
Abstract
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a hematopoietic cytokine responsible for the proliferation, differentiation, and maturation of cells of the myeloid lineage, which was cloned more than 20 years ago. Here we uncovered a novel function of GM-CSF in the central nervous system (CNS). We identified the GM-CSF α-receptor as an upregulated gene in a screen for ischemia-induced genes in the cortex. This receptor is broadly expressed on neurons throughout the brain together with its ligand and induced by ischemic insults. In primary cortical neurons and human neuroblastoma cells, GM-CSF counteracts programmed cell death and induces BCL-2 and BCL-Xl expression in a dose- and time-dependent manner. Of the signaling pathways studied, GM-CSF most prominently induced the PI3K-Akt pathway, and inhibition of Akt strongly decreased antiapoptotic activity. Intravenously given GM-CSF passes the blood—brain barrier, and decreases infarct damage in two different experimental stroke models (middle cerebral artery occlusion (MCAO), and combined common carotid/distal MCA occlusion) concomitant with induction of BCL-Xl expression. Thus, GM-CSF acts as a neuroprotective protein in the CNS. This finding is remarkably reminiscent of the recently discovered functionality of two other hematopoietic factors, erythropoietin and granulocyte colony-stimulating factor in the CNS. The identification of a third hematopoietic factor acting as a neurotrophic factor in the CNS suggests a common principle in the functional evolution of these factors. Clinically, GM-CSF now broadens the repertoire of hematopoietic factors available as novel drug candidates for stroke and neurodegenerative diseases.
Keywords
Stroke and chronic neurodegenerative disorders have a complex pathophysiology with multiple mechanisms active simultaneously. Several pharmacological strategies for neuroprotection such as counteracting excitotoxicity by blocking NMDA (N-methyl-
One characteristic of endogenous neuroprotectants is their genomic regulation by cerebral ischemia or related paradigms. This feature can therefore be used to identify novel candidates. A particular interesting area for studying gene expression changes in relation to ischemia is the infarct adjacent zone (penumbra), where neurons face the decision to live on or die. Infarct enlargement over time occurs at the expense of the penumbral zone, and neuroprotective approaches ideally target events in this area at risk. Using a sensitive fragment-display technique, we have previously conducted expression profiling experiments in the peri-infarct rat sensorimotor cortex after induction of defined ischemic lesions with the photothrombotic model (Kruger et al, 2006). Among approximately 70 genes found in this screen as being upregulated, we surprisingly identified the receptor for another hematopoietic factor, granulocyte-macrophage colony-stimulating factor (GM-CSF).
Granulocyte-macrophage colony-stimulating factor (CSF2) is a glycoprotein of 127 amino acids traditionally viewed as a growth and differentiation factor necessary for the development of hematopoietic progenitor cells into granulocytes, macrophages, and dendritic cells (Metcalf, 1989). Granulocyte-macrophage colony-stimulating factor was cloned initially more than 20 years ago as a factor stimulating the growth of macrophage/granulocyte-containing colonies in soft agar cultures (Wong et al, 1985). Granulocyte-macrophage colony-stimulating factor exerts its function via the action on the GM-CSF receptor, which is composed of the GM-CSF receptor α- and the β-subunit which is not directly involved in binding GM-CSF. Granulocyte-macrophage colony-stimulating factor receptors are known to be expressed on the cell surface of myeloid cells and also on non-hematopoietic cells such as endothelial cells and small-cell lung carcinoma cells. Granulocyte-macrophage colony-stimulating factor is clinically employed for the physiological reconstitution of hematopoiesis in all diseases characterized either by an aberrant maturation of blood cells or by a reduced production of leukocytes (Dale, 2002). Granulocyte-macrophage colony-stimulating factor is generally well tolerated with rare serious complications.
Here, we report that, after EPO and G-CSF, GM-CSF is the third hematopoietic factor that has a protective function in neurons.
Materials and methods
Cloning of the Rat Granulocyte-Macrophage Colony-Stimulating Factor α-Receptor
Restriction-mediated differential display was performed on rat cortical tissue biopsies as described previously in detail (Schneider et al, 2004). Full-length sequences were obtained by using a polymerase chain reaction (PCR)-based cloning approach (Shepard and Rae, 1997) with the following primer combinations from the 3′-fragment obtained from the restriction-mediated differential display screening procedure: CGGGATCCGGGACCGCGTATCT GATGACGAGCGTGTCAA, CTCGGAGACGCTGAGGAAG GACCTG, and CTGCGGCCCTAGACCACGCCCACCGCTC CCCGTGACGTCG. Clones were sequenced on both strands using an ABI3700 sequencer.
Quantitative Polymerase Chain Reaction
Quantitative polymerase chain reaction was performed using the Lightcycler system (Roche Diagnostics, Basel, Switzerland) with SYBR-green staining of DNA double strands on threefold serially diluted cDNA samples. Cycling conditions were as follows: 5 secs 95°C, 10 secs 63°C, and 30 secs 72°C with a measuring temperature of 86°C for GM-CSF (primers CTGGAGAACGAAAAGAAC GAAGAC, TCAAAAGGGATATCAAACAGAAAG), and 80°C for GM-CSF receptor α (primers ACGTCGTTGGCT CAGTTATGTC and ATTTATGTCAGAGATGGAGGATGG). Specificity of product was ensured by melting point analysis and agarose gel electrophoresis. cDNA content of samples was normalized to the expression level of cyclophilin (primers: ACCCCACCGTGTTCTTCGAC and CATTTGCCATGGACAAGATG). Relative regulation levels were derived after normalization to cyclophilin, and compared to the sham-operated animals.
BCL-Xl Expression In Vivo
Adult Wistar rats were subjected to 90 mins middle cerebral artery occlusion (MCAO) and subsequent reperfusion for 6 h. Granulocyte-macrophage colony-stimulating factor (10 μg/kg body weight intravenously) was applied 1 h after occlusion. Animals were transcardially perfused with ice-cold Hanks balanced salt solution, dissected on ice, and RNA isolated from ischemic hemispheres. The following primers were used for BCL-Xl amplification: BCL-xL-290s: CAGTTTGGATGCGC GGGAGGTAAT; BCL-xL-520as: AGTGCCCCGCCAAAGGA GAAGAAG.
Immunohistochemistry
After microwave treatment, 2-μm paraffin sections were incubated over night with antibodies against GM-CSF (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA), GM-CSF α-(1:100; Santa Cruz Biotechnology) or β-receptor (1:100; Upstate Biotechnology, Lake Placid, NY, USA), and the appropriate biotinylated secondary antibody (1:200; Dianova, Hamburg, Germany). Staining was visualized using the ABC technique (DAKO, Hamburg, Germany). For immunofluorescence, sections of paraffin-embedded tissues (2 μm) were deparaffinated and microwaved (citrate buffer at 600 W for 15 min). Afterwards, sections were incubated simultaneously with the GM-CSF α-receptor antiserum (1:100; Santa Cruz Biotechnology) and the NeuN antibody (1:100; Chemicon Europe Ltd, UK), the glial fibrillary acidic protein (GFAP) antibody (1:100; Chemicon Europe Ltd) or the CNPase antibody (1:100; Chemicon Europe Ltd) at 4°C over night. After adding a biotinylated anti-rabbit secondary antibody (1:200; Dianova), sections were incubated with Streptavidin-coupled fluorophores (1:200; Invitrogen, Karlsruhe, Germany) and a fluorescence-coupled anti-mouse secondary antibody (1:200; Dianova). For double-immunofluorescence with the GM-CSF ligand and NeuN, GFAP, and CNPase, respectively, the same staining protocol as described for the GM-CSF α-receptor was used. As primary antibodies for the co-staining of the receptors served GM-CSF α-receptor antibody (1:100; Chemicon Europe Ltd) and the G-CSF receptor antibody (1:100; Santa Cruz Biotechnology). The nuclei were counterstained with Hoechst 33342 (1:10,000; Molecular Probes, Invitrogen, Karlsruhe, Germany). Controls for all stainings shown included omission of primary antibodies, fluorophor swapping, and single-fluorescence stainings. Images were obtained with an Olympus IX-81 microscope with narrow-bandwidth monochromator excitation (Polychrome IV, Till Photonics, Gräfelfing, Germany) and appropriate filters.
