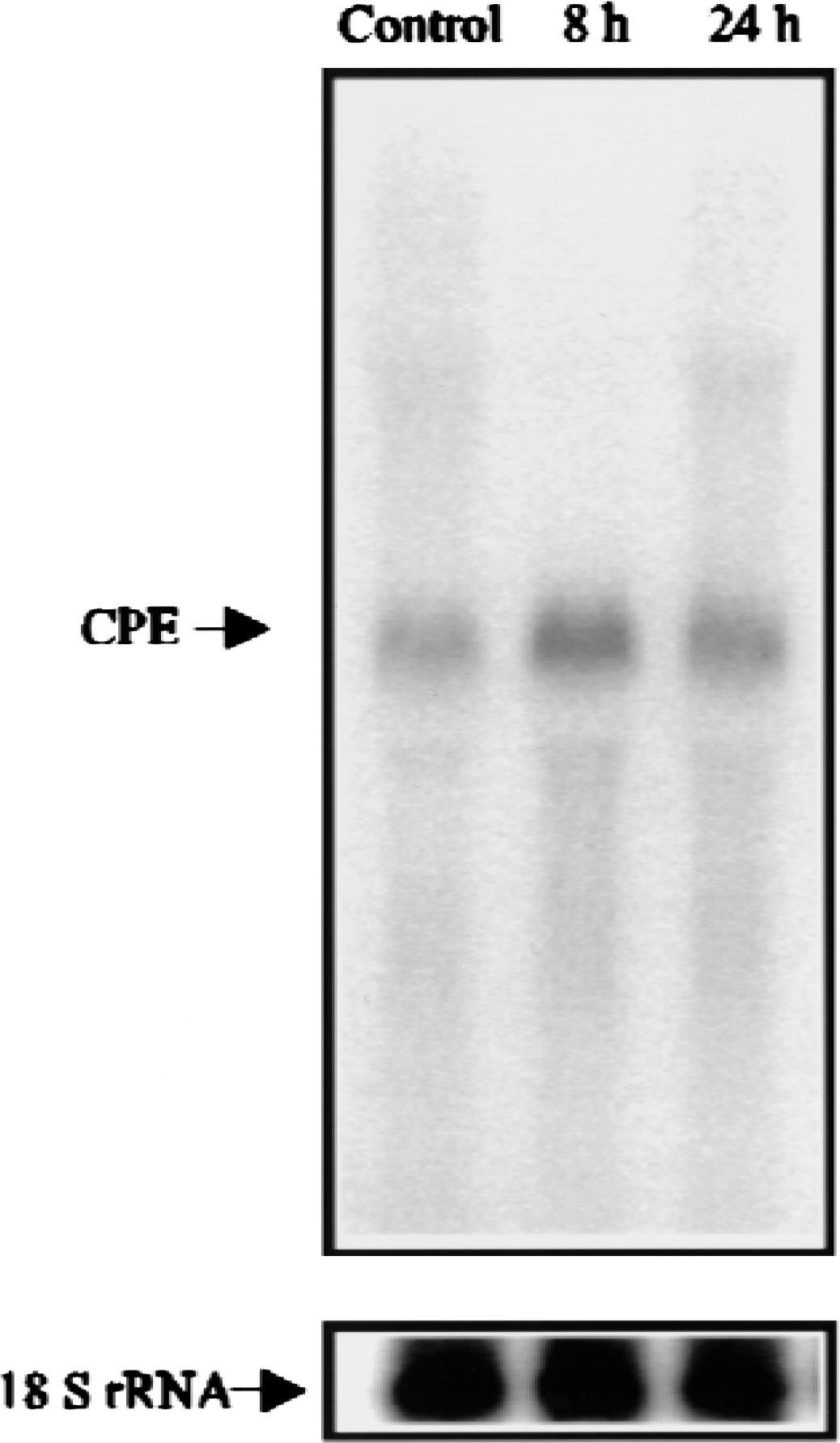
Northern blot analysis of carboxypeptidase E (CPE) expression in the hippocampus. Total RNA (20 mg/lane) from hippocampal tissue removed from sham-operated rats (control) or at the indicated reperfusion times following 15 minutes of global cerebral ischemia was analyzed by Northern blot analysis using a CPE complimentary DNA probe (
Distribution of carboxypeptidase E mRNA
The distribution of CPE mRNA was studied in sham-operated and ischemic rat brain sections by in situ hybridization. The CPE mRNA was expressed in the thalamus, striatum, and cerebral cortex of control brains, as observed previously (MacCumber et al., 1990; Schafer et al., 1993). The CPE mRNA expression was also evident in the control hippocampal CA1 and CA3 regions, but little expression was seen in the granule cells of the dentate gyrus (Fig. 2A). By 8 hours of reperfusion after global ischemia, there was significantly increased expression in CA1 and CA3 (Figs. 2A and B). Although the increase in CA1 diminished at 24 hours and disappeared at 72 hours of reperfusion, CPE mRNA remained increased