Cerebral Ischemic Models
Photothrombotic cerebral ischemia was performed as described previously (Kruger et al, 2006). Combined common carotid artery/distal MCAO with 180 min occlusion followed by 72 h reperfusion was done as described (Aronowski et al, 1997) in adult male Long-Evans (Harland Long-Evans, Indianapolis, Ind). Neurological deficite score (0 to 18) in this model was calculated by combining the score on the following 4 tests: Forelimb placing (whisker and forward), footfault, and cylinder test (Schallert et al, 2000). Proximal MCAO with 90 min occlusion and 72 h reperfusion was performed by using the filament occlusion model as described previously (Schabitz et al, 2003) in adult male Wistar rats (Charles River, Sulzfeld, Germany). Treatment was started at indicated times after occlusion onset with 10 μg/kg bodyweight GM-CSF (Leukine®, Immunex/Schering, Berlin, Germany) intravenously in the original buffer in a 2 mL volume over a time period of 20 mins, control animals were treated with buffer solution only. For determining the infarct volume, animals were killed 72 h after MCAO, brains cut into 2 mm-thick coronal slices, and stained with 2% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich, Seelze, Germany). Slides were scanned from both sides, and areas of the contralateral, ipsilateral hemisphere, and the infarct determined using ImageJ (http://rsb.info.nih.gov/ij/). Edema-corrected infarct area was obtained by calculating left hemisphere area—(right hemisphere area—infarct area). Animals with signs of subarachnoid hemorrhage, no, or minimal infarcts (<60 mm3) were excluded before unblinding. Experiments and analyses were conducted in a fully randomized and masked manner. All experimental protocols were approved by the adequate authorities.
Biodistribution In vivo
Granulocyte-macrophage colony-stimulating factor and bovine serum albumin were labeled with 131I (Amersham, Buckinghamshire, UK) by the Iodogen (Sigma) method and purified on Sephadex G-25 (Amersham) columns. The purified proteins co-eluted with the unlabeled standard compounds as a single peak of correct molecular weight when analyzed by size exclusion chromatography. The radiolabeled compounds were injected via the tail vein of female Sprague—Dawley rats (250 to 300 g). Shortly before dissection, rats were anesthetised with Rompun/Ketanest and perfused with saline via the inferior abdominal aorta (at 1, 4, and 24 h after injection). The radioactivity was measured in tissues and serum with a γ-counter (LB 951G, Berthold, Bad Wildbad, Germany) along with a sample of the injectate to calculate %ID/g (percentage injected dose/g) of the tissues.
Primary Neuronal Cultures
Cortices were dissected from rat E18 embryos. The tissue was dissociated using 10 mg/mL trypsin, 5 mg/mL EDTA/DNase (Roche Diagnostics) in Hanks balanced salt solution. Cells were plated at a density of 250,000 cells/well of a 24-well plate or at equivalent density on 6-well plates on glass coverslips coated with poly-
Western Blots
Blots containing 100 μg of neuronal protein were incu bated for 1 h at room temperature with the respective primary (BCL-2 antibody; BD Biosciences, Heidelberg, Germany; all other antibodies from Cell Signaling Technology, Danvers, MA, USA) and secondary antibodies (Dianova). Signals were detected using the supersignal chemiluminescence system (Pierce, Rockford, IL, USA).