in CA3 at 24 hours, and returned to basal levels at 72 hours of reperfusion. Sections hybridized with a CPE sense probe showed only background activity. In contrast to findings in the hippocampus, expression of CPE mRNA in the cerebral cortex, striatum, and thalamus was not significantly altered by ischemia, as determined by optical density measurements on hybridized sections (data not shown).
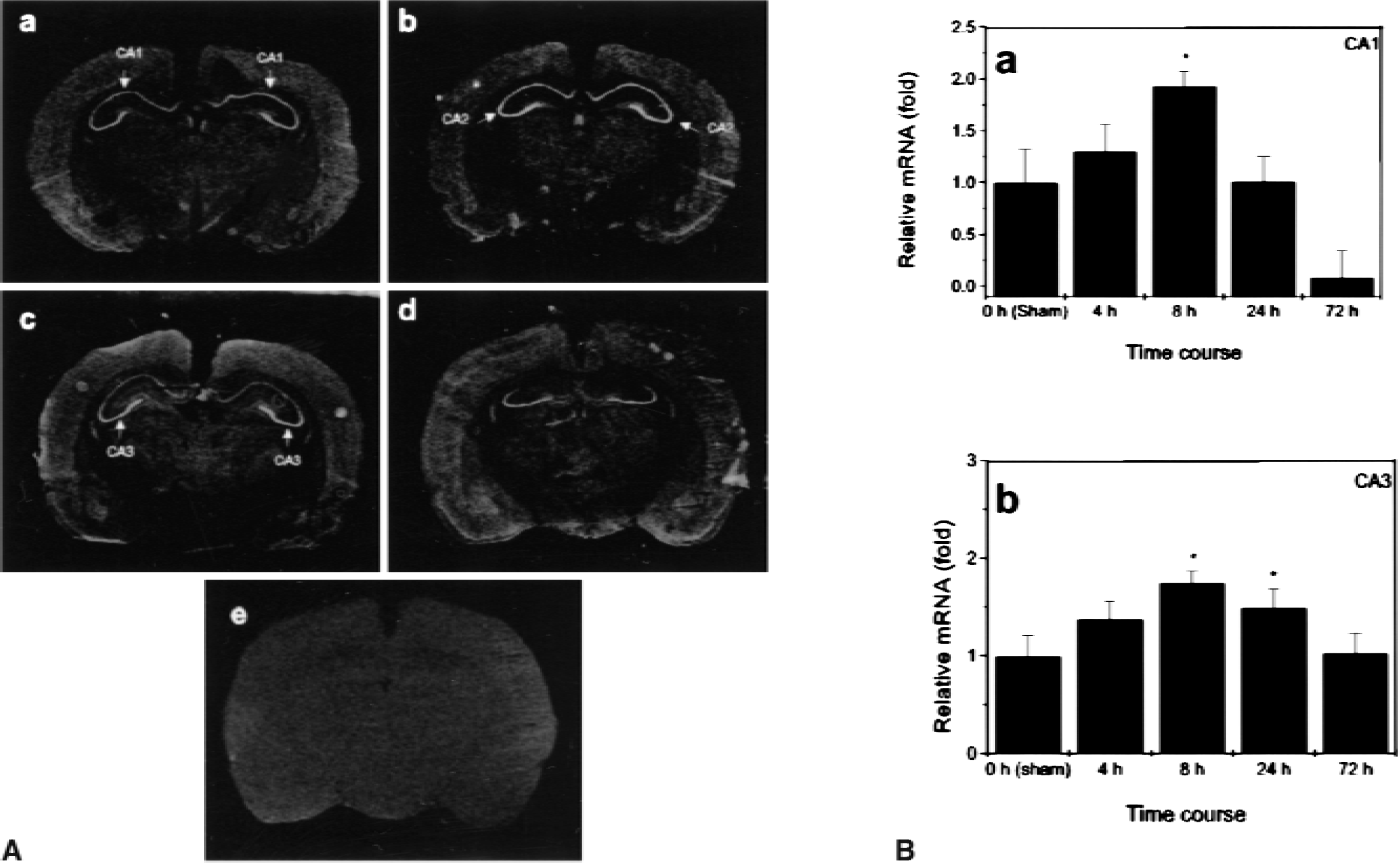
In situ hybridization analysis of carboxypeptidase E (CPE) messenger RNA (mRNA). In situ hybridization was performed using CPE complimentary RNA probes in sham-operated control (a) and post-ischemic brains at 8 (b), 24 (c), and 72 hours (d) of reperfusion following 15 minutes of global cerebral ischemia. Sense probe control hybridized with post-ischmic brain at 24 hours (e) (
Expression of carboxypeptidase E protein
The increased level of CPE mRNA after ischemia suggested that CPE protein might also increase in the ischemic brain. Expression of CPE protein after cerebral ischemic injury was investigated by Western blot analysis using a monoclonal antibody against human CPE (amino acids 49–200). In hippocampi of sham-operated animals, the predominant form of CPE protein showed a relative molecular mass of 50 kDa (Fig. 3). In ischemic hippocampi, a higher level of CPE protein was detected at 8 hours of reperfusion. At 72 hours, the level of CPE protein was decreased, which is similar to the changes in CPE mRNA levels observed by Northern blot analysis,. Interestingly, the CPE protein in the ischemic hippocampi appeared to be approximately the size of the proenzyme form of CPE (58kDa).
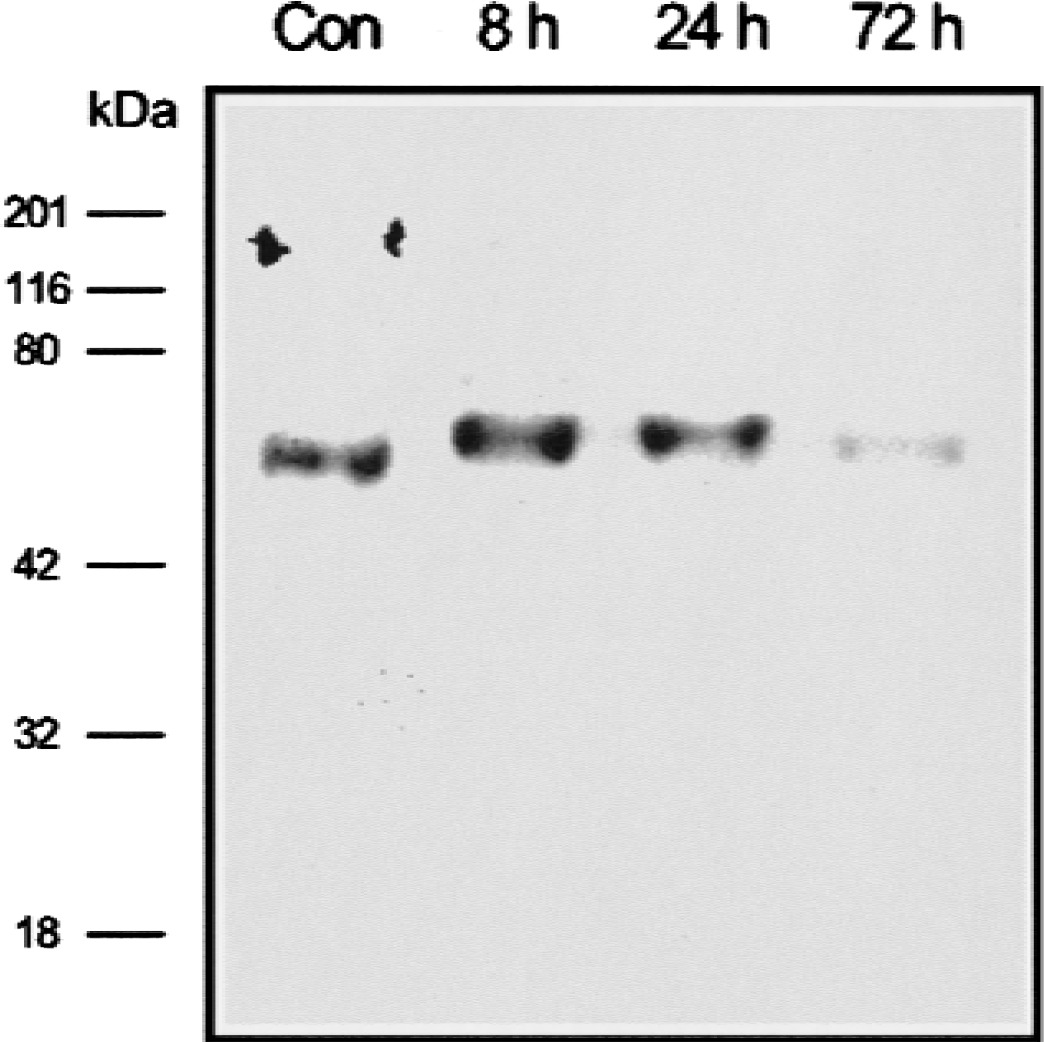
Western blot analysis of carboxypeptidase E (CPE) protein expression in a whole hippocampus. Protein was extracted from the hippocampus of nonischemic control rat brains or from ischemic rat brains reperfused for 8, 24, or 72 hours (100 mg/lane, pooled from 4 animals). Western blot analysis was performed using a mouse monoclonal antibody against CPE.
Distribution of CPE protein
Carboxypeptidase E protein expression in the rat brain was detectable by immunocytochemistry in the hippocampus, cerebral cortex, thalamus, and striatum with the same antibody used for Western blot analysis. In control hippocampi, expression of CPE was mainly seen in fibers extending from CA1 neurons; a similar pattern was seen in neurons in the internal pyramidal layer of the cerebral cortex (Fig. 4). After 4 to 8 hours of reperfusion, the intensity of CPE immunostaining increased in the cytosol and processes of CA1 pyramidal cells; however, at 24 and 72 hours after ischemia, staining intensity was markedly decreased (Fig. 4). The expression pattern of CPE protein in CA3 was similar to that in CA1 at 4 to 8 hours of reperfusion, with a more dramatic increase in expression level at 8 hours; staining was decreased at 72 hours, but was still detectable (Fig. 4). The CPE protein was also detectable in apical dendrites of pyramidal cells in the internal pyramidal layer of the cerebral cortex from sham-operated brains, and was increased at 4 and 8 hours before declining (Fig. 4). Double-labeling sections for CPE and NF-68 (Fig. 5A) or CPE and glial fibrillary acidic protein (Fig. 5B) showed that CPE protein was expressed mainly in neurons rather than in astrocytes.