Caspase Assay
For Caspase 3/7 assays, we used the human neuroblastoma cell line SHSY-5Y, or primary cortical rat neurons. Cells were seeded in 96-well plates (5 × 104 cells/well) for 2 (SHSY-5Y) or 14 days (neurons). Cells were treated with 150 μM NOR-3 or 20 μM camptothecin for 5 h with or without human (Leukine®, Immunex/Schering) or mouse (R&D Systems, Minneapolis, MN, USA) GM-CSF (20 ng/mL). Caspase 3/7 activity was determined by the Caspase-Glow Assay (Promega, Mannheim, Germany) and luminescence measured with a plate reader (Mithras, Berthold).
Statistical Evaluation
The two-sided t-test, Mann—Whitney rank test, or analysis of variance followed by Student-Newman-Keuls test were used to determine significance as appropriate. A P-value <0.05 was considered statistically significant.
Results
The Receptor for Granulocyte-Macrophage Colony-Stimulating Factor is Induced in Neurons by Cerebral Ischemia and Expressed in many Areas in the Brain
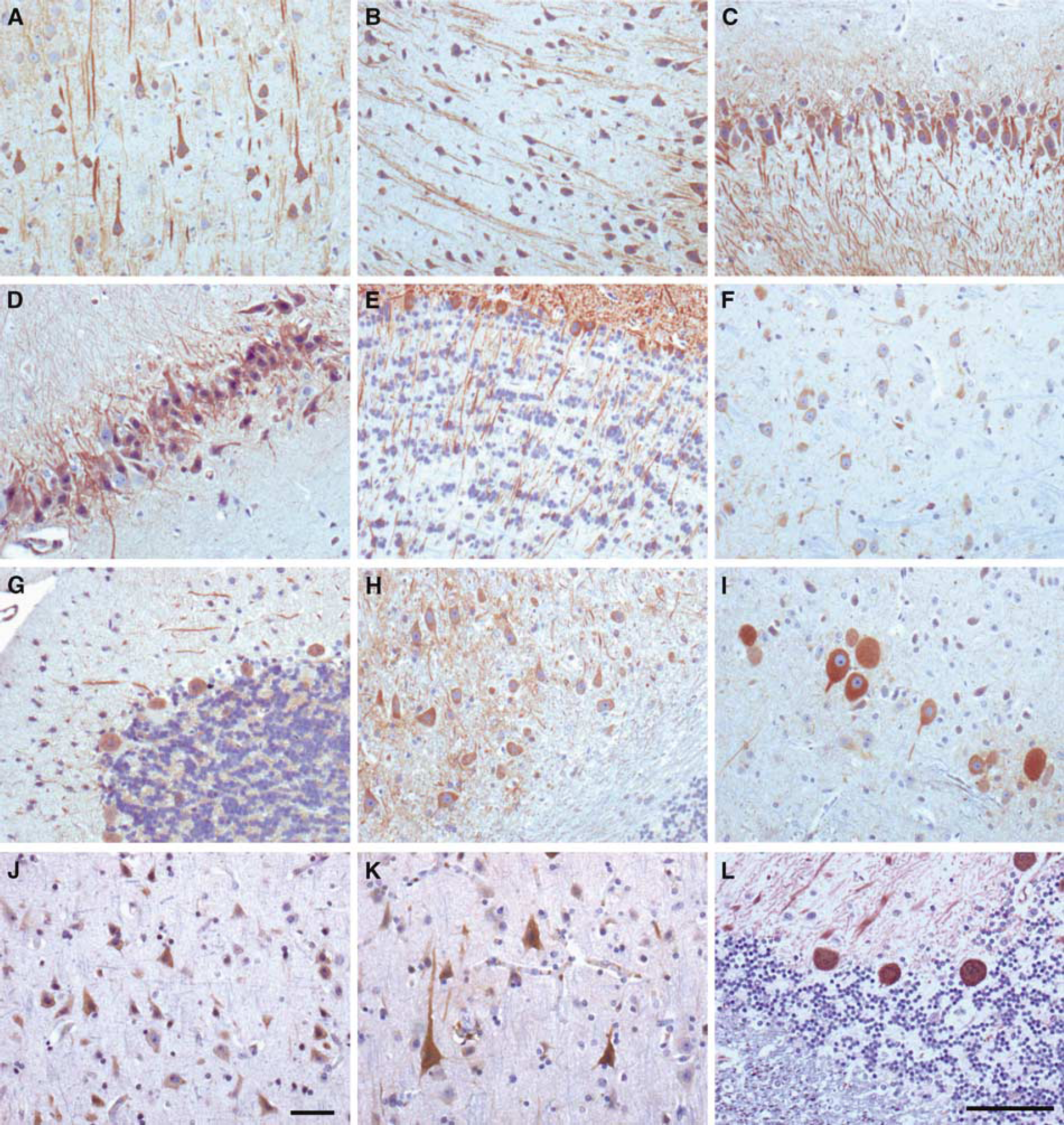
Applying the restriction-mediated differential display transcription profiling technique, we have searched for differentially regulated genes in the perilesional sensorimotor cortex of small cortical infarcts generated with the photothrombotic model (Kruger et al, 2006; Figure 1A). Among a number of other genes, we identified an unknown sequence upregulated at 6 and 48 h after induction of ischemia in the infarct-adjacent cortex. Verification by quantitative PCR revealed induction ratios of approximately 4.5-fold and 3-fold at 6 and 48 h after ischemia, respectively, in the infarct-adjacent cortex (Figure 1B). Full-length cloning revealed this expressed sequence tag to be the rat homologue of the GM-CSF receptor a (69% identity to the mouse GM-CSF receptor a (Figure 1C); submitted to EMBL database, accession number AJ628424). This finding raised our immediate interest in view of the function of two other hematopoietic factors in the brain, EPO (Brines et al, 2000) and G-CSF (Schneider et al, 2005). In contrast to the homo-oligomeric G-CSF receptor, the GM-CSF receptor is composed of an α-subunit that confers specificity, and a common β-subunit, that is also used by the factors IL-3 and IL-5, and classifies as a type 1 transmembrane non-tyrosine kinase receptor. We conducted immunohistochemistry of brains from rats subjected to cortical photothrombotic ischemia in search of the origin of the mRNA signal, and surprisingly detected prominent expression of the α-receptor on large pyramidal neurons in the surrounding area of the infarct (Figures 1D and 1E). Neurons in this area (‘penumbra’) are known to be at risk of dying causing the infarct to enlarge over time. Upregulation of the receptor might therefore signify an adaptive response of these neurons. However, we also noted a broad baseline neuronal expression of the receptor in the non-ischemic brain. Expression mapping of the GM-CSF α-receptor in the uninjured CNS revealed a broad, predominantly neuronal localization in many areas, such as pyramidal cells in the cortex, particularly in layer V (Figure 2A) and layer I/II, the entorhinal cortex (Figure 2B), hippocampus CA2 and CA3 fields (Figures 2C and 2D), mitral cells in the olfactory bulb (Figure 2E), neurons in all thalamic nuclei (Figure 2F), Purkinje cells in the cerebellum (Figure 2G), cerebellar nuclei (Figure 2H), and many brainstem nuclei (e.g., mesencephalic tract V, Figure 2I). This neuronal expression pattern was confirmed both in rat and mouse (not shown), and also in the human brain (Figures 2J to L). The GM-CSF receptor β-chain was also neuronally expressed and co-localized to neurons positive for the α-receptor (Supplementary Figure 1). Apart from neurons (Figure 3A), GM-CSF α-receptor expression in the rat brain was detected in oligodendrocytes by co-immunofluorescence (Figure 3D), whereas no signal was obtained from microglia in the rodent brain (Figure 3C). The vast majority of astrocytes was completely negative for receptor staining (Figure 3B) but few GFAP + cells showed expression (see upper right quadrant in Figure 3B). The preferential neuronal expression of the receptor was also confirmed in the mouse brain by double-fluorescent immunohistochemistry (Supplementary Figure 2). In addition, we controlled this staining pattern by using an independent antibody for the GM-CSF α-receptor (Abcam ab 10670, Cambridge, UK; Supplementary Figure 3). We therefore conclude that the receptor for GM-CSF is preferentially expressed by neurons in a wide variety of brain regions.
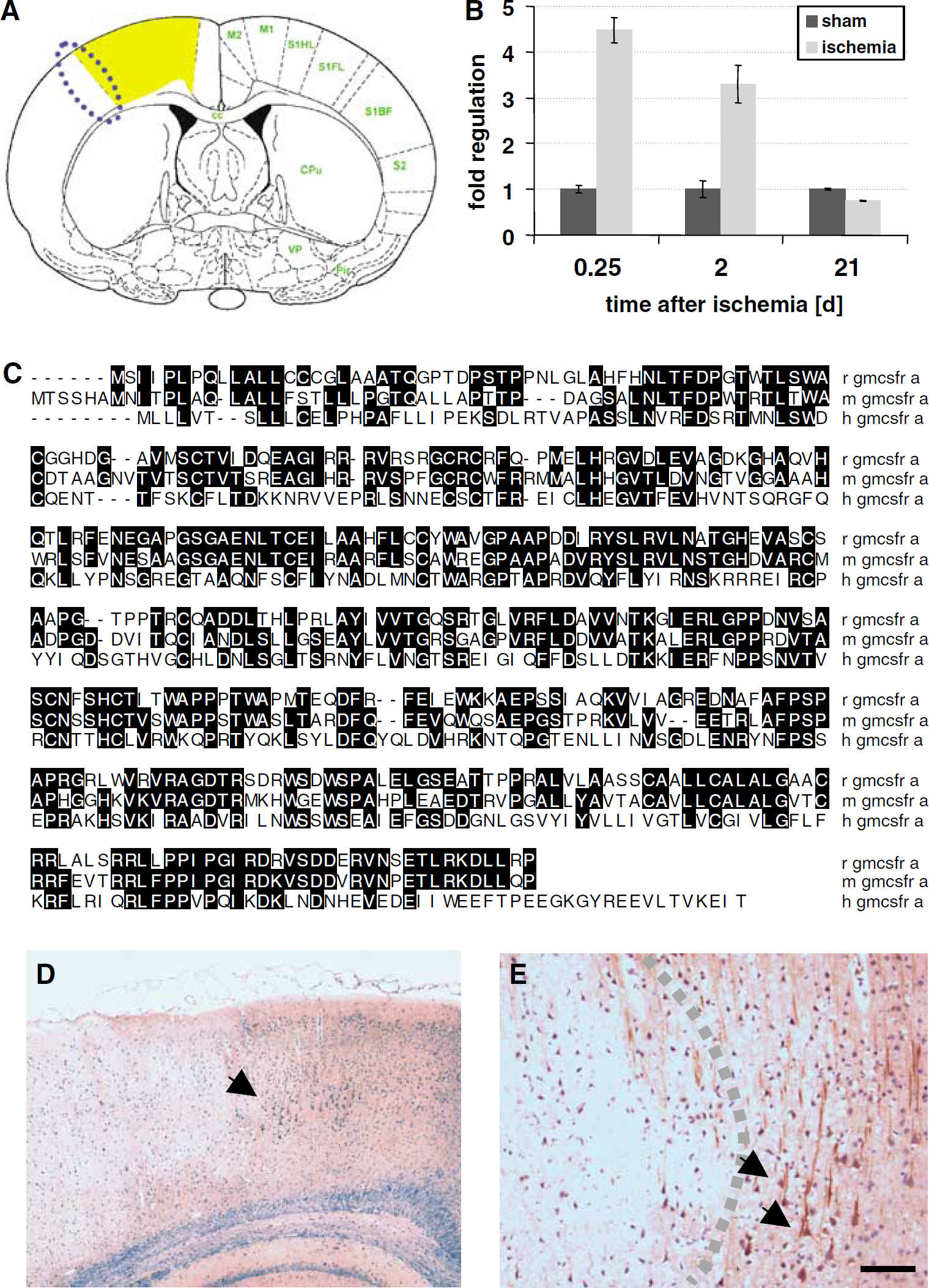
Discovery of the GM-CSF α-receptor as an upregulated gene in the rat photothrombotic model of cerebral ischemia. (
The GM-CSF α-receptor is neuronally expressed in many regions in the CNS in non-ischemic rats: (
Granulocyte-macrophage colony-stimulating factor α-receptor is expressed on neurons and oligodendrocytes in the rat brain. (
Granulocyte-Macrophage Colony-Stimulating Factor is Co-Expressed in Neurons with its Receptor
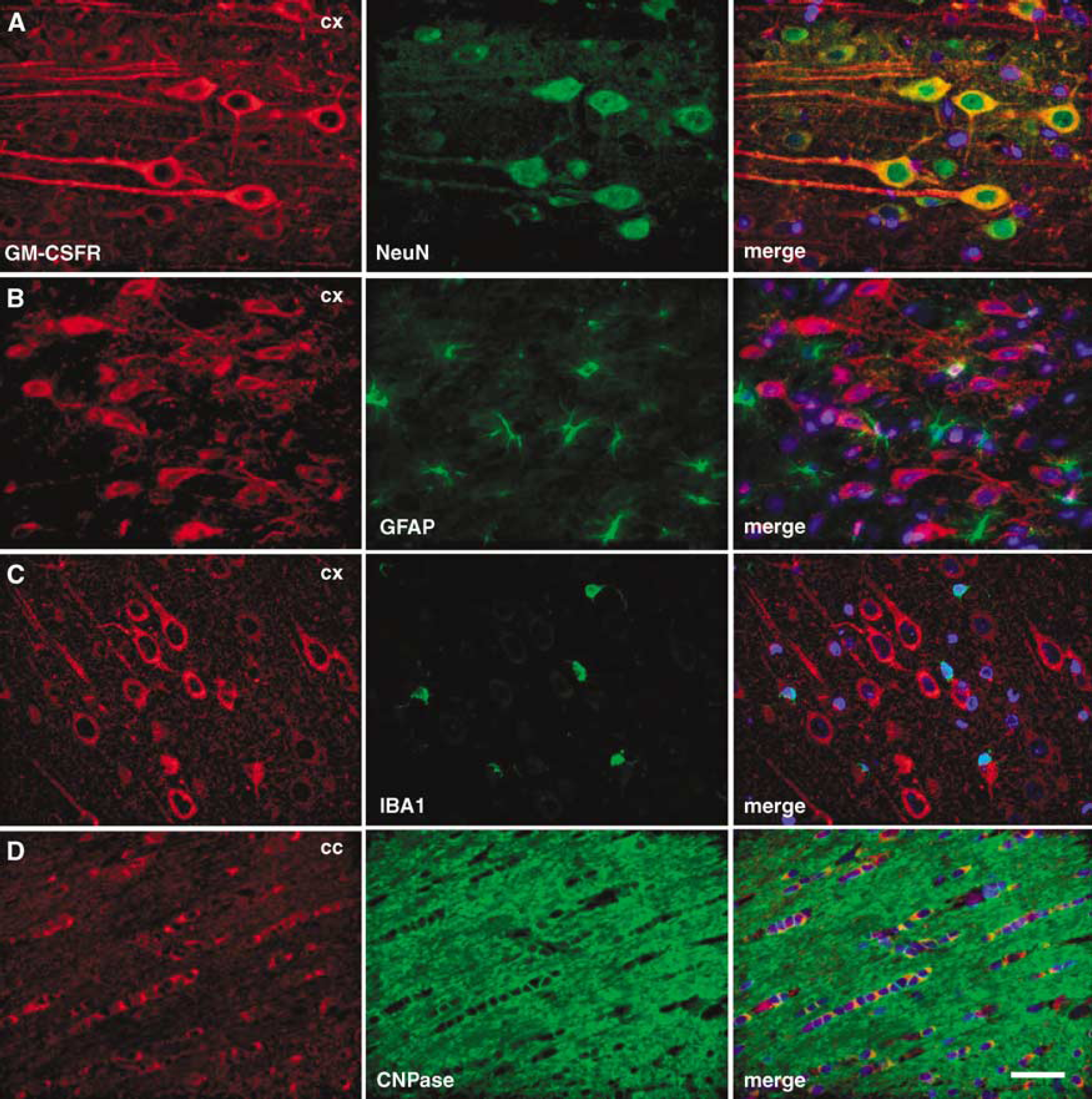
In search of a possible source of the ligand in the CNS, we discovered that GM-CSF was also expressed by morphologically clearly identifiable neurons in the same regions as its receptor (Figures 4A to D). Double-fluorescence co-staining with the neuronal marker NeuN confirmed the predominant neuronal expression of GM-CSF (Figures 4E and 4F). There was no overlap of the GM-CSF signal with microglial cells identified by the marker IBA-1 (Figure 4G). We were not able to detect any staining for GM-CSF in astrocytes in all brain regions examined (Figure 4H). Granulocyte-macrophage colony-stimulating factor was, however, expressed by oligodendrocytes (Figure 4I), but the signal was weaker than the neuronal staining. In summary, GM-CSF is a predominantly neuronally expressed protein in the CNS, with detectable expression in oligodendrocytes. This pattern parallels the expression of the GM-CSF α-receptor. Double immunohistochemistry indeed showed expression of the ligand in neurons expressing the GM-CSF receptor α (Figure 5A), suggesting a partially autocrine neuronal signaling mechanism. This notion was further supported by reactive induction of the ligand in response to cerebral ischemia such as in the rat MCAO model.