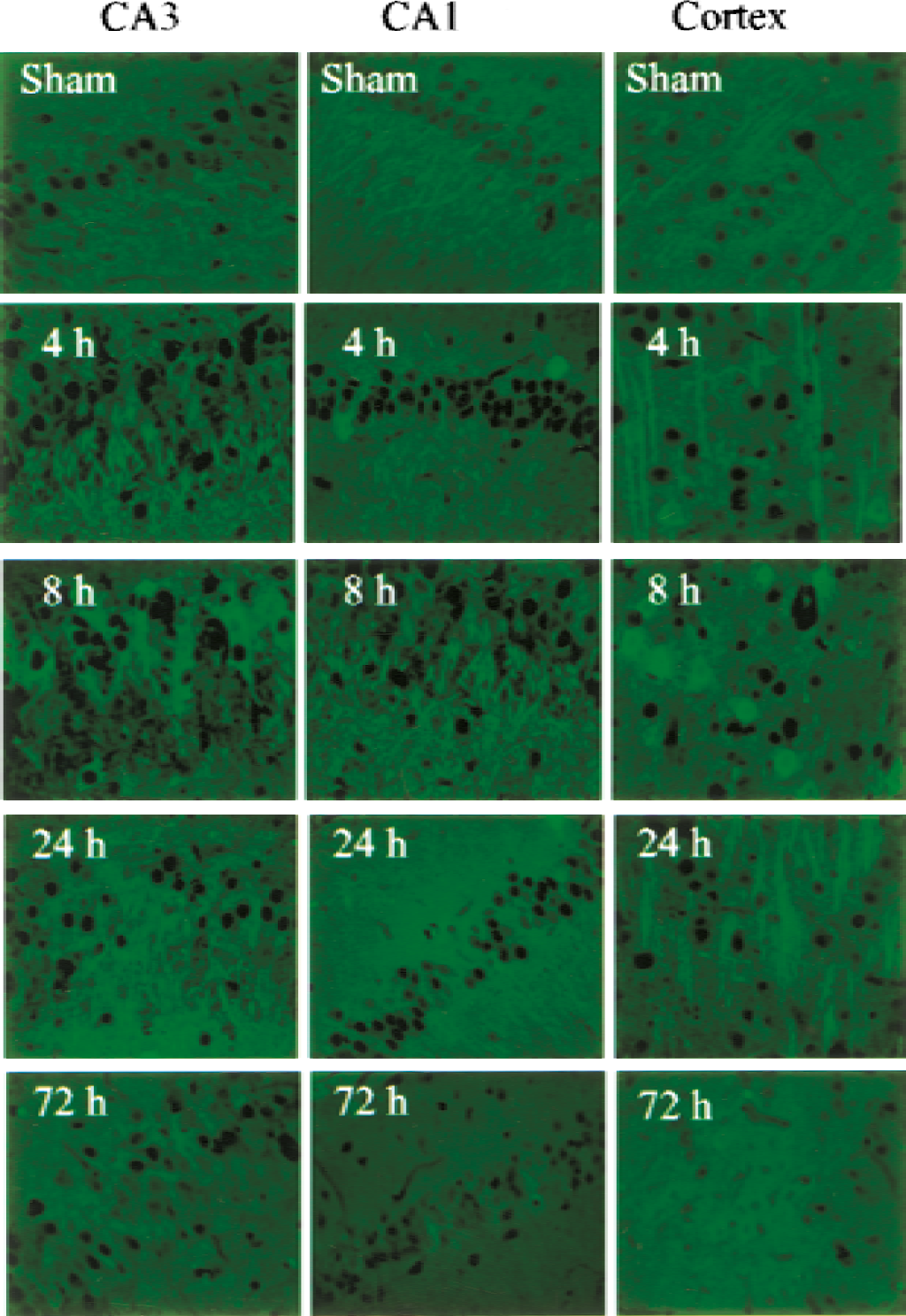
Immunocytochemical analysis of CPE protein expression. Paraffin-embedded sections were prepared from different regions of sham-operated rat brains or from brains after 15 min of ischemia followed by reperfusion for the indicated times. Immunostaining was performed using the same antibody used for Western blotting in Fig. 3, at a dilution of 1:100. The scale bars represent 50 μm.