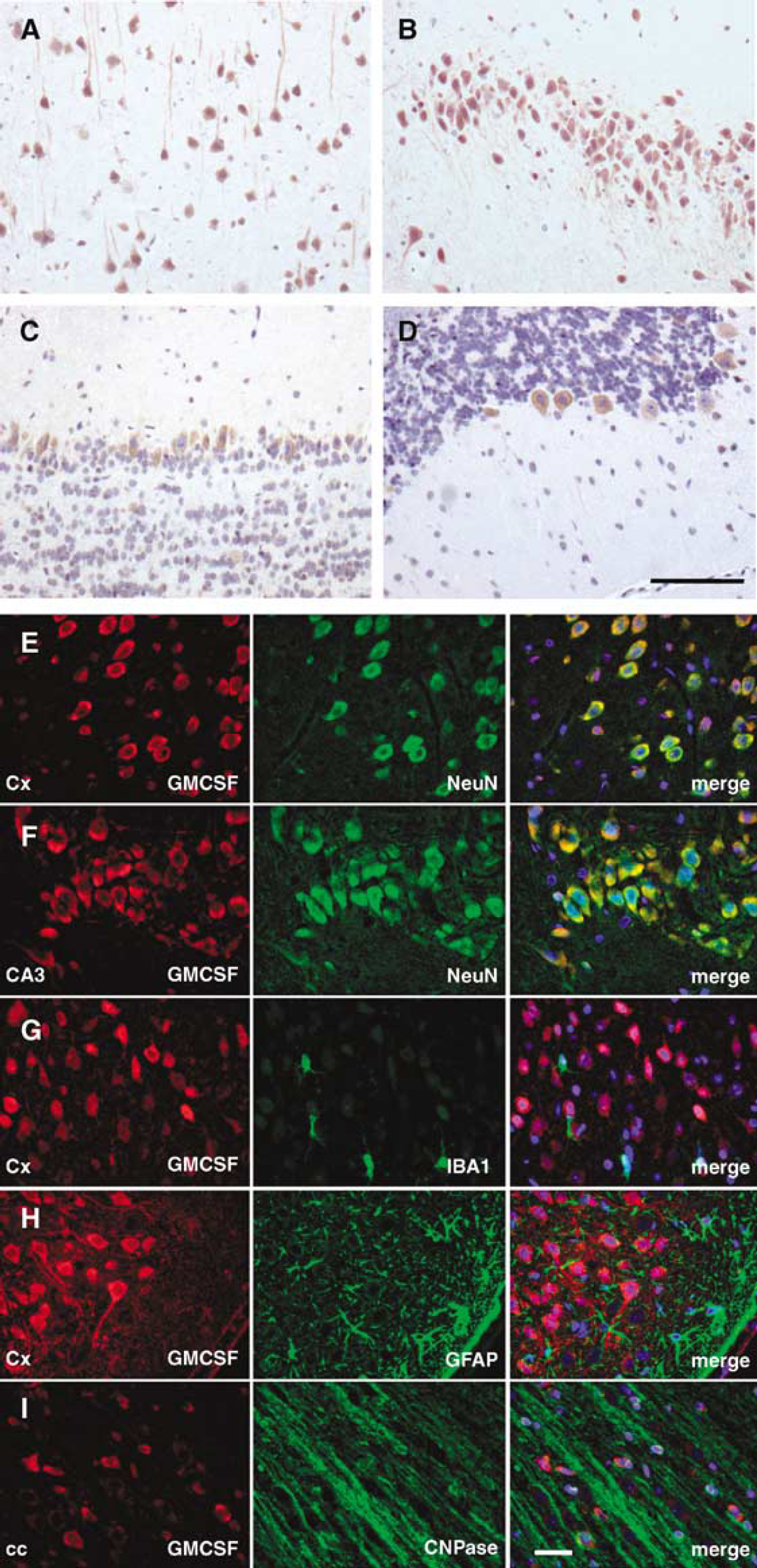
Granulocyte-macrophage colony-stimulating factor is a predominantly neuronally expressed protein in the rat CNS. Granulocyte-macrophage colony-stimulating factor staining was detected in morphologically identifiable neurons in identical CNS regions as its receptor, for example, cortex (
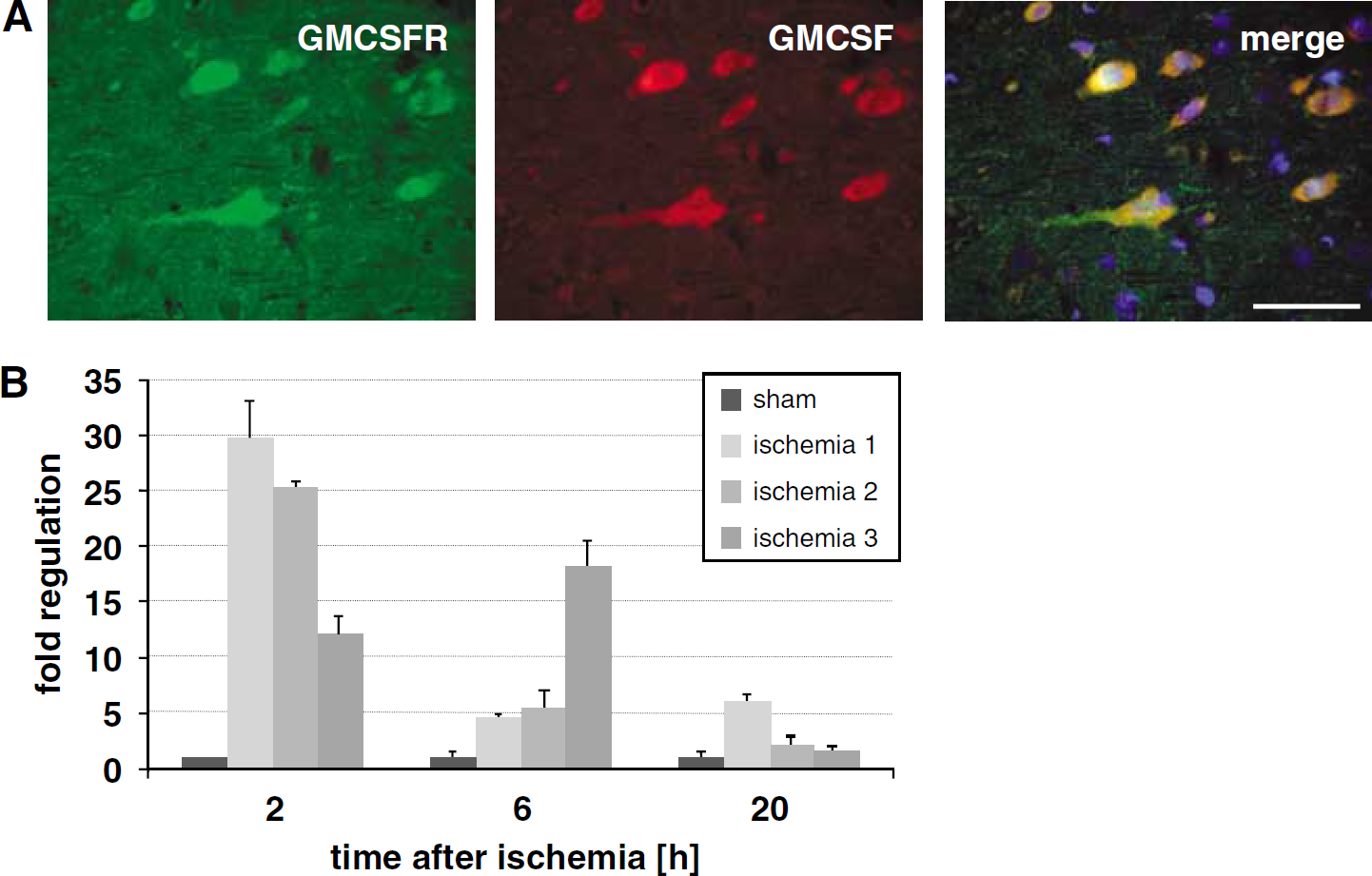
Granulocyte-macrophage colony-stimulating factor co-localizes with its receptor on neurons in the rat brain, and is induced by ischemic stimuli. (A) Granulocyte-macrophage colony-stimulating factor is predominantly expressed by neurons that also express the GM-CSF α-receptor (e.g., cortex, original magnification × 40; scale bar = 50 μm). (
Granulocyte-macrophage colony-stimulating factor induction was time-dependent after ischemia (90 mins filament occlusion) with an initial upregulation after 2 h reperfusion of approximately 20-fold, which dropped strongly after 6 and 20 h (Figure 5B).
Granulocyte-Macrophage Colony-Stimulating Factor Counteracts Programmed Cell Death in Neurons
One of the most important mechanisms in stroke pathophysiology is delayed neuronal cell death. As GM-CSF upregulation in the brain possibly indicates an endogenous adaptive response to harmful ischemic stimuli, and one of its prominent modes of action in the myeloid lineage is inhibition of programmed cell death, we hypothesized that this ligand might be interfering with apoptotic cascades in neurons. In primary neurons from the rat, that were found to express the GM-CSF receptor in culture (not shown), GM-CSF of both mouse or human origin at 20 ng/mL counteracted apoptosis measured by caspase 3/7 activity evoked by the DNA topoisomerase I inhibitor camptothecin (Figure 6A). In view of the broad expression of the GM-CSF receptor in the human brain, we also assayed the human neuroblastoma cell line SHSY-5Y, where GM-CSF also reduced apoptosis, for example, evoked by the nitric oxide donor NOR-3 (Figure 6B). The antiapoptotic activity of GM-CSF was also confirmed in rat primary neurons by direct detection of caspase 3 cleavage by Western blot after challenge with toxic doses of nitric oxide (Figure 6C). Thus, GM-CSF displayed a stable antiapoptotic activity in neuronal cells of rodent or human origin.
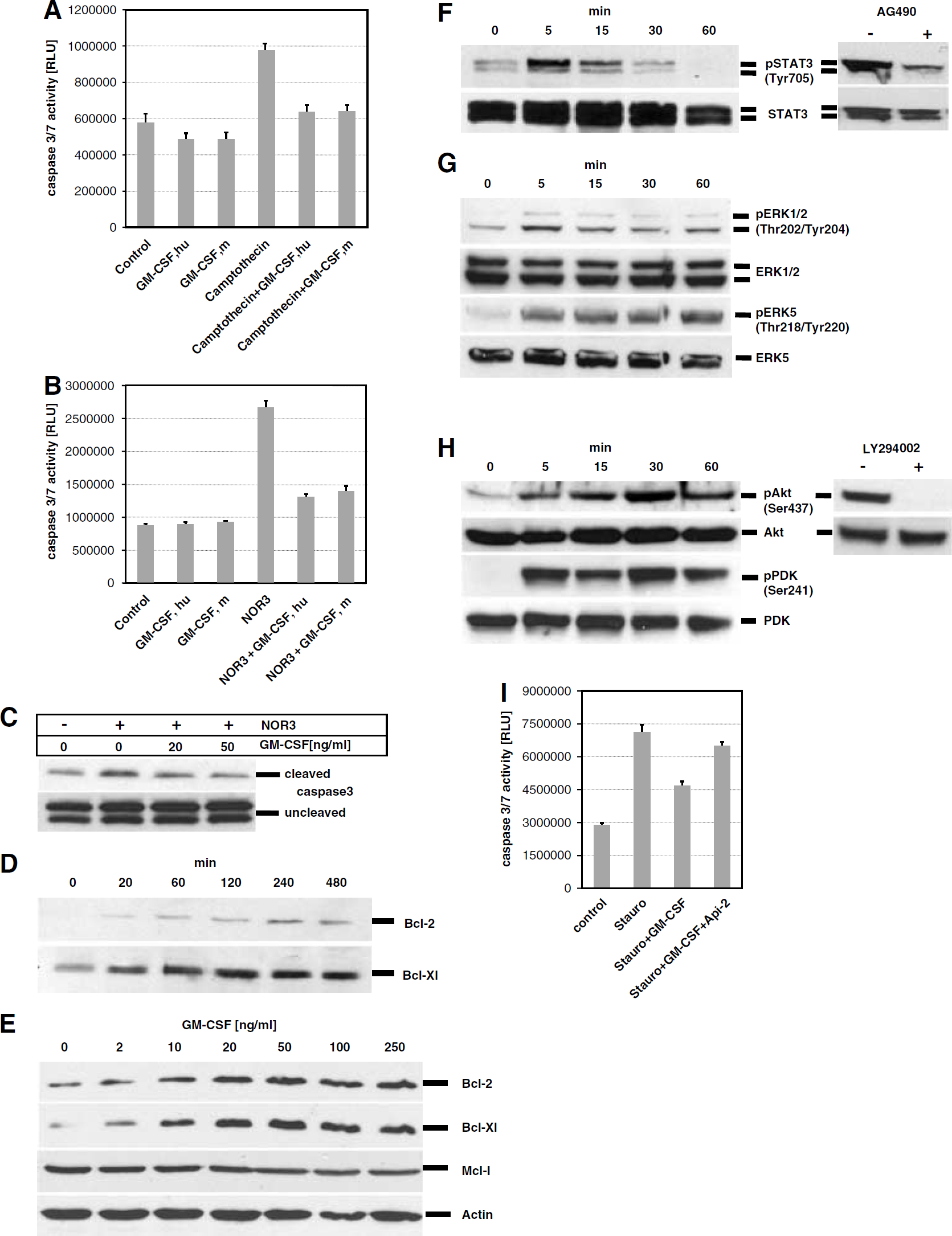
Granulocyte-macrophage colony-stimulating factor acts via its receptor on neurons to induce anti-apoptotic pathways. (
Next, we determined mechanisms responsible for the antiapoptotic profile of GM-CSF on rat primary neurons. We have shown previously that the hematopoietic factor G-CSF induces antiapoptotic proteins of the BCL-family in neurons (Schneider et al, 2005). Addition of GM-CSF to neuronal cultures also led to a substantial time-dependent increase on the protein level of both BCL-Xl and BCL-2, two potent antiapoptotic proteins in neurons with relevance to cerebral ischemia (Figure 6D). We then used BCL-protein induction to determine the dose-dependency of GM-CSF on neuronal induction of antiapoptotic pathways. Increasing doses from 0 to 250 ng/mL, GM-CSF led to an increase in BCL-2 and BCL-xl levels after 24 h with the maximal response reached at 20 and 50 ng/mL, and a subsequent plateau reached at higher levels (Figure 6E). In contrast, another antiapoptotic protein of the BCL family, Mcl-1, did not respond to GM-CSF treatment in a similar way, at least at the time point examined (Figure 6E).
We have previously detected several pathways activated by G-CSF in neurons: STAT3, Erk 1/2 and 5, and PI3K-Akt (Schneider et al, 2005), and therefore asked whether these were also candidates for GM-CSF-induced signaling in neurons. In cells of the hematopoietic lineage, the GM-CSF receptor also activates Janus kinase 2 (JAK2) and signal transducer and transactivator (STAT) proteins (JAK-STAT pathway) (Epling-Burnette et al, 2001). STAT3 was indeed rapidly and strongly phosphorylated 5 mins after the addition of GM-CSF in rat cortical neurons in a time course typical of the JAK-STAT kinetics in other cell types (Figure 6F). The STAT3 tyr705 phosphorylation evoked by GM-CSF was specifically mediated via the GM-CSF receptor/JAK2 pathway, as blocking of the JAK2 kinase by the inhibitor AG490 led to drastically reduced signals for phosphorylated STAT3 in the presence of GM-CSF (Figure 6F, right side). Granulocyte-macrophage colony-stimulating factor also induced rapid but transient activation of Erk 1/2 by phosphorylation (Figure 6G). In contrast, activation of Erk 5 appeared stronger and stable for at least 1 h after GM-CSF addition (Figure 6G, lower panels).
One of the most potent antiapoptotic signal transduction pathways in neurons that can be activated by a number of growth factor receptors is the PI3-kinase/Akt pathway. Granulocyte-macrophage colony-stimulating factor led to a rapid and drastic increase in Ser473-phosphorylated active Akt, which was stable for at least 1 h after GM-CSF addition (Figure 6H). Parallel to the induction of Akt, we observed phosphorylation of the upstream kinase PDK1 (Figure 6H, lower panels). The phosphorylation of Akt 5 mins after GM-CSF addition could be completely blocked by the PI3-kinase inhibitor LY294002 (Vlahos et al, 1994), suggesting that activation of Akt by GM-CSF is mediated by the canonical PI3K—PDK1 pathway (Figure 5H, right side). Among the different pathways activated by GM-CSF in neurons, Akt appears as the strongest and least disputed antiapoptotic signal. Indeed, blocking activated Akt kinase function by the specific inhibitor Api-2 (Yang et al, 2004) led to a loss of 75% of GM-CSF antiapoptotic potency after challenge with the broad-spectrum apoptosis inducer staurosporine (relative protection GM-CSF: 58%, relative protection GM-CSF + Api-2: 14%; Figure 6I).