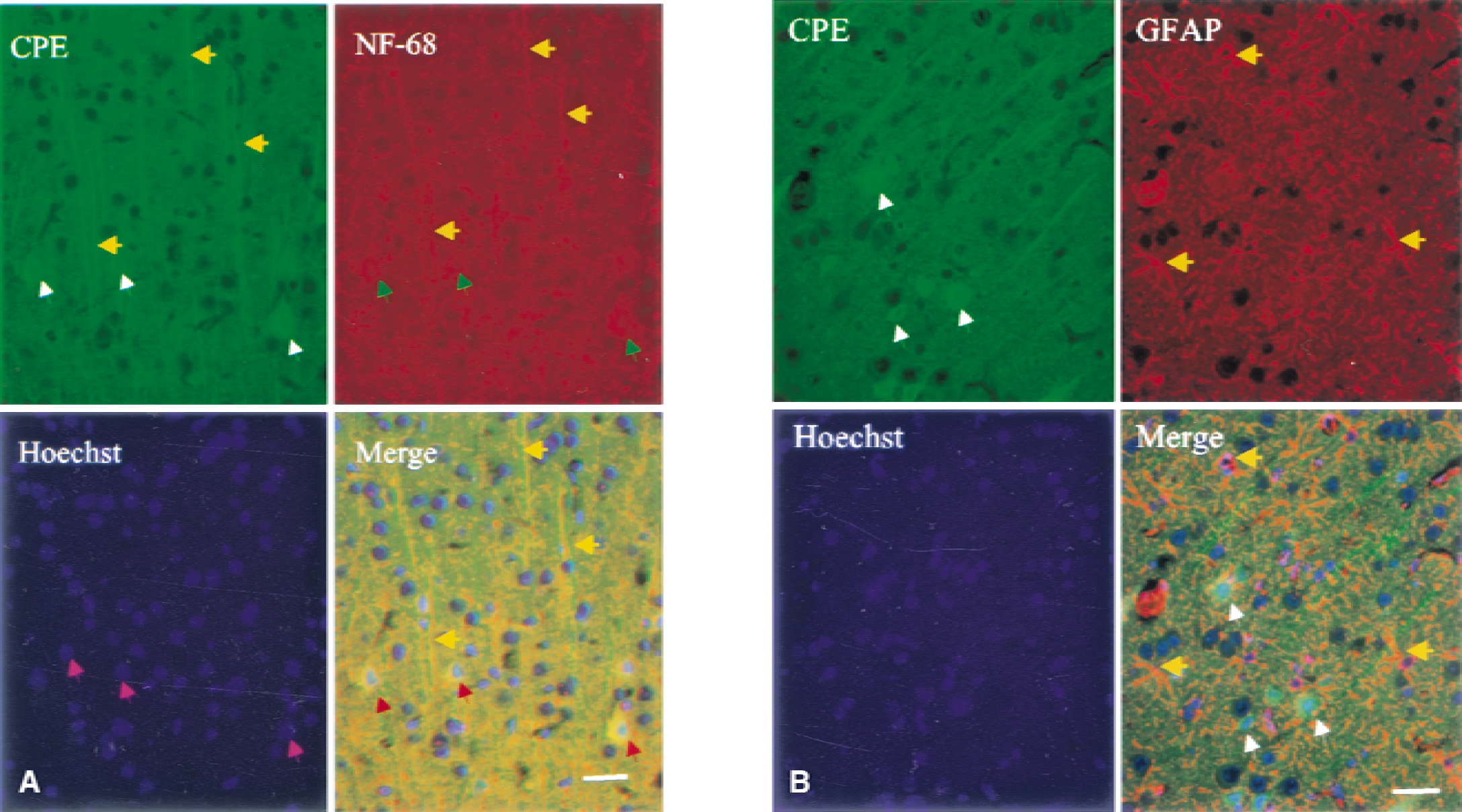
Identity of carboxypeptidase E (CPE) protein-expressing cells. Triple fluorescence labeling with antibodies against CPE, NF-68 (to identify neuronal cells) and Hoechst 33258 (to label nuclei) was performed on sections prepared from cerebral cortex obtained after ischemia and 8 hours of reperfusion. The CPE was expressed in the cytosol (white arrows) and apical dendrites (yellow arrows) of pyramidal neurons (red arrows) in cortical layer V. Pink arrows indicate pyramidal cell nuclei. The merged image shows the expression of CPE protein in NF-68-positive (neuronal) cells (
DISCUSSION
In the present study, we isolated a CPE gene from a subtractive ischemic rat brain cDNA library using differential screening. Northern and Western blot analyses showed that the expression of CPE mRNA and protein was increased in ischemic brains. In situ hybridization showed that CPE mRNA was increased most prominently in the CA3 region of the hippocampus at 8 hours of reperfusion. However, CPE expression was barely detectable in the hippocampal CA1 region at 72 hours. Immunocytochemical analysis showed predominant expression of CPE protein in neurons, and progressively increased expression in hippocampal CA3 pyramidal cells and pyramidal neurons of the cortical layer V for up to 8 hours after ischemia. The discordance between CPE mRNA (unchanged) and protein expression (increased) in cortex suggests the existence of additional mechanisms for regulating CPE protein expression that affect, for example, mRNA nuclear export, subcellular distribution or stability, or protein translation or degradation (Pradet-Balade et al., 2001). The increased CPE protein expression was observed in neuronal cell bodies, processes, and fibers. These results suggest that a neuropeptide processing enzyme, CPE, is responsive to brain ischemia.