Collectively, our results indicate that GM-CSF has substantial antiapoptotic activity in neurons against a number of stimuli, which is partially mediated by activation of the Akt pathway.
Granulocyte-Macrophage Colony-Stimulating Factor is Neuroprotective In vivo
In view of this potent antiapoptotic activity, we sought to determine whether GM-CSF had neuroprotective efficacy in experimental stroke. To test whether we could apply this protein by a clinically feasible route, we determined permeability of the intact blood—brain barrier (BBB) in healthy, nonischemic rats by measuring distribution rates of iodinated GM-CSF versus serum albumin, which does not cross the BBB. A significantly higher portion of the GM-CSF present in the serum was found in the brain after perfusion compared with bovine serum albumin (n = 4 per group, P <0.05; Figure 7A) suggesting that GM-CSF passes the intact BBB, a result in concordance with a previous report that shows saturable transport of GM-CSF across the BBB (McLay et al, 1997). Thus we chose to apply GM-CSF intravenously.
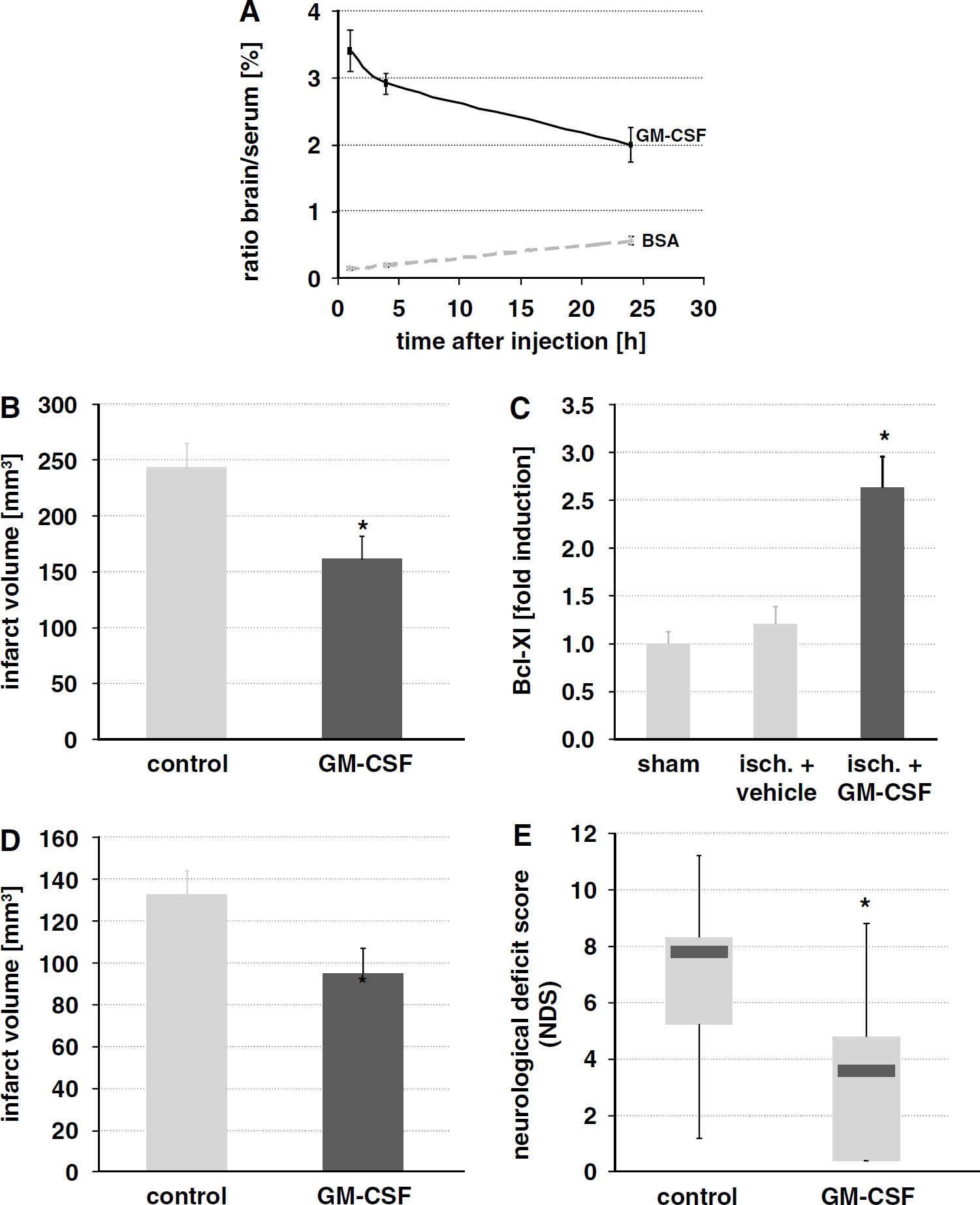
Granulocyte-macrophage colony-stimulating factor passes the intact blood—brain barrier (BBB) in healthy rats, and protects against focal cerebral ischemia. (
We performed MCAO in the rat, the most standard model of human stroke in the MCA territory, with 90 mins occlusion and 72 h reperfusion time, to exclude possible transient effects on infarct volume after evaluation at 24 h after stroke. We applied a dose of 10 μg/kg GM-CSF intravenously at 1h after onset of ischemia as a short infusion (20 mins). Granulocyte-macrophage colony-stimulating factor treatment led to significantly smaller infarcts (vehicle: n = 22; GM-CSF: n = 18; 34% reduction, Figure 7B). To link this effect to the antiapoptotic function of GM-CSF in vitro, we determined mRNA levels of BCL-Xl 6 h after MCAO induction in the ischemic hemisphere. Granulocyte-macrophage colony-stimulating factor treatment triggered a significant increase in BCL-Xl expression (approximately 2.5-fold increase by quantitative PCR; sham: n = 6; ischemia + vehicle: n = 3; ischemia + GM-CSF: n = 6; P <0.05 by analysis of variance and SNK post hoc test; Figure 7C).
Also in the combined common carotid artery/distal MCAO model in the rat (180 mins occlusion time) (Aronowski et al, 1997), GM-CSF diminished infarct size to a similar degree when treatment was initiated 1 h after onset of ischemia, and infarct size determined after 72 h (vehicle: n = 10; GM-CSF: n = 9; 34% reduction, Figure 7D). Moreover, sensorimotor performance in the common carotid artery/distal MCAO model was significantly improved as determined by the neurological deficit score (Schallert et al, 2000) (median control: 7.8; median GM-CSF group: 3.6; P < 0.05 by Mann—Whitney rank-sum test; Figure 7E). There was no influence of GM-CSF treatment on mortality, systemic arterial blood pressure, or cerebral blood flow as determined by laser-Doppler flowmetry (Supplementary Table 1). In conclusion, intravenously given GM-CSF has a robust neuroprotective activity in two different stroke models.
Discussion
Here, we have shown that GM-CSF and its receptor are co-expressed by a wide variety of neurons in the CNS, induced on ischemic events in neurons at risk, and provide protection against experimental stroke. In neurons, GM-CSF counteracts programmed cell death. Our findings thus place the hematopoietic factor GM-CSF among a number of important neurotrophic factors, and suggest that expression and functioning in the CNS is a common feature of many hematopoietic factors.