Carboxypeptidase E is expressed widely in brain tissue (Lynch et al., 1990; MacCumber et al., 1990; Shafer et al., 1993). In addition to its function as a carboxypeptidase to remove C-terminal basic residues from neuropeptide intermediates (Fricker and Herbert, 1988; Fricker et al., 1991), CPE has also been shown to play a role in directing neuropeptide routing into the regulated secretory pathway of neuroendocrine cells (Cool et al., 1997). A point mutation in the CPE gene in mice (cpefat/cpefat) yields an unstable and inactive CPE protein, resulting in defective processing of a number of neuropeptides and peptide hormones, including neuropeptides that may have protective effects in cerebral ischemia (Antonawich et al., 1999; Gustafson et al., 1999; Huh et al., 1997; Yasui and Kawasaki, 1995; Yu et al., 1997). These neuropeptides include proopiomelanocortin-derived peptides (Cool et al, 1997; Shen and Loh, 1997), growth hormone (Shen and Loh, 1997), substance P (Perloff et al., 1998), and cholecystokinin (Wang et al., 1998). Thus, changes in CPE expression following cerebral ischemia may lead to changes in the production and secretion of protective neuropeptides in brain, and thereby help to mediate the response of neuronal cells to ischemia. Studies from other laboratories have shown that the expression of both brain-derived neurotrophic factor and its receptor, tyrosine kinase B, is increased after global ischemia, and that postischemic intracerebroventricular infusion of brain-derived neurotrophic factor prevents neuronal death in the vulnerable CA1 region of the hippocampus (Kiprianova et al., 1999).
Putative mediators of ischemic neuronal death, such as nitric oxide, can reduce cellular CPE activity in vitro (Devi et al., 1994). In PC12 cells, nerve growth factor markedly induces the expression of CPE (Laslop and Tschernitz, 1992), whereas depletion of nerve growth factor from differentiated PC12 cells leads to programmed cell death (Mayumi-Matsuda et al., 1999). One mechanism underlying cell death following withdrawal of nerve growth factor could, therefore, involve impaired neuropeptide processing by CPE. The observation that after transient ischemia, the increase in CPE expression in the hippocampal CA3 region was greater and more sustained than in the more vulnerable CA1 region, suggests a role for CPE in promoting the survival of ischemic neurons.
In summary, the neuropeptide-processing enzyme CPE shows increased expression in hippocampal CA3 and cortical neurons, which survive global cerebral ischemia. Carboxypeptidase E also shows an initial increase in expression in CA1 neurons, which are preferentially vulnerable to ischemia; however, this increase is less sustained than those in the CA3 and cortex and is followed by a pronounced decrease. Because CPE is involved in the processing of potentially neuroprotective peptides, and because its expression is induced by neurotrophic factors and impaired by cytotoxic factors such as nitric oxide, local levels of CPE expression and activity may help to regulate delayed cell death in the ischemic brain.