A number of literature data support our data on the neuronal expression of the GM-CSF receptor in the CNS. Granulocyte-macrophage colony-stimulating factor receptor expression has been noted in neurons in the fetal human brain (Dame et al, 1999), supporting the notion that GM-CSF likely also has a neurodevelopmental role. Neuronal staining has been reported in the adult spinal cord (Ha et al, 2005). Recently, presence of the GM-CSF receptor on hypothalamic neurons in the adult brain has been described (Reed et al, 2005). The low-level expression of GM-CSF α-receptor observed on oligodendrocytes in our study is in line with one paper where radioligand binding on cultured glial cells was performed (Baldwin et al, 1993). In contrast to our findings, earlier reports also suggest expression of the GM-CSF receptor on astrocytes (Guillemin et al, 1996), or production of GM-CSF by stimulated astrocytes (Malipiero et al, 1990). Likewise, studies have shown reactivity of microglia in vitro to GM-CSF (Suzumura et al, 1990). In our stainings of rodent or human brain, we could not detect significant astrocytic or microglial expression of the receptor except for very few astrocytes. It is therefore possible that in vitro cultivation induces expression of the receptor, or that some of the observed in vitro effects may have been indirect. Alternatively, the level of expression may be below the detection limit of our immunohistochemical protocol.
The GM-CSF ligand had an identical expression profile, and indeed co-localized to neurons expressing its receptor. Also, GM-CSF was upregulated on ischemia to more than 20-fold in the standard MCAO model after 2 h of reperfusion. Induction by ischemic stimuli is supported by a report of a sixfold induction in the photothrombotic model (Kleinschnitz et al, 2004), and parallels comparable behavior of two other hematopoietic factors in the brain, EPO (Hasselblatt et al, 2006), and G-CSF (Schneider et al, 2005). In the MCAO model, the maximal 20-fold induction observed remains below the induction of G-CSF (> 150-fold, Schneider et al, 2005), however, it is difficult to derive conclusions as to the relative significance of both factors from this observation. Together with the antiapoptotic action of GM-CSF in vitro, and the localization to GM-CSF receptor expressing neurons this upregulation suggests a partially autocrine protective signaling mechanism in neurons, as has been suggested for a number of neurotrophins such as FGF-2 (Desire et al, 2000), BDNF, or NT-3 (Horton et al, 2001). The pathways activated by GM-CSF in neurons are both reminiscent of the basic function of GM-CSF in inhibiting apoptosis in cells of the myeloid lineage as well as to signals evoked by G-CSF in neurons (Schneider et al, 2005), underlining the similar profiles of both factors in neurons. Granulocyte-macrophage colony-stimulating factor most strongly induced the PI3kinase-Akt pathway, which is also considered critical for the antiapoptotic effects of GM-CSF in the myeloid lineage (Klein et al, 2000).
Our findings of efficacy of GM-CSF in experimental stroke are supported by a recent report by Nakagawa et al (2006). The authors applied GM-CSF at onset of ischemia by intracarotid injection in a milder rat MCAO model (60 mins occlusion), and find substantial infarct reduction. Support for a neuroprotective action of GM-CSF in the CNS also comes from another disease model, spinal cord contusion (Ha et al, 2005). The authors could show effects of GM-CSF treatment (20 μg/day for 5 days) on the improvement of the BBB score, and show decreased apoptosis in the spinal cord.
The protective effect of GM-CSF in the CNS is most likely caused by its direct action on neurons and possibly oligodendrocytes: GM-CSF has direct antiapoptotic activity on neurons in vitro, its receptor is predominantly expressed on neurons in the brain, GM-CSF passes the BBB, and induces BCL-Xl expression in the brain within 6 h after application. Moreover, neuronal co-expression of ligand and receptor strongly suggests that induction of the ligand after injury serves as a protective autocrine mechanism. However, we cannot exclude at present that there are other mechanisms, which also have a role in the observed effects. One is modulation of the systemic inflammatory response and immunodeficiency that play an important pathophysiological role in cerebral ischemia (Meisel et al, 2005), and which GM-CSF may influence positively. Another potential mechanism of action may be the mobilization of bone-marrow derived stem cells that can invade the infarcted brain and have beneficial activity (Li et al, 2001). A recently discovered activity of GM-CSF that is not of relevance in the acute effects studied here but in long-term recovery effects of GM-CSF after stroke is enhancement of arteriogenesis (Buschmann et al, 2003; Schneeloch et al, 2004).
We describe a function for GM-CSF in a pathophysiological setting. The physiological function(s) of GM-CSF in the healthy CNS remain obscure at present. Interestingly, a recent report suggests involvement of GM-CSF in the regulation of food intake behavior, and body fat content (Reed et al, 2005). The authors describe effects of centrally, but not peripherally administered GM-CSF on food intake owing to the hypothalamic expression of the GM-CSF receptor a. The reason for the apparent failure of peripherally given GM-CSF are not fully clear in light of the intact BBB passage of GM-CSF, but may possibly reflect dependency on rather high GM-CSF doses of this effect. The authors also report that only GM-CSF of murine, but not human origin was effective. This is in contradiction to our data, and to data from a number of other labs that show efficacy of human GM-CSF in rodents in vivo (Buschmann et al, 2003; Ha et al, 2005; Schneeloch et al, 2004). A possible explanation may be the use of non-glycosylated human GM-CSF by the authors, whereas we and others have used glycosylated GM-CSF. The described effects on food intake and body weight regulation are unlikely a hindrance to novel clinical applications of GM-CSF, for example, in stroke. In the clinical situation, even long-term application of GM-CSF does not to influence food intake or body weight negatively, as exemplified by a recent trial in Crohn's disease, a condition especially prone to weight loss (Korzenik et al, 2005). Finally, the paraventricular nucleus does not display a particularly strong staining for the receptor in contrast to neurons in other brain areas, for example, in the cortex (data not shown). Taking together the above points and our data, we would therefore argue that the role in food intake regulation is unlikely the sole or major physiological function of GM-CSF and its receptor in the CNS.
Another hematopoietic factor, EPO, has a broad expression in the CNS, crosses the BBB, and has been established as a potent neuroprotective factor (Hasselblatt et al, 2006), thus paralleling characteristics of GM-CSF established here. It has been suggested that a specific neuronal receptor may be responsible for the neuroprotective action of EPO that consists of a heterodimer of the EPO receptor and the GM-CSF β-receptor (Brines et al, 2004), suggesting a possible inherent connection of the neuroprotective properties of both proteins. Recently, we have uncovered the neuroprotective potential of G-CSF (Meuer et al, 2006; Schabitz et al, 2003; Schneider et al, 2005, 2006; Schabitz and Schneider, 2007). The parallels in expression, reactivity, and function of these two factors are surprising: Both react to injury by upregulation, have an antiapoptotic function on neurons, trigger an identical subset of intracellular signaling pathways, and are co-expressed with their receptors. Thus, these proteins and their receptors have apparently co-evolved for distinct roles in the hematopoietic and nervous system.
Hematopoietic growth factors are attractive drug candidates for stroke treatment, mostly because of their long history of safe use in humans, and their multimodal acitivities as neurotrophic factors (Rogalewski et al, 2006). Indeed, multiple clinical studies in stroke patients are ongoing for both EPO and G-CSF (Bath and Sprigg, 2006; Schabitz and Schneider, 2006). It is highly probable that GM-CSF plays a similar role in the human brain as in the rodent system: the GM-CSF receptor is expressed in the human brain in a similar pattern to the rodent, GM-CSF has antiapoptotic activity on human neuroblastoma cells, and, finally, GM-CSF appears induced by stroke intrathecally in human patients (Tarkowski et al, 1997). Granulocyte-macrophage colony-stimulating factor is a protein that is in clinical use for many years now, and is well tolerated. Moreover, GM-CSF acts protective at dose ranges in animals that appear realistic from the clinical point of view, especially relating to tolerance issues. Granulocyte-macrophage colony-stimulating factor therefore provides a valuable complementation of the repertoire of hematopoietic factors in development for stroke treatment, as all of these proteins have a complex set of potential advantages and disadavantages for stroke treatment.
Further pre-clinical and clinical studies are needed to define the profile of each protein in stroke, and select the most promising candidate(s) for further development.